Embed Size (px)
Citation preview

Affections de l’artère pulmonaireS MagnierJM LupoglazoffA Casasoprana
Résumé. – Les affections de l’artère pulmonaire regroupent deux types d’anomalies, les unesphysiopathologiques avec pour conséquence l’hypertension artérielle pulmonaire, les autres anatomiquesavec les obstacles sur l’arbre artériel pulmonaire, les agénésies partielles de la voie artérielle pulmonaire et ladilatation idiopathique du tronc pulmonaire. Les plus fréquentes de ces affections sont les pathologiesprimitives ou secondaires, responsables du développement d’une l’hypertension artérielle pulmonaire. Lepronostic de celle-ci, variable selon son étiologie lorsqu’elle est connue, reste très sévère du fait de lésionsartériolaires distales souvent définitives. Ces 20 dernières années ont été marquées par les progrès de lachirurgie précoce, avant l’âge de 2 ans, des cardiopathies malformatives avec débit pulmonaire élevé,génératrices autrefois de l’évolution des ces malformations vers le développement d’une l’hypertensionartérielle pulmonaire fixée irréversible. Plus récemment, l’apparition de vasodilatateurs artériels pulmonaires,parfois très puissants et sélectifs (prostacycline) permet de traiter des patients atteints d’hypertensionartérielle pulmonaire, notamment primitive, et ces traitements peuvent constituer une alternative à latransplantation cardiopulmonaire dont les complications à court et moyen termes sont encore nombreuses etsévères.Les anomalies anatomiques de l’artère pulmonaire de type malformatif ou acquis après chirurgie palliativedes cardiopathies congénitales sont de diagnostic simple, avec les progrès de l’imagerie par scanner,résonance magnétique nucléaire et angiographie. Leur traitement reste difficile chirurgicalement, mais lesprogrès des techniques interventionnelles d’angioplastie laissent envisager de meilleurs résultatsthérapeutiques encore à évaluer.© 2000 Editions Scientifiques et Médicales Elsevier SAS. Tous droits réservés.
Mots-clés : hypertension artérielle pulmonaire, hypertension artérielle pulmonaire primitive, cardiopathiescongénitales, maladie thromboembolique, prostacycline, sténoses des branches artériellespulmonaires.
IntroductionLes pathologies de l’artère pulmonaire (AP) sont variées, intéressantla physiologie artériolaire pulmonaire pour la plupart d’entre elles,et l’anatomie artérielle proximale dans un nombre assez rare de cas.Les anomalies de la physiologie artériolaire pulmonaire sont àl’origine de l’hypertension artérielle pulmonaire primitive (HTAPP),ou plus souvent secondaire soit à des anomalies malformativescardiaques, soit à des anomalies acquises parenchymateusespulmonaires, soit en rapport avec la circulation pulmonairetransitionnelle néonatale modifiée par des conditions pathologiquesspécifiques à cette période de la vie.Les anomalies anatomiques artérielles pulmonaires sont proximaleset plus souvent congénitales malformatives qu’acquises.La circulation pulmonaire permet l’artérialisation du sang veineuxmêlé grâce aux échanges alvéolocapillaires.
Anatomie normale de la circulationpulmonaireLes poumons ont une double vascularisation, artérielle pulmonaireà basse pression et bronchique à pression systémique [17, 34].
Suzel Magnier : Praticien hospitalier.Jean-Marc Lupoglazoff : Attaché des Hôpitaux, assistant des Universités.Antoine Casasoprana : Professeur des Universités, praticien hospitalier.Service de cardiologie infantile, hôpital Robert Debré, 48, boulevard Sérurier, 75019 Paris, France.
L’arbre artériel pulmonaire s’étend des valves pulmonaires auxcapillaires.
ARTÈRE PULMONAIRE
¶ Artère pulmonaire élastiqueElle comprend deux segments extrapulmonaire et intrapulmonaire.
¶ Segment extrapulmonaireLe calibre du tronc pulmonaire est voisin du calibre aortique, avecune épaisseur pariétale qui est de l’ordre de 60 à 75 % de l’épaisseurde la paroi aortique. Le tronc et les branches artériellesextrapulmonaires sont de type élastique. L’intima, à ce niveau,comporte une seule couche cellulaire, et l’adventice est riche en vasavasorum. L’athérome peut toucher le tronc pulmonaire et descalcifications peuvent survenir, mais de façon moins rapide que surl’aorte.À l’âge fœtal, la structure de l’AP est identique à celle de l’aorte,mais après la naissance, le tissu élastique est remplacé par ducollagène en 4 à 12 mois et la lame élastique interne se fragmente.
¶ Segment intrapulmonaireLes branches artérielles pulmonaires secondaires gardent unestructure identique mais n’ont pas jusqu’à 1 000 µm defragmentation de la lame élastique interne. La transition entre artèresde types élastique et musculaire se fait entre 1 000 et 500 µm decalibre normal.
Ency
clop
édie
Méd
ico-
Chi
rurg
ical
e1
1-3
12
-A-1
0
11-312-A-10
Toute référence à cet article doit porter la mention : Magnier S, Lupoglazoff JM et Casasoprana A. Affections de l’artère pulmonaire. Encycl Méd Chir (Editions Scientifiques et Médicales Elsevier SAS, Paris, tous droits réservés),Cardiologie, 11-312-A-10, 2000, 15 p.

ARTÈRES MUSCULAIRES ET ARTÉRIOLES
Entre 500 et 70 µm les artères et artérioles pulmonaires sont de typemusculaire, comportant une média faite de petits muscles arrangésde façon circulaire de proche en proche. Entre les muscles serépartissent des fibres de collagène et de réticulum visibles aumicroscope optique et électronique. Le calibre de la média estexprimé en pourcentage du diamètre externe du vaisseau,normalement 5 % pour les artérioles musculaires pulmonaires et15 à 25 % pour les artérioles systémiques. L’adventice est faite detissu fibreux dont l’épaisseur est deux à trois fois celle de la média.Il y a peu de différences de structure suivant le lobe pulmonaire oule sexe. Le passage aux artérioles pulmonaires, ne comportant pasde muscles, se fait progressivement à partir de 70 µm. Pendant lavie fœtale, les artérioles pulmonaires sont de type systémique, avecune média dont l’épaisseur est de 15 à 20 %. Après la naissance, lorsde l’ouverture de la lumière vasculaire, l’épaisseur de la médiadiminue.
CAPILLAIRES PULMONAIRES
Ils réalisent l’aspect d’un filet dont les mailles décrivent un hexagonede 10 à 15 µm de côté et de 8 µm de diamètre. Plus large que lecapillaire systémique, le capillaire pulmonaire est séparé de l’alvéoleadjacente par une paroi très fine (1,8 µm), faite de cinq couches dontles deux extrêmes sont formées de membranes séparées par ducollagène.L’arbre vasculaire pulmonaire peut être le siège d’anastomoses :l’existence de connexions entre les branches artérielles précapillaireset les veines pulmonaires est controversée. En revanche, il existe desanastomoses entre la vascularisation bronchique et pulmonaire, etdes anastomoses de petite taille (inférieures à 250 µm) à l’étageartériel ; elles seraient plus fréquentes chez le petit enfant que chezl’adulte.La croissance pulmonaire [26] répond à un processus de maturationdébutant précocement et se faisant en quatre étapes : embryonnaire(0-8 semaines), fœtale (9-40 semaines), périnatale, et pendant lapetite enfance. Les bourgeons pulmonaires proviennent del’endoderme et du mésendyme, et aboutissent à la 24e semaine,après formation glandulaire et canaliculaire, aux sacs terminaux.Dans le même temps, les voies respiratoires se forment etdébouchent sur les alvéoles qui constituent les unités fonctionnellesrespiratoires. Les pneumocytes recouvrent les alvéoles et produisentle surfactant. À la naissance, le nombre d’alvéoles fonctionnellesreprésente un dixième du nombre présent à l’âge adulte. Ce nombreaugmente jusqu’à l’âge de 8 ans environ, et au-delà la croissancealvéolaire, ne se fait plus que par hypertrophie. Pendant la viefœtale, le poumon est un organe doué d’une activité métabolique(production de surfactant, production de liquide intrapulmonaire),avec un système lymphatique développé et fonctionnel [84].
ENDOTHÉLIUM VASCULAIRE PULMONAIRE
L’endothélium vasculaire pulmonaire existe dès la période fœtale,avec une immaturité à exercer un pouvoir relaxant sur les vaisseaux(fig 1).Le rôle central de la cellule endothéliale [27] dans la régulation de lavasomotricité vasculaire a été récemment reconnu. Cette cellulelibère divers médiateurs vasodilatateurs, comme la prostacycline etle monoxyde d’azote (NO) [54].La biosynthèse du NO [33, 90] est sous la dépendance d’une familled’enzymes, les NO-synthétases (NOS), dont il existe trois isoformesqui diffèrent entre elles par leurs fonctions, leurs localisationscellulaires et leurs caractéristiques biochimiques. Certaines NOS sontconstitutives, normalement présentes à l’état physiologique. Leurexpression est régulée par une augmentation transitoire de calciumintercellulaire et sont à l’origine de la production de faibles quantitésde NO pendant une brève période.L’isoforme inductible de NOS ne se manifeste qu’à l’occasion d’étatspathologiques, tels que le choc septique, par l’intermédiaire
d’endotoxines bactériennes et de cytokines. Les isoformes de NOSsont codées par trois gènes distincts localisés sur les chromosomes7, 12 et 17.Le NO diffuse rapidement à la face luminale de l’endothélium enempêchant l’adhésion des plaquettes à la paroi musculaire etl’agrégation plaquettaire. Il diffuse également vers la cellulemusculaire lisse où, par une suite de réactions biochimiques faisantintervenir des phosphodiestérases et l’ouverture de canauxpotassiques, cela aboutit à une vasodilatation artérielle et veineuse.Pendant la vie fœtale, le tonus vasculaire pulmonaire est élevé dufait d’une activité importante des phosphodiestérases et de laprésence, dans le poumon fœtal, de médiateurs vasoconstricteurss’opposant à la vasodilatation pulmonaire induite par l’activité NOendogène. Les médiateurs ont un effet dominant en milieu privéd’oxygène.À la naissance, la capacité fonctionnelle respiratoire s’établit enquelques cris s’accompagnant d’une augmentation du débitpulmonaire de huit à dix fois en quelques systoles, et d’unediminution progressive des résistances vasculaires pulmonaires pourpermettre au poumon d’assurer les échanges gazeux. De multiplesstimuli contribuent à la diminution normale des résistancesvasculaires pulmonaires [89] : distension pulmonaire rythmique,drainage du liquide intrapulmonaire, interface liquide-gaz etaugmentation de l’oxygénation alvéolaire et artérielle. Le stimulusprépondérant de la libération d’endogène (EDNO) semble bien êtrel’augmentation du débit sanguin pulmonaire et les contraintesmécaniques induites sur la cellule endothéliale.
Moyens d’explorationde la circulation pulmonaire
HÉMODYNAMIQUE NORMALE
¶ CathétérismeL’étude de référence de la circulation pulmonaire se fait par lecathétérisme, dont les indications ont été réduites par l’apport del’échocardiographie couplée au doppler dans l’hémodynamique noninvasive.La circulation pulmonaire se fait à basse pression, de l’oreillettedroite (OD) à l’oreillette gauche (OG), avec pour intermédiaire lepassage capillaire pulmonaire.Les travaux de Forssman (1929) et Cournand (1941) ont permis, parcathétérisme veineux, de mesurer les pressions depuis les veinescaves jusqu’au capillaire bloqué.
Pressions
Les pressions droites normales sont en millimètres de mercure(mmHg) :
cellule endothéliale
Médiateurs circulants
Facteurs de la celluleendothéliale
Facteurs dilatateurs Facteurs constricteurs
nitrite d'azote
prostacycline
endothéline
canaux potassiques
thromboxane
Thromboxane
cellule musculaireCa2+
Sérotonineplaquettes
1 Mécanismes possibles de la pathogénie de l’hypertension artérielle pulmonaire [30].
11-312-A-10 Affections de l’artère pulmonaire Cardiologie
2

– pression OD : moyenne 0
– pression ventricule droit (VD) 20/0-1
– pression AP (PAP) 20/5 10
– pression capillaire pulmonaire (PCP) : 5
– pression OG : 5Elles sont basses en regard des pressions gauches mesurées parcathétérisme artériel rétrograde :
– ventricule gauche (VG) 120/0-5
– aorte 120/70 85.La PAP diastolique (PAPd) est, normalement, au niveau de lapression télédiastolique du VG, elle-même au niveau de la PCP.L’existence d’un obstacle fonctionnel ou anatomique situé dans le litvasculaire pulmonaire se traduit par la présence d’un gradient depression entre le chiffre diastolique artériel pulmonaire et la PCP,auriculaire gauche et télédiastolique ventriculaire gauche, selon lesiège de l’obstacle.Seule la pression diastolique artérielle pulmonaire reflète fidèlementl’état des résistances au débit pulmonaire, et l’obstacle est :
– précapillaire si la pression capillaire est normale et la pressiondiastolique de l’AP supérieure à la PCP ;
– postcapillaire si la pression capillaire est augmentée et la pressiondiastolique de l’AP égale à la PCP ;
– mixte si la pression capillaire est augmentée et la pressiondiastolique de l’AP supérieure à la PCP.Le degré de réversibilité de l’élévation des résistances pulmonairesdépend du gradient de pressions artérielle pulmonaire diastoliqueet capillaire pulmonaire.
Débit pulmonaire
Il est mesuré par application du principe de Fick et/ou desméthodes de dilution. Il est égal au débit systémique si l’on ne tientpas compte, en l’absence de cardiopathie, de la minime circulationbronchique qui l’accroît physiologiquement de 1 %. Le débitpulmonaire indexé à la surface corporelle IP (index pulmonaire) estégal à 3 à 4 L/min/m2, et la traversée pulmonaire de l’ondesystolique se fait en 6 secondes.Le débit capillaire pulmonaire est passif en l’absence de sphincterpréartériolaire et de type pulsatile, le lit capillaire étant peudistensible. La distribution du flux capillaire est inégale, variantdans l’espace, plus importante à la base qu’au sommet pulmonaireen position debout, et dans le temps, le cycle respiratoire imprimantdes variations du débit capillaire. La pression partielle d’oxygèneintra-alvéolaire (PAO2) intervient dans la régulation locale du débitcapillaire, toute réduction de PAO2 entraînant une vasoconstrictionlocale. Le volume sanguin pulmonaire représente environ 10 % duvolume sanguin total, variant très peu au cours de l’exercice et del’hypertension artérielle pulmonaire (HTAP).
Résistances artériolaires pulmonaires (RAP)
Elles sont chiffrées en application de la loi de Poiseuille selonlaquelle P = QR, où le débit (Q) dans un conduit est proportionnel àla pression (P) régnant dans le conduit et inversement proportionnelà la résistance (R).
Ainsi RAP =PAP Ð PCP
ICUI/m2 exprimées en dyne·s−1·cm−5, si RAP
est corrigé par le facteur 1,332. Les RAP normales sont de l’ordre de150 dyne·s−1·cm–5, quatre à cinq fois inférieures aux résistancessystémiques.Cette formule donne une appréciation plus qu’une mesure précisedes RAP, l’index cardiaque (IC) mesuré par thermodilution ne tenantpas compte de la circulation bronchique, et la mesure des pressionsvasculaires pulmonaires ne prenant pas en compte les facteursextravasculaires (insufflation pulmonaire, pressions pleurale etalvéolaire).
La distensibilité de l’arbre artériel pulmonaire total est importanteau niveau des gros vaisseaux pulmonaires et à l’étage précapillaire,alors que les capillaires sont en revanche peu distensibles. Ladistension du lit vasculaire pulmonaire, en réponse à de faiblesvariations de pression, résulte de l’ouverture de nouveaux territoiresvasculaires et de la dilatation de vaisseaux déjà fonctionnels.
¶ Opacification distale de l’arbre artériel pulmonairepar angiographie « bloquée »Elle complète le cathétérisme. De connaissance ancienne, elleconsiste à injecter de façon manuelle du produit de contraste enposition « capillaire » ou « bloquée » de la sonde. Le passage rapide,en moins de 1 seconde, du produit de contraste vers les veinespulmonaires, atteste d’un lit vasculaire normal ou peu altéré.Inversement, un ralentissement important du passage du produitopaque, associé à un aspect tortueux en « arbre mort » des artériolespulmonaires, témoigne d’une obstruction artériolaire pulmonairesévère. Une étude morphocinétique, avec mesure du temps depassage du produit de contraste et du caractère rapide ou progressifde la diminution du calibre artériolaire, permet, lors de cetteopacification, une bonne appréciation de l’état du lit artériolairepulmonaire, avec une assez bonne corrélation anatomique.L’angiographie sélective artérielle pulmonaire proximale permetd’analyser la distribution artérielle pulmonaire et de rechercherd’éventuels thrombi.
Variations physiologiques de la circulation pulmonaire
Physiologiquement, la circulation pulmonaire varie avec l’exercice,l’âge et l’altitude.
• ExerciceIl provoque une augmentation du débit cardiaque proportionnelle àl’intensité de l’effort en fonction du sexe, de l’entraînement, du tauxd’hémoglobine et de la masse sanguine. L’exercice entraîne uneaugmentation modérée de la pression systolique.
• ÂgeÀ la naissance, les artérioles pulmonaires sont de structure fœtaleavec hypertrophie de la média ; les résistances pulmonaires sontélevées et la pression pulmonaire est de niveau systémique. Cettestructure fœtale est réversible et, entre 3 et 7 jours de vie, la PAP senormalise.
• AltitudePar hypoxémie, elle est responsable d’une vasoconstrictionpulmonaire avec, pour conséquence, une augmentation de la PAP etdes résistances pulmonaires.
¶ Échocardiographie et dopplerL’apport des ultrasons, enrichis par l’effet doppler [18, 39, 49, 50] et lecodage couleur, a permis l’évaluation et le suivi del’hémodynamique artérielle pulmonaire de façon non invasive.Initialement, l’approche du niveau des pressions pulmonaires étaitindirecte, basée sur la refermeture prématurée de la valvepulmonaire, la disparition de l’onde a sur l’enregistrement Tmvalvulaire pulmonaire et les variations des intervalles de tempssystolique, associés ou non en bidimensionnel aux modifications dela courbure septale, d’autant plus plate ou concave vers le VG quela pression ventriculaire droite, en l’absence d’obstacle proximal surla voie droite, est élevée.Le développement du doppler a permis une approche plus exactedes pressions pulmonaires [37]. La mesure au doppler est basée sur lecalcul du gradient de pression de part et d’autre, soit d’unerégurgitation valvulaire (tricuspide ou pulmonaire), soit d’unecommunication intercavitaire (CIV, canal artériel). Le gradient depression D est mesuré selon l’équation simplifiée de Bernouilli (D =4 V2 max). Cette méthode est très fiable, surtout chez l’enfant, pourles normes des pressions ventriculaires droites systoliques et desPAPs ou PAPd.
Cardiologie Affections de l’artère pulmonaire 11-312-A-10
3

En présence d’une HTAP, la courbe de vélocité du flux pulmonairese modifie. La vélocité du flux maximal pulmonaire ne se modifiepas en cas d’élévation des PAP et des résistances artériellespulmonaires. Cependant, le pic de vélocité est plus précoce et leflux peut être le siège d’une encoche mésosystolique. La mesuredirecte de la PAP en l’absence d’obstruction peut être obtenue enprésence d’une insuffisance tricuspide ; selon l’équation deBernouilli, le gradient de pression systolique VD-OD est quatre foisla valeur au carré du pic de vélocité maximal de régurgitation, et lapression systolique ventriculaire droite et artérielle pulmonaire estmesurée par ce gradient, additionnée à la pression auriculaire droite.Cette dernière est basse en l’absence de pathologie du cœur droit,estimée de 10 à 15 mmHg. La vélocité maximale du flux derégurgitation tricuspide est au mieux enregistrée en incidenceapicale et la pression OD intervient peu si l’alignement est bon. Àl’état normal, elle n’excède pas 2 à 2,5 m/s. D’excellentescorrélations existent entre la mesure de la PAP par cette technique etles données du cathétérisme [48]. L’insuffisance tricuspide est trèsfréquente chez l’enfant et en cas d’HTAP.Les travaux de Heatle ont montré qu’à partir de la vélocité du fluxde régurgitation pulmonaire, fréquente à l’état physiologique et dansles HTAP, il était possible d’évaluer les PAPd, et ainsi d’approcherle niveau des résistances pulmonaires. La vélocité maximale enprotodiastole du flux de régurgitation pulmonaire est d’autant plusélevée que les pressions pulmonaires sont élevées, et permet unebonne évaluation de la PAPd. La fuite pulmonaire est retrouvée dansplus de 80 % des HTAP, recueillie au doppler à codage couleur dansl’infundibulum pulmonaire ; la PAP systolique (PAPs) est estiméeen se servant d’une formule empirique utilisée pour le calcul de lapression artérielle moyenne (PAPm) :PAPm = (1/3 × PAPs) +(2/3 × PAPd)d’oùPAPs = 3 PAPm − 2 PAPdEn pratique, le chiffre de PAPs calculé par la fuite tricuspide estsupérieur à celui calculé par l’insuffisance pulmonaire. En revanche,un chiffre de pression pulmonaire mesuré sur l’insuffisancepulmonaire plus élevé que sur l’insuffisance tricuspide doit inciter àreprendre cette dernière mesure avec une meilleure enveloppe defuite tricuspide.La fiabilité de la mesure des pressions pulmonaires au doppler,notamment systoliques, est largement admise. Si une erreurd’appréciation de ± 7 mmHg (OD) peut être négligée pour unepression systolique de l’ordre de 35 mmHg, elle l’est difficilementpour une pression diastolique et moyenne de 15 mmHg, d’où uneprécision moindre pour ces dernières.
¶ Imagerie de résonance magnétique et scanneren haute résolutionCes techniques non invasives permettent d’évaluer les morphologiesventriculaire et auriculaire droites, la morphologie artériellepulmonaire et la fonction ventriculaire droite.Le scanner thoracique en haute résolution permet en outrel’évaluation du parenchyme pulmonaire et détecte la présence d’uneéventuelle maladie veino-occlusive pulmonaire.La valeur de ces techniques d’imagerie dans le suivi des patientsayant une pathologie hypertensive artérielle pulmonaire n’est pasencore établie.
¶ Scintigraphie pulmonaire [78]
Elle permet de visualiser des irrégularités de distribution du traceuren cas de maladie veino-occlusive, et de plus importants défauts deperfusion pulmonaire en cas de maladie thromboemboliqueartérielle pulmonaire.La scintigraphie de perfusion doit être couplée à une scintigraphiede ventilation.
¶ Épreuves pharmacodynamiquesLa mesure par cathétérisme droit de la PAP et le calcul desrésistances vasculaires pulmonaires à l’état de base ne suffisent pas
à l’appréciation exacte de l’état du lit vasculaire pulmonaire. Si leschiffres des pressions et des résistances sont élevés, il importe dedéterminer si ces modifications sont réversibles ou s’il existe uneartériolite pulmonaire fixée.Des épreuves dynamiques (test d’effort) [23] ou pharmacodynamiques(hyperoxie, NO, prostacycline et, autrefois, plusieurs vasodila-tateurs) [4, 61] tentent une approche plus fine du degré de réversibilitédes lésions obstructives artériolaires pulmonaires.Le test d’effort peut être utilisé pour suivre la réponse autraitement [60] et permet de préciser les caractéristiques de lalimitation à l’effort avec une réduction de la consommationmaximale d’oxygène et une réponse ventilatoire à l’effortaugmentée. Le test de marche de 6 minutes complète les donnéeshémodynamiques de repos et donne une appréciation du pronosticde survie à long terme. Le test d’hyperoxie est sans danger, sauf encas d’hypercapnie chronique. S’il fait baisser la PAP par rapport à lapression systémique, il traduit la réversibilité de l’HTAP.Ce test, peu précis, a été supplanté dans les 15 dernières années parles épreuves pharmacodynamiques utilisant autrefois desvasodilatateurs tels que la tolazoline, l’hydralazine, le diazoxide etla phentolamine. De nombreuses publications en ont fait état, avecdes molécules, d’effets variables sur les pressions et les RAP, et lestravaux plus récents sur le mécanisme endothélial de l’HTAP ontconduit à utiliser, pour ces tests, le NO inhalé, les prostaglandines etles inhibiteurs calciques (nifédipine, diltiazem). Les mécanismesexacts de l’HTAP ne sont pas encore tous connus, et les effets de cesdifférentes drogues sur le lit vasculaire pulmonaire ne sont pasunivoques et peuvent être fonction de l’affection cardiaque oupulmonaire responsable de l’HTAP.
¶ Biopsie pulmonaire
Elle reste actuellement le procédé qui apparaît le moins incertainpour établir la réversibilité des lésions du lit artériolaire pulmonaire,les lésions retrouvées sur un fragment de poumon étant en règlereprésentatives de l’ensemble du lit vasculaire pulmonaire.Bien que l’histologie pulmonaire, et en particulier artériolairepulmonaire, permette de caractériser le niveau de l’atteinteartériolaire pulmonaire, la biopsie pulmonaire comporte un risqueet cette étude des structures artériolaires n’est que d’un faible apporten regard des informations cliniques et hémodynamiques, invasivesou non.Son indication n’est pas retenue en routine dans le bilan des HTP etn’est discutée qu’en situation spécifique, faisant évoquer notammentune éventuelle vascularite inflammatoire.Les modifications anatomopathologiques de l’arbre artérielpulmonaire dans l’HTAP ont été étudiées en détail par Heath etEdwards [36], auteurs d’une classification de ces lésions, confirméepar les travaux de Wagenvoort [95, 96]. Cette classification établit sixstades évolutifs (fig 2) :
– stade 1 : c’est le stade de la persistance du « type fœtal » desvaisseaux pulmonaires ; les lésions intéressent les artérioles et lesartères musculaires ; il apparaît alors une extension distale de lamédia des artères vasculaires vers les artérioles ; cette médias’épaissit, atteignant 25 % du diamètre externe du vaisseau ;l’adventice devient épaisse et fibreuse ;
– stade 2 : l’hypertrophie de la média s’associe, au niveau des petitesartères musculaires (inférieures à 300 µm), à une proliférationcellulaire de l’intima dont l’importance peut conduire à l’obstructiondu vaisseau ;
– stade 3 : il se caractérise par l’apparition de la fibrose intimaleréalisant le stade de l’« occlusion vasculaire fibreuse progressive »,où la progression de la fibrose intimale se fait des petites artèresmusculaires vers l’arrière, à l’origine du vaisseau, et s’accompagne,lorsqu’elle est très développée, d’une rupture de la lame élastiqueinterne. À un stade 3 évolué, la fibrose intimale aboutit à l’occlusiondes artères musculaires et des artérioles ; à ce stade, l’hypertrophiede la média atteint sa valeur maximale et, plus tardivement,
11-312-A-10 Affections de l’artère pulmonaire Cardiologie
4

apparaissent des lésions d’amincissement secondaire de cette couchemusculaire, annonçant des lésions de dilatation qui caractérisent lesstades 4, 5 et 6 de cette classification ;
– stade 4 : ce stade est caractérisé par l’existence de lésions dedilatation de type plexiforme, où les artérioles forment des sacs dontles parois sont fragiles, constituées d’une seule couche de muscleentre deux lames élastiques mal définies ; des canaux aux paroisminces naissent de ces sacs dilatés et se terminent en capillaires quiaboutissent aux parois alvéolaires ;
– stade 5 : les lésions de dilatation de type plexiforme ouangiomatoïde caverneux sont nombreuses et s’accompagnent d’unehémosidérose pulmonaire ; le poumon a un aspect très vasculairerecouvert de vaisseaux de type sinusoïde ;
– stade 6 : peu de patients porteurs d’une maladie pulmonairehypertensive atteignent ce stade d’« artérite nécrosante », où lesartères musculaires sont le siège d’une nécrose fibrinoïde,accompagnée parfois d’une réaction inflammatoire faite surtout deleucocytes polynucléaires neutrophiles avec peu d’éosinophiles.Cette classification a constitué une référence, notamment dansl’évaluation pronostique des cardiopathies congénitales, les lésionsd’artériolite pulmonaire étant réversibles jusqu’au stade 3 inclus et,pour quelques auteurs, le stade 4 est susceptible d’un certain degréde réversibilité.Les corrélations entre les étiologies ou la maladie hypertensivepulmonaire et les aspects histologiques font apparaître que :
– l’hypoxémie conduit à un grade 1 ;
– les obstacles gauches sont longtemps responsables d’un grade 1réversible ;
– dans les cardiopathies malformatives avec shunt gauche-droite,les lésions sont d’autant plus marquées que le shunt est important,la pression partielle en oxygène du sang artériel pulmonaire élevéeet la durée d’évolution plus longue.Les progrès dans la compréhension des mécanismes cellulaires(notamment endothéliaux) de l’HTAP conduisent à modifierl’utilisation de la classification de Heath et Edwards, parfoisrestrictive, et à préférer une analyse histologique plus descriptiveque normative [65], à la lumière des informations cliniques ethémodynamiques. Les différentes structures sont examinées. Auniveau des vaisseaux, la lumière vasculaire est décrite et la présencede thrombi anciens ou récents notée.Les composants du vaisseau sont analysés avec, au niveau del’endothélium, la cellule endothéliale et de petites cellulesmusculaires, ainsi que le tissu de soutien (élastine, collagène,mucopolysaccharides). Au niveau de la média est étudiée ladistribution musculaire excentrique ou concentrique, les composantscellulaires et le tissu de soutien ; au niveau de l’adventice, le type decomposants cellulaires (fibroblastes) et le tissu de soutien. Deslésions vasculaires complexes peuvent être présentes, telles quedilatations complexes, lésions plexiformes, nécrose fibrinoïde,artérite, hémosidérose et granulomes. La présence de cellulesinflammatoires, leur type et leur siège sont notés.
Le tissu pulmonaire est lui-même analysé en précisant la source duprélèvement (post mortem, poumon entier ou biopsie), le siège duprélèvement, la préparation et le mode de fixation du tissu. Unequantification de l’atteinte artérielle pulmonaire peut être faite.
Hypertension artérielle pulmonaireprimitive
L’HTAPP est un syndrome clinique, anatomique et hémodynamiquedont l’étiologie n’est pas connue à ce jour.Le premier cas documenté a été publié par Dresdale en 1951 [25], bienqu’il y ait plus d’un siècle Romberg [81] ait rapporté l’observationd’un patient ayant une insuffisance cardiaque droite où l’autopsiene montrait aucune évidence d’artériosclérose pulmonaire. En 1967,une épidémie d’HTAPP en Europe est attribuée à l’utilisationétendue d’anorexigènes contenant du fumarate d’aminorex. En 1973,l’Organisation mondiale de la santé (OMS) organise, à la lumière decette augmentation des cas d’HTAPP, une réunion internationale quipermet de revoir et discuter les mécanismes physiopathologiques etépidémiologiques de l’HTAPP. En 1994, le groupe internationald’étude sur l’HTAPP clarifie le rôle des réducteurs d’appétit dans cesyndrome. Les 25 dernières années sont marquées par une meilleurecompréhension de la pathologie, de la pathobiologie des facteurs derisque et de la génétique de la maladie. Les progrès thérapeutiquesavec l’apparition de la prostacycline dominent cette dernière périodeen améliorant le pronostic de cette affection.
DÉFINITION
L’HTAPP se caractérise par une élévation soutenue des pressionsartérielles pulmonaires sans cause démontrable [41, 82]. Les critèresdiagnostiques retenus par l’Institut national du registre des maladiessont une PAPm supérieure ou égale à 25 mmHg au repos et/ou àplus de 30 mmHg à l’exercice. Cette définition hémodynamiqueimplique une PCP bloquée normale inférieure ou, au plus, égale à12 mmHg. Ces critères excluent toute association à une éventuellemaladie valvulaire gauche, une atteinte myocardique ou unecardiopathie malformative. Sont également éliminées les affectionsrespiratoires, la maladie thromboembolique et les connectivites[30, 66].
ÉTIOLOGIE, ÉPIDÉMIOLOGIE, FACTEURS CLINIQUES
L’HTAPP est une maladie rare dont aucune cause précise n’a puêtre démontrée [19].Son incidence [1, 83] est de 1/500 000 habitants, selon une étudemulticentrique européenne récente, identique aux États-Unis. Laprévalence féminine [10] est une donnée habituelle, bien que variable,avec un sex-ratio de 1,7/1 aux États-Unis (extrêmes : 1,3-3,5). Cetteprévalence féminine est particulièrement nette dans la race noire.Le pic de fréquence du diagnostic de la maladie se situe à 36 ans,mais la maladie peut se voir à tout âge ; la moyenne d’âge audiagnostic est légèrement plus élevée chez l’homme que chez lafemme.
2 Classification de Heath et Edwards.A. Stade 1. B. Stade 2. C. Stade 3.
*A *B *C
Cardiologie Affections de l’artère pulmonaire 11-312-A-10
5

Les formes familiales [46] d’HTAPP sont retrouvées dans 6 à 10 % descas. Le développement de la maladie dans ces formes est identiqueà celui des formes sporadiques, avec cependant une apparition dessymptômes de plus en plus jeune dans les générationssuccessivement atteintes. Ce phénomène d’anticipation génétique estcaractéristique de la forme familiale de la maladie.
Si la cause de l’HTAPP n’est pas connue, l’apparition de celle-ciserait facilitée par des facteurs de risque, pour une part certains,pour d’autres possibles, et pour les derniers peu probables. De façoncertaine, la prise d’aminorex, de fenfluramine, de dexfenfluramine[1, 30, 83] et de certaines huiles de colza frelatées augmente le risqued’HTAPP.
La prise d’anorexigènes multiplie ce risque par 6, et par 20 si cetteprise a dépassé 3 mois, consécutifs ou non. Les conséquencestardives de ces traitements sur plusieurs mois ou années ne sont pasencore connues, mais la fenfluramine a été retirée du marché enFrance. La prise d’anorexigènes est retrouvée chez 3 à 7 % despatients ayant une HTAPP. En 1981, l’HTAPP a été rapportée commeune complication qui a touché 20 000 personnes en Espagne [31, 41]
après consommation d’huile de colza frelatée (toxic-oil syndrome).Les symptômes initiaux ont été marqués par un syndromeinflammatoire avec pneumopathie interstitielle transitoire et, 2 à3 mois plus tard, apparition, surtout chez les femmes, dans 2 à 3 %des cas, d’un syndrome de Raynaud et d’une HTAPP, dont certainscas ont été décrits au sein d’une même famille.
L’association d’une sérologie du virus de l’immunodéficiencehumaine (VIH) positive [52, 64, 88] au développement d’une HTAPP estconnue depuis 1987. L’HTAPP se développe indépendamment dustade de l’immunosuppression et de l’évolution clinique del’infection à VIH. L’existence d’une sérologie positive au VIH a éténotée chez 8 % d’une série française de 125 patients [10] suivis pourune HTAPP. La survie des patients ayant une sérologie positive n’estpas différente de ceux dont la sérologie est négative.
Le risque de développement d’une HTAPP est probable avec la prised’amphétamines ou de L-tryptophane, l’hypertension portale et lesmaladies du tissu collagène.
La prévalence de l’HTAPP est augmentée dans les cirrhoses,démontrée dans des séries autopsiques où elle est retrouvée chez0,73 % environ des cirrhotiques, avec comme facteur favorisant laprésence d’un shunt portosystémique, spontané ou chirurgical. Lemécanisme essentiel et probable de ce facteur, mais non démontré,est peut-être la présence de substances vasoactives en excès, non ouincomplètement métabolisées du fait de l’absence de passagehépatique. Le rôle évoqué de l’hyperdébit et d’éventuelles emboliespulmonaires répétées n’a pas été confirmé. Dans une série françaisede 125 patients suivis pour HTAPP, 13 % avaient une cirrhose. Ledélai entre l’apparition des signes de cirrhose et des signes d’HTAPPest de 4,8 ans, avec un âge moyen de 40 ans et une prédominancemasculine.
L’association de l’HTAPP à des maladies systémiques a fait discuterun mécanisme auto-immun. Le syndrome de Raynaud [77] estretrouvé chez près de 10 % des patients, essentiellement des femmesatteintes d’HTAPP. La positivité des facteurs antinucléaires à tauxmodéré est retrouvée chez 25 % des patients. Le mécanisme dudéveloppement de l’HTAPP dans ces pathologies n’est pasclairement connu. Ce développement est particulièrement fréquentdans la sclérodermie, le lupus érythémateux disséminé, le syndromede Sjögren et apparentés. Il est plus rare dans la polymyosite, ladermatomyosite et la polyarthrite rhumatoïde. Enfin, l’associationHTAPP et dysfonction thyroïdienne a été signalée avec un tauxinhabituellement élevé, dans ces pathologies, d’anticorpsantithyroïdiens. Le risque d’HTAPP est possible lors de la prise deméta-amphétamines, de cocaïne ou d’agents chimiothérapeutiques,ainsi que lors de la grossesse ou d’HTA systémique. Le rôle destraitements antidépresseurs, œstrogéniques ou contraceptifs,l’obésité et le tabac n’a pas été prouvé comme constituant desfacteurs de risque d’HTAPP.
GÉNÉTIQUE ET PHYSIOPATHOLOGIE DE L’HTAPP
Le locus du gène atteint dans les formes familiales d’HTAPP a étéidentifié sur le chromosome 2q31-32. La faible pénétrance de ce gèneexplique la probabilité de 10 à 20 % de développer la maladie.Quand une personne dans la famille a développé une HTAPP, laprobabilité pour un autre membre de la famille de développer lamaladie est estimée de 0,6 à 1,2 % [47]. Si deux personnes sontatteintes, ce risque s’élève de 5 à 10 %. La physiopathologie del’HTAPP reste peu claire, mais fait intervenir deux éléments :l’obstruction de la lumière vasculaire par prolifération fibreuse del’intima et musculaire de la média d’une part, et d’autre part lavasoconstriction. Ces deux éléments impliquent un dérèglement dela croissance et de la différenciation musculaire, et un déséquilibreau niveau de la cellule endothéliale entre facteurs vasoconstricteurset vasodilatateurs [16]. Des études immunohistochimiques suggèrentque l’expression endothéliale des NOS est diminuée dans les AP despatients atteints d’HTAPP et l’excrétion urinaire des métabolites dela prostacycline est également basse chez ces patients. Inversement,les concentrations en endothéline-1 (ET1) sont particulièrementélevées dans les AP de ces patients. Le thromboxane A2,vasoconstricteur, activateur des plaquettes, a été retrouvé augmentédans le sang et dans les urines des patients atteints d’HTAPP. Laprésence de stimuli vasoconstricteurs chez ces patients favoriseraitla prolifération du muscle lisse artériolaire, modification précoce del’histologie artériolaire pulmonaire.
ANATOMOPATHOLOGIE
Au sein des HTAPP, trois types principaux d’altérations vasculairessont décrits [65, 75] :
– l’artériopathie pulmonaire plexogénique : cet aspect n’est passpécifique de l’HTAPP. Il associe une hypertrophie de la média parprolifération et augmentation de la taille des cellules musculaireslisses, une fibrose concentrique de l’intima qui réduit la lumièrevasculaire par prolifération cellulaire essentiellement demyofibroblastes, et des lésions plexiformes qui traduisent un stadeavancé de la maladie avec une lumière vasculaire remplacée par unentrecroisement de canaux séparés par du tissu fibreux. En certainspoints, il peut être noté des dilatations vasculaires dans les zones oùl’hypertrophie de la média est absente. Peuvent se constituer enoutre des lésions de nécroses fibrinoïdes avec destruction desstructures pariétales et constitution de caillots dans la lumièrevasculaire. Cette forme est retrouvée chez 30 à 60 % des HTAPP ;
– l’artériopathie pulmonaire thrombotique ne comporte pas de lésionsplexiformes et se caractérise par une prolifération intimale avecfibrose et la présence de thrombi d’âge différent dans la lumièrevasculaire, parfois recanalisés. Les thrombi sont probablement dus àune thrombose vasculaire in situ. Cette forme intéresse 40 à 50 %des HTAPP [15, 28] ;
– la maladie veino-occlusive se définit par une fibrose intimale diffusedes veines et des veinules possiblement due à une thrombose localeavec recanalisation des thrombi. Une atteinte artériolaire s’associe àl’atteinte retrouvée dans de nombreux cas. Beaucoup plus rare, elleest retrouvée chez moins de 10 % des patients avec HTAPP ;
– plus récemment, une nouvelle entité a été décrite : l’héman-giomatose pulmonaire [49], très rare, caractérisée par une proliférationde capillaires dans l’interstitum pulmonaire. Ces vaisseaux infiltrentles parois des veines pulmonaires entraînant l’HTAPP.
SYMPTÔMES ET DIAGNOSTIC CLINIQUE
L’obstacle principal à un diagnostic clinique précoce est lié au peude spécificité des signes fonctionnels initiaux et, lors de l’apparitionde ceux-ci, la maladie est le plus souvent déjà évoluée. Le délaimoyen entre l’apparition des symptômes et le diagnostic de lamaladie est de 2 ans et, chez 10 % des patients, ce délai peutatteindre 3 ans.Le plus souvent, dans 60 % des cas, les patients consultent pour unedyspnée qui s’aggrave progressivement. La fatigabilité est un signe
11-312-A-10 Affections de l’artère pulmonaire Cardiologie
6

précoce et fréquent. Les douleurs thoraciques, parfois de caractèreangineux, et les syncopes, particulièrement à l’effort, indiquent unelimitation sévère du débit cardiaque et se voient dans 50 % des cas.Un syndrome de Raynaud, le plus souvent chez des femmes, peutêtre révélateur, dans 10 % des cas, de l’HTAPP. Parfois, une irritationdu nerf récurrent par un tronc artériel pulmonaire dilaté peutconduire à la survenue d’un syndrome d’Ortner. Hémoptysies, enrègle de faible abondance, et palpitations sont rares. L’insuffisanceventriculaire droite, rarement révélatrice, émaille l’évolution dans43 % des cas. À ce stade, l’interrogatoire recherche des antécédentsfamiliaux d’HTAPP ou la prise d’anorexigènes récente ou ancienne,en précisant le type de l’anorexigène et la durée du traitement. Lessignes d’une hypertension portale sont recherchés ainsi qu’uneéventuelle infection à VIH.L’examen physique est pauvre, en l’absence de signes d’insuffisancecardiaque droite se limitant à un éclat du deuxième bruitpratiquement constant et parfois associé à un souffle systolique douxd’insuffisance tricuspidienne. Mais le plus souvent, l’examenclinique est normal au début.
EXAMENS COMPLÉMENTAIRES
¶ Radiographie pulmonaire, électrocardiogramme,échographie
La radiographie pulmonaire est très fréquemment anormale,montrant une dilatation du tronc et des branches pulmonairesproximales avec une saillie de l’infundibulum et une cardiomégalieinitialement modérée. Le parenchyme pulmonaire est le plussouvent normal, voire clair et, exceptionnellement, le siège d’unaspect de syndrome interstitiel (fig 3).L’électrocardiogramme (ECG) montre très habituellement un rythmesinusal auquel s’associe un aspect d’hypertrophie auriculaire droiteet de surcharge ventriculaire droite.Ces examens restent utiles, mais ont une valeur plus qualitative quequantitative ; l’apport de l’échographie cardiaque couplée audoppler est important pour le diagnostic de l’HTAP. L’examenbidimensionnel permet d’éliminer une cardiopathie malformative ouune cause d’HTAP postcapillaire, telles qu’une sténose mitrale ouune dysfonction ventriculaire gauche. L’échographie cardiaqueretrouve, en outre, les signes indirects de l’HTAP, sous la formed’une dilatation des cavités droites et du tronc pulmonaire, auxquelss’ajoutent une hypertrophie myocardique ventriculaire droite et un
mouvement paradoxal du septum interventriculaire. Les cavitéscardiaques gauches sont refoulées par les cavités droites dilatées,mais la cinétique ventriculaire gauche est normale, attestant d’unefonction systolique conservée. Le doppler permet d’estimer la PAPpar la mesure de la précocité du pic du flux systolique au travers dela valve pulmonaire, et surtout la vélocité du flux de régurgitationtricuspide pratiquement constante dans l’HTAPP. Ces mesurespermettent de suivre l’évolution du niveau des PAP sous traitementvasodilatateur. Quelques patients ont un shunt droite-gauche sefaisant par un foramen ovale perméable. Les explorationsfonctionnelles respiratoires, avec mesure des débits et volumesrespiratoires, sont dans les limites de la normale, permettant ainsid’éliminer une anomalie parenchymateuse ou ventilatoire. Quelquespatients ont un discret syndrome restrictif ou un trouble dediffusion, mais qui n’est pas corrélé avec la sévérité de la maladieartérielle pulmonaire. La gazométrie artérielle peut montrer unealcalose respiratoire chronique et une hypoxémie en rapport avecun trouble de ventilation-perfusion, ou un shunt droite-gaucheauriculaire.L’épreuve cardiorespiratoire d’effort [23, 60, 72] permet de préciser lescaractères de la limitation à l’effort, en montrant une diminution dela consommation d’oxygène et une réponse ventilatoire exagérée àl’effort. Le test de 6 minutes de marche est un test simple, apportantdes informations utiles sur l’hémodynamique et le pronostic. Ce testest utilisé pour le suivi des patients sous traitement vasodilatateur,mais il s’agit d’un test subjectif imposant la compréhension et lacoopération du patient.
¶ Scintigraphie pulmonaire
La scintigraphie pulmonaire couplant la ventilation à la perfusionpermet d’éliminer une maladie thromboembolique. Elle esthabituellement normale dans l’HTAPP, mais peut évoquer, lorsd’une distribution inégale du traceur, une maladie veino-occlusive.Lorsque la scintigraphie n’est pas normale, il est nécessaire de faireune angiographie sélective pulmonaire montrant, dans l’HTAP, devolumineuses AP proximales libres, avec une vascularisationpériphérique grêle et tortueuse [78].
¶ Cathétérisme cardiaque
Le cathétérisme cardiaque reste l’examen de référence dans l’HTAPP.Il permet d’éliminer une malformation cardiaque, de chiffrer leniveau de l’HTP, de confirmer son caractère précapillaire et de tester,en cours d’exploration, les drogues vasoactives, soit NO, soitprostacycline.Une PAPm supérieure à 25 mmHg, avec une pression capillairenormale, confirme le diagnostic d’HTAP. Le plus souvent, lors dudiagnostic, la PAPm est supérieure à 50 mmHg, avec un IC diminuépermettant d’évaluer des résistances artériolaires à plus de dix foisla normale. La pression dans l’OD est normale ou augmentée.Les paramètres hémodynamiques sont identiques selon le sexe,l’âge, l’existence ou non d’un syndrome de Raynaud. Les patientsayant une hypertension portale ont des chiffres de PAP moins élevésque les autres. Aucun facteur hémodynamique ou clinique nepermet de prévoir la réponse des patients à un vasodilatateur donné.Il est donc indispensable, lors du cathétérisme initial, de tester lesvasodilatateurs efficaces pour envisager le traitement au long cours,soit par voie orale, soit par voie veineuse, permettant ainsid’individualiser les patients répondeurs aux vasodilatateurs [70]. Cespatients ont un meilleur pronostic et une survie plus prolongée [5]
que les non-répondeurs. Il n’y a pas de critère uniforme de réponsepositive aux vasodilatateurs, mais sont considérés commerépondeurs les patients dont les résistances pulmonaires totalesdiminuent de 20 % sans chute du débit cardiaque, sans qu’il y ait demodification constante de la PAPm.Les agents les plus souvent utilisés sont le NO inhalé et laprostacycline injectée.De nombreux vasodilatateurs ont été testés avec des résultats parfoiscontradictoires pour un même produit. La définition de l’effet
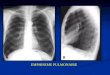
3 Radiographie pulmonaire : hypertension artérielle pulmonaire primitive.
Cardiologie Affections de l’artère pulmonaire 11-312-A-10
7

vasodilatateur est elle-même imprécise, car on ne peut pas tenircompte de la variabilité hémodynamique spontanée de la maladie.Parmi les vasodilatateurs testés : prostacycline, isoprostérénol,trinitrine, diltiazem, phentolamine, hydralazine [9, 61, 62, 86, 87]. Laprostacycline ou isoprostérénol, vasodilatateur endogène, a l’effet leplus puissant chez les répondeurs, provoquant une baisse desrésistances vasculaires pulmonaires, une chute significative de lapression artérielle systémique, et une augmentation significative dudébit cardiaque et de la fréquence cardiaque. La PAPm reste stable.La réponse à la prostacycline a un effet prédictif sur l’existence d’unecomposante réversible de la maladie artériolaire pulmonaire.Le NO inhalé [53 , 54] a une demi-vie courte. Inactivé parl’hémoglobine, il n’agit que sur les artérioles pulmonaires. Il a uneaction reproductible. Il est donné sous forme gazeuse, inhalé à raisonde 10 à 20 ppm (parts par million) pendant 15 à 20 minutes. Il nefait pas varier le débit cardiaque et diminue la fréquence cardiaque.Les inhibiteurs calciques sont également testés en coursd’exploration [73, 74], leur prescription thérapeutique pouvant être faitepar voie orale. Les plus utilisés sont la nifédipine et le diltiazem.L’effet souhaité est une réduction de la PAP, sans chute du débitcardiaque, mais ces produits s’accompagnent souvent, du fait deleur mécanisme d’action, d’une baisse des résistances systémiques.Le cathétérisme cardiaque dans l’HTAPP est indispensable,essentiellement pour évaluer les possibilités thérapeutiques. Il estcependant grevé [13], dans ce contexte, d’une mortalité évaluée de 5 à10 %, d’autant plus élevée que l’examen est prolongé et que lepatient est porteur d’un patent foramen ovale. Les accidents sévèrespeuvent survenir au cours de l’examen ou dans les heures quisuivent, et en l’absence d’opacification angiographique. Lemécanisme de ces accidents n’est pas parfaitement connu,probablement multifactoriel.
¶ Biopsie pulmonaireCet examen non dénué de risques est très rarement nécessaire audiagnostic d’HTAPP et n’est prescrit qu’en cas d’incertitudediagnostique.
TRAITEMENT DE L’HTAPP
L’HTAPP n’a pas de traitement curateur. Des rémissions spontanéesont été rapportées, mais sont exceptionnelles, pouvant se rencontrerchez les patients ayant pris des anorexigènes et après arrêt de cettethérapeutique. Ces 15 dernières années ont été marquées par lesprogrès des thérapeutiques vasodilatatrices médicales dans cetteaffection [22, 29].
¶ Traitement médical
Vasodilatateurs
Ce sont essentiellement les inhibiteurs calciques, le NO et laprostacycline par voie veineuse continue, après test lors ducathétérisme de la réponse à ces différents produits.
• Inhibiteurs calciquesLes inhibiteurs calciques peuvent être prescrits par voie orale, sousla forme essentiellement de nifédipine, nicardipine et, plus rarement,le diltiazem. Leur efficacité chez les répondeurs a été confirmée parune survie de 94 % à 5 ans contre 55 % chez les non-répondeurs.Mais 30 % des patients répondent en aigu au NO et à laprostacycline, et 10 % d’entre eux seulement aux inhibiteurscalciques. Des doses supérieures aux posologies prescrites dansl’HTA systémique sont nécessaires et la sensibilité individuelle esttrès variable. L’arrêt brutal des inhibiteurs calciques peut êtreresponsable d’un effet rebond sur l’HTAPP, et fatal. Les patients avechypotension, débit cardiaque abaissé ou désaturation systémique,ne peuvent bénéficier de ce traitement [76].
• ProstacyclineChez les répondeurs en aigu, la perfusion continue par cathétercentral de prostacycline a maintenant montré son efficacité, en la
faisant passer d’une solution d’attente de transplantation cœur-poumons à une alternative à cette dernière. Les effets secondairesde cette thérapeutique sont liés au produit, telles les douleursosseuses et articulaires, la diarrhée et les éruptions. Lescomplications plus graves sont en rapport avec la nécessité d’uncathéter central. La perfusion de prostacycline permet une netteamélioration fonctionnelle, avec augmentation des capacités àl’exercice et contrôle des symptômes cliniques. Habituellement, laPAP se modifie peu [51].La posologie initiale est déterminée lors du cathétérisme, puisaugmentée régulièrement avec le temps. Le mécanisme des besoinsaccrus en prostacycline avec le temps n’est pas connu, mais peutêtre lié à une accélération de la dégradation du produit et de laproduction endogène de thromboxane vasoconstricteur [98]. Laperfusion de prostacycline est contre-indiquée chez les patientsayant une maladie veino-occlusive, en raison du risque d’œdèmepulmonaire. La prostacycline a des propriétés d’inhibitiond’agrégation plaquettaire et d’adhésion leucocytaire à l’endothéliumvasculaire. Les patients mis sous prostacycline sont aux stades IIIou IV de la New York Heart Association (NYHA), sans améliorationpar le traitement conventionnel et/ou non répondeurs auxinhibiteurs calciques. Des études sont encore nécessaires pourclarifier le mécanisme d’action de la prostacycline sur le tissucardiaque et vasculaire. Une meilleure compréhension de cesmécanismes devrait permettre de prévoir la dose optimale efficacepour les patients. Des formes d’administration de prostacycline parvoie sous-cutanée, inhalée ou orale, sont en attente.
• Monoxyde d’azote
Par son métabolisme, le NO est un puissant et sélectif vasodilatateurartériel pulmonaire, mais qui ne peut être administré qu’eninhalation, limitant son utilisation de façon chronique, avec deseffets dans la durée encore mal connus.
Traitements associés
• Traitement anticoagulant
Il est recommandé, en raison du risque accru de thromboses in situ,confirmé par les études anatomopathologiques montrant desthromboses extensives dans les petits vaisseaux pulmonaires et dansles gros troncs, en l’absence de maladie thromboembolique [15, 28].Quelques études font état de l’amélioration de la survie des patientsporteurs d’HTAPP, sous anticoagulants au long cours.L’anticoagulant de choix est la warfarine, prescrite par voie oralepour maintenir l’international normalized ratio (INR) entre 2 et 3.
• Traitement de l’insuffisance cardiaque
Sont indiqués les diurétiques lors de poussées d’insuffisancecardiaque, plus volontiers sous forme d’épargnants potassiques àl’effet plus progressif.Seuls les troubles du rythme auriculaire bénéficient des glycosidestonicardiaques. Le maintien d’une systole auriculaire en rythmesinusal est indispensable au remplissage diastolique du VG, souventlimité par la dilatation des cavités droites.L’oxygénothérapie améliore la tolérance de l’effort, mais a un effetlimité lors d’un shunt droite-gauche par un foramen ovale réouvert.
¶ Traitements chirurgicaux
Atrioseptostomie
Par méthode interventionnelle par cathétérisme, cette création decommunication interauriculaire a été proposée dans quelquesobservations. Cette méthode est en cours d’évaluation, indiquée encas de syncopes non contrôlées, d’insuffisance cardiaque réfractaire,en attente de transplantation avec échappement au traitementmédical maximal, et en l’absence d’autre possibilité thérapeutique.Les candidats à la septostomie doivent avoir une saturation artériellede base supérieure à 90 % et le bénéfice de ce geste est attendu pour
11-312-A-10 Affections de l’artère pulmonaire Cardiologie
8

une saturation réduite de 5 à 10 %. L’effet à long terme et lesconséquences défavorables de la septostomie ne sont pas encoreétudiés.
Transplantation cœur-poumonsLa transplantation cœur-poumons est pratiquée depuis une dizained’années, remplacée plus récemment par des transplantations uni-ou bipulmonaires [32, 73]. La mortalité opératoire est chiffrée entre16 et 20 %, apparaissant plus élevée lors d’une transplantationmonopulmonaire. La survie à 1 an est de 70 à 75 %, de 55 à 60 % à3 ans, et de 40 à 45 % à 5 ans. Les indications de transplantationsont retenues chez les patients qui sont en classes fonctionnelles IIIet IV de la NYHA malgré le traitement médical, ou chez lesquels laprostacycline est inefficace ou responsable d’effets secondairesincontrôlables. Il n’y a pas encore de consensus pour le choix dutype de transplantation, la procédure de transplantation uni- oubipulmonaire étant pratiquée lorsqu’il n’y a pas d’altération majeurede la fonction ventriculaire droite. Les délais d’attente de greffonsont longs et bon nombre de patients décèdent durant cette période.
ÉVOLUTION ET PRONOSTICLe pronostic de l’HTAPP est mauvais, malgré de rares descriptionsde formes régressives [7], avec une espérance de vie moyenne de2,5 ans après le diagnostic de la maladie [9, 22]. Ce chiffre n’estfonction ni de l’âge, ni du sexe, ni de la durée des symptômes, ni dela présence d’anticorps antinucléaires, ni d’une HTAP familiale, nide la prise de contraceptifs, de la survenue d’une grossesse ou d’untabagisme. Seule la gravité des perturbations hémodynamiquesrecueillies au cathétérisme a été corrélée avec la durée de la survie.Les patients qui répondent aux inhibiteurs calciques ont 95 % dechances de survie à 5 ans, quand un traitement anticoagulant estassocié. La prostacycline améliore la survie des patients nerépondant pas aux inhibiteurs calciques, qui ont un taux de surviecomparable à 5 ans, voire plus élevé qu’après transplantationcardiopulmonaire (fig 4).L’échappement au traitement médical de ces patients aboutit audécès par insuffisance cardiaque, mort subite, surinfectionspulmonaires, choc cardiogénique ou troubles du rythme. Bien quela grossesse ne constitue pas un facteur prouvé influant la surviedes patients atteints d’HTAPP, celle-ci peut être très mal tolérée,notamment en post-partum. La contraception oraleœstroprogestative aggrave le risque de thrombose de la maladie etest contre-indiquée.
Hypertension artérielle pulmonairedu nouveau-né ou persistancede circulation fœtaleEntité rare, réalisant une détresse respiratoire aiguë néonatale,caractérisée par une hypoxémie systémique avec un shuntextrapulmonaire du sang veineux et mise en évidence du niveau
élevé de la PAP en l’absence de malformation cardiaque congénitale[44, 55], ce syndrome se voit habituellement chez des enfants à termeayant une inhalation méconiale, une infection, une hypoplasiepulmonaire ou une souffrance fœtale. Il peut être idiopathique,intéressant un enfant pour 1 500 naissances.Les mécanismes physiopathologiques expliquant ce défautd’adaptation postnatale de la circulation pulmonaire sont encore malcompris, faisant intervenir une dysfonction pulmonaire endothélialeavec libération diminuée de NO et accrue d’ET1. À cette dernières’associe une dysfonction musculaire lisse par mécanismesbiomoléculaires et enzymatiques.Le tableau clinique est dominé par la cyanose intense, non ou malcorrigée par l’oxygène. L’examen cardiaque est normal. L’ECG et laradiographie pulmonaire sont peu contributifs. L’échographiecardiaque permet d’éliminer une anomalie des structures cardiaques.La courbure septale, plate ou droite-gauche, donne une appréciationindirecte du niveau des pressions ventriculaire droite et artériellepulmonaire. Celle-ci est chiffrée à partir de la vélocité du fluxd’insuffisance tricuspide, pratiquement constante. Les pressionspulmonaires artérielles sont de niveau systémique ousuprasystémique, s’accompagnant d’une dilatation importante descavités droites et d’une fonction diastolique ventriculaire gauche,parfois altérée par la dilatation du cœur droit. La mesure desrésistances pulmonaires à l’échographie et au doppler est trèsdifficile dans ce contexte. Le foramen ovale perméable et le canalartériel ouvert dans ce syndrome sont le siège d’un shunt droite-gauche authentifié par le doppler à codage couleur. L’évolution dece syndrome est variable, se faisant vers la régression en quelquesjours. Dans certains cas, notamment lors d’une inhalation méconialeou d’une hernie diaphragmatique malformative, l’hypoxie persisteet conduit au décès. Dans certaines formes, l’hypoxie peut nes’installer que quelques jours après la naissance, évoquant, aprèsune évolution normale néonatale des résistances pulmonaires, unretour de ces dernières à leur niveau fœtal. Le traitement des formesgraves a été transformé par l’utilisation du NO.Le traitement symptomatique comporte l’oxygénothérapie élevée,avec hyperventilation mécanique, source de toxicité pour leparenchyme pulmonaire par baro- et volotraumatismes.L’utilisation de vasodilatateurs, telle la tolazoline, a permis uneamélioration de ces enfants, mais au risque d’une hypotension avecréduction du débit systémique. L’échec de ces traitements avantl’apparition du NO a fait utiliser les techniques d’oxygénationextracorporelle (AREC/ECMO), efficaces mais invasives, coûteuseset grevées de risques de morbidité élevés. Le NO inhalé [90] atransformé le traitement et le pronostic de ce syndrome, permettant,dans les cas graves, de réduire notablement les indicationsd’oxygénation extracorporelle. Cependant, des incertitudespersistent quant à une éventuelle toxicité du NO, notamment chezle nouveau-né. Cette inquiétude est tempérée par la mise enévidence récente de concentrations significatives de NO au niveaudes fosses nasales, notamment chez le nouveau-né.
Hypertension artérielle pulmonairesecondaire aux cardiopathies
L’HTAP est la complication la plus sévère qui puisse être observéeau cours de l’évolution des cardiopathies congénitales, notammentdes shunts gauche-droite, car elle aggrave le risque chirurgical et lepronostic ultérieur à moyen et long termes.L’existence, quelle que soit la cardiopathie, d’une HTAP indique ledéveloppement de lésions artériolaires pulmonaires dont lecaractère, réversible ou non, conditionne les indicationsthérapeutiques et le pronostic.
CARDIOPATHIES MALFORMATIVES
Lorsque le lit vasculaire pulmonaire et les RAP sont normaux, unecommunication intracardiaque entre les circulations gauche et droite
Testing en aiguNO inhalé
Diminution au moins 20%Rpt et PAPm
Traitement 3 moisinhibiteur calcique
Absence de critèrede réponse
Amélioration Pas d'amélioration
HTAPP modérée(classe NYHA I ou II)
HTAPP sévère(classe NYHA III ou IV)
HTAPP modérée(classe NYHA I ou II)
Traitementconventionnel
+ inhibiteur calcique
Traitementconventionnel
seul
Traitement conventionnel± autre vasodilatateur
oral
Traitement conventionnel+ prostacycline IV
et/ou transplantation
4 Schéma thérapeutique de l’hypertension artérielle pulmonaire primitive(HTAPP) [87].NO : monoxyde d’azote ; Rpt : résistances pulmonaires totales ; PAPm : pression arté-rielle pulmonaire moyenne ; NYHA : New York Heart Association.
Cardiologie Affections de l’artère pulmonaire 11-312-A-10
9

induit une augmentation du débit pulmonaire, puis, en fonction del’importance de cette augmentation, une élévation des pressionsventriculaire droite et artérielle pulmonaire. Les facteurspathogéniques des lésions artériolaires pulmonaires ne sont pas tousconnus, faisant intervenir un probable facteur génétique et desmécanismes cellulaires [67, 68]. Les lésions artériolaires pulmonairesfont intervenir l’élévation de la PAP, la gêne fonctionnelle ouorganique au retour veineux pulmonaire, l’augmentation du débitpulmonaire, l’hyperoxie du sang artériel pulmonaire, le temps et desfacteurs génétiques probables. L’hypoxie du sang artériel systémiqueet l’hyperviscosité sanguine des cardiopathies cyanogènes jouentégalement un rôle [94].Les travaux de M Rabinovitch [69] ont permis de distinguer à labiopsie pulmonaire trois stades d’altération du lit vasculairepulmonaire, corrélés avec les données hémodynamiques.
– Le stade A se caractérise par une expansion anormale du tissumusculaire dans les petites AP périphériques.
– Le stade B associe à cette expansion musculaire une hypertrophiede la média des artères normalement muscularisées. Le mécanismede cette hypertrophie est mal connu ; elle s’accompagne d’unedégradation de la lame élastique interne facilitant peut-être letransfert de facteurs de croissance mitogènes pour le muscle lisse.
– Au stade C, l’hypertrophie musculaire s’aggrave d’une densité etd’un calibre artériels qui se réduisent. Ce stade, au-delà de l’âge de2 ans, peut être irréversible.
¶ « Shunts » gauche-droite unidirectionnels
Communication interauriculaire
L’élévation des RAP dans la communication interauriculaire est rare(6 %) et le plus souvent tardive, après 20 ans, plus fréquente chez lafemme que chez l’homme, dans le rapport 4/1. Cette évolution peutégalement, de façon exceptionnelle, se voir chez le nourrisson etl’enfant jeune [38, 92].Cependant, l’absence d’HTAP chez la grande majorité des sujetsporteurs de communication interauriculaire est attribuée à ladilatation des AP proximales élastiques. Les variations individuellesde réponse artérielle pulmonaire à un shunt gauche-droiteauriculaire ne sont pas totalement expliquées, probablementmultifactorielles et faisant peut-être intervenir un facteur génétique.Chez l’enfant, ces formes font intervenir la possibilité d’une maladievasculaire autonome ou la persistance d’une circulation pulmonairede type fœtal. L’élévation des résistances artérielles pulmonairesdans la communication interauriculaire contre-indique la fermeturechirurgicale du défaut septal.
Communication interventriculaire, canal artériel,fistule aortopulmonaire, canal atrioventriculaire
Ces patients développent une HTAP plus rapidement et plusfréquemment que ceux atteints d’un shunt gauche-droiteauriculaire [20]. Les lésions artériolaires pulmonaires ont été décritesdans la classification de Heath et Edwards [36], comportant aux stadesinitiaux une hypertrophie de la média, puis une proliférationintimale, et enfin des lésions plexiformes. Dans le canalatrioventriculaire complet, la prolifération intimale est plus sévèreet plus précoce chez l’enfant, présente parfois dès les premiers moisde vie, et plus rapide chez les enfants trisomiques atteints, dans 40 %des cas, de cette malformation [39].L’artériolite pulmonaire obstructive se développe dans 8 à 10 % descommunications interventriculaires isolées et est exceptionnellementirréversible avant l’âge de 2 ans. Le risque de développement del’HTAP avec artériolite obstructive dans les shunts gauche-droite àl’étage artériel apparaît plus important que dans les communicationsinterventriculaires isolées. Le caractère définitif et fixé des lésionsartériolaires pulmonaires, contre-indiquant la fermeture chirurgicaledes shunts gauche-droite post-tricuspidiens, est difficile à prévoirpar l’exploration hémodynamique, même si parfois celle-ci estcomplétée par des épreuves pharmacodynamiques, notamment au
NO, et par l’oxygène. Certains cas justifient l’indication d’unebiopsie pulmonaire préopératoire. Celle-ci ne permet pas toujoursd’affirmer avec certitude la réversibilité des lésions, le prélèvementen un site n’étant pas le reflet complet du lit artériolaire pulmonaire.
¶ Cardiopathies cyanogènes
Elles sont susceptibles de se compliquer d’une HTAP avec lésionsobstructives artériolaires pulmonaires, même lorsqu’il existe unesténose, voire une atrésie, sur la voie artérielle pulmonaire.Les facteurs du développement des lésions histologiques sontmultiples et parfois mal expliqués. Ceux-ci comprennentl’augmentation du débit pulmonaire, l’hyperoxie du sang artérielpulmonaire, le rôle des anastomoses bronchiques systémico-pulmonaires avec du sang désaturé et l’obstacle fonctionnel auretour veineux pulmonaire massif.
Transposition des gros vaisseaux
Sans communication interventriculaire (CIV) ou canal artérielperméable, la transposition vasculaire peut se compliquerd’artériolite pulmonaire obstructive chez 8 % des enfants [43, 59]. Chezles patients ayant une CIV large associée ou un canal artérielperméable, le risque de développement d’une artériolite pulmonaireobstructive atteint 40 %, celle-ci pouvant être très précoce, dès l’âgede 3 mois. Les facteurs de la vulnérabilité artériolaire pulmonaire,dans ces cas, ne sont pas clairs, faisant intervenir outre l’hyperdébit,la circulation bronchique, la polyglobulie et la « toxicité » pourl’endothélium vasculaire du sang très oxygéné [43, 59]. Les lésionshistologiques intimales sont particulièrement excentriques,s’accompagnant de microthrombi. Ce risque évolutif destranspositions vasculaires a conduit à proposer une chirurgie trèsprécoce dans les premières semaines de vie.
Tronc artériel commun
Comme dans la transposition des gros vaisseaux avec CIV, le litvasculaire pulmonaire est exposé à un risque précoce dedéveloppement d’artériolite obstructive en règle avant l’âge de 1 an,justifiant une chirurgie réparatrice dans les premiers mois de vie.Les cardiopathies cyanogènes vieillies, à poumons protégés par unesténose ou une atrésie pulmonaire, sont susceptibles de secompliquer d’artériolite pulmonaire obstructive. Celle-ci peut êtrefavorisée par une chirurgie palliative d’anastomosesystémicopulmonaire faite dans la petite enfance, avec un risqued’autant plus important que l’anastomose est généreuse (atrésiepulmonaire avec CIV). Outre les possibles anastomoses, d’autresfacteurs peuvent intervenir, tels le développement d’une circulationcollatérale systémicopulmonaire, les thromboses distales, lapolyglobulie et les anomalies du retour veineux pulmonaire parobstacle mitral ou dysfonction myocardique (double discordance)avec CIV et fuite auriculoventriculaire systémique.
¶ Cardiopathies malformatives gauches
Les cardiopathies gauches malformatives peuvent déterminer uneHTAP. Ce sont essentiellement les obstacles auriculaire gauche,mitral et sur la voie d’éjection aortique. À l’exception durétrécissement mitral malformatif pouvant être rapidement évolutif,les autres obstacles peuvent déterminer une HTAP qui restelongtemps réversible avec, histologiquement, des lésionsd’hypertrophie de la média et de prolifération intimale modérée.
CARDIOPATHIES ACQUISES
Les cardiopathies gauches acquises, responsables d’une HTAP, sontles valvulopathies fuyantes et sténosantes, les valvulopathiesaortiques et les atteintes par augmentation des pressions deremplissage ventriculaire gauche [35]. Ces différentes pathologiespeuvent déterminer une HTAP postcapillaire avec des lésionshistologiques modérées et longtemps réversibles. La sténose mitraleserrée rhumatismale, rencontrée chez l’enfant souvent encore jeune
11-312-A-10 Affections de l’artère pulmonaire Cardiologie
10

en pays d’endémie rhumatismale, peut déterminer une HTAPsévère, avec des lésions histologiques plus sévères qu’à obstacleidentique chez un adulte. Ces lésions se situent au niveau capillaireet dans les vaisseaux immédiatement proximaux et distaux du litcapillaire. Les vaisseaux à parois épaissies sont noyés dans du tissuconjonctif qui enveloppe et comprime les branches vasculairespériphériques.L’hypertrophie artérielle pulmonaire, secondaire aux cardiopathiesmalformatives ou acquises, expose, lors de la chirurgie, quand elleest possible en l’absence d’artériolite obstructive évoluée, au risquepostopératoire immédiat de crises d’HTAP. Celles-ci sont définieshabituellement par une élévation brutale de la PAPs à plus de 75 %de la pression artérielle systémique [12, 97]. Ces crises surviennentprécocement après chirurgie correctrice, entre la fin de la circulationextracorporelle et les tout premiers jours postopératoires. Elles sontimprévisibles. Les facteurs intervenant dans le déclenchement de lavasoconstriction artériolaire sont mieux appréhendés avec, pendantl’intervention, un rôle de circulation extracorporelle responsabled’altération de l’endothélium vasculaire, et des cellules sanguinesresponsables de la libération de substances vasoactives. Enpostopératoire immédiat, l’hypoxie alvéolaire et toute caused’élévation des cathécholamines endogènes constituent des facteursfavorisant la vasoconstriction artériolaire. Le traitement a pour butde supprimer les facteurs aggravants (sédation, lutte contre ladouleur, curarisation, équilibre ionique, maintien d’une normoxiealvéolaire par la ventilation) et d’agir directement sur lavasoconstriction artériolaire. Les drogues les plus régulièrementefficaces sont l’oxygène, le NO et la prostacycline [84].
HYPERTENSION ARTÉRIELLE PULMONAIRETHROMBOEMBOLIQUE
L’HTAP dans le cœur pulmonaire embolique survient quandl’obstruction ampute le lit vasculaire pulmonaire de 60 % de sasurface, et le tableau réalisé est alors celui du cœur pulmonairechronique embolique.Dans les suites d’une embolie pulmonaire aiguë, l’évolutionnaturelle des caillots est de disparaître en 3 à 6 mois [24]. Dans 0,1 %des cas, et pour des raisons inconnues, les caillots persistent etconduisent à la constitution d’une HTAP. Le cœur pulmonaireembolique est maintenant accessible, dans certains cas, à untraitement chirurgical. L’HTAP postembolique est rare chez lespatients ayant des antécédents d’embolies pulmonaires, mais plusfréquente chez les patients associant la survenue d’emboliespulmonaires à une maladie cardiaque ou respiratoire [56]. Le pointde départ embolique est, dans 75 % des cas, d’origine phlébitique,chez l’adulte au-delà de la quatrième décennie. Les facteursfavorisant la survenue de l’HTAP sont l’importance de l’emboliepulmonaire initiale, la récidive de l’accident embolique et laprésence d’une pathologie cardiaque ou pulmonaire associée.On distingue deux formes anatomopathologiques :
– les thromboembolies pulmonaires chroniques des gros troncsartériels pulmonaires : les thrombi d’âges différents, chamois,oblitèrent la lumière artérielle pulmonaire, parfois incorporés à laparoi sous forme de zone de fibrose intimale excentrée ; ces formespeuvent être accessibles à une désobstruction chirurgicale parthromboendartériectomie ;
– les thromboembolies pulmonaires chroniques distales detopographie artériolaire sont moins bien individualisées ; cesembolies sont souvent reperméabilisées par des capillairesnéoformés induisant des lésions de type plexiforme, posant leproblème du diagnostic différentiel avec certaines formes d’HTAPP.
¶ Clinique
Le tableau clinique est celui d’une HTAP avec une dyspnée nonobstructive progressivement aggravée. Parfois, au stade d’HTPévoluée, peuvent survenir des syncopes ou lipothymies d’effort, etrarement des hémoptysies. L’anamnèse est importante à préciser,notamment à la recherche de phlébites ou pneumopathies
récidivantes, mais dans 30 % des cas, il n’est retrouvé aucunantécédent précis de manifestations thromboemboliques. Parfoisexistent, lors de la première consultation, des signes d’insuffisancecardiaque droite. L’examen clinique est initialement peu informatif.La radiographie pulmonaire apporte des signes indirects d’HTAP,avec une saillie du tronc pulmonaire, une dilatation des branchesproximales et parfois une artère lobaire inférieure droite dilatée seterminant en « queue de radis ». L’ECG montre une onde P ample,avec un aspect de surcharge ventriculaire droite.L’échographie cardiaque permet au doppler de chiffrer, sur lavélocité du flux d’insuffisance tricuspide, le niveau de l’HTAP etd’apprécier le retentissement ventriculaire et auriculaire droits decelle-ci [57].
¶ Diagnostic
Il repose sur le cathétérisme, complété par une angiographiepulmonaire.L’exploration hémodynamique confirme l’HTAP précapillaire élevée,parfois de niveau systémique. L’élévation de la pression moyenneauriculaire droite atteste d’une mauvaise adaptation du VD. L’IC austade évolué est diminué et les RAP totales peuvent atteindre deschiffres supérieurs à 20 unités/m_.L’angiographie pulmonaire sélective permet d’affirmer lesobstructions multiples d’origine embolique. Au niveau des grostroncs, elle donne des images de sténoses, parfois excentrées. Lesoblitérations distales au-delà des branches sous-segmentaires sontrarement visibles, se manifestant alors seulement par des zonesavasculaires avec un prolongement du temps artériel et un retardd’opacification capillaire.La scintigraphie pulmonaire, à condition qu’il n’y ait pas depathologie bronchopulmonaire associée, objective la présence detroubles de distribution sanguine, sous forme de zones avasculairesdisséminées et souvent bilatérales. Dans certains cas difficiles, uneangioscopie pulmonaire [42] ou une échographie endovasculaire ontété proposées. Les explorations fonctionnelles respiratoiresconfirment l’absence de pathologie respiratoire pouvant expliquerl’HTAP et montrent, à l’analyse des gaz du sang artériel, unehypoxémie et une hypocapnie. Le bilan diagnostique est complétépar une angiographie veineuse des membres inférieurs et cave, etpar une recherche d’anomalie de l’hémostase (déficit en protéines C,S, antithrombine III, ou en plasminogène) ou la présence d’unanticoagulant circulant.
¶ Évolution et pronostic
Le pronostic de l’HTAP postembolique est sévère, mais très variabled’un patient à l’autre. Des survies prolongées à 10 ans del’apparition des symptômes ont été décrites, ainsi que certainesaméliorations peut-être en rapport avec des fibrinolyses endogènes.L’évolution se fait habituellement cependant vers un tableaud’insuffisance cardiaque droite progressive, parfois interrompue parun accident terminal brutal.
¶ Traitement
Il est préventif par le traitement des embolies pulmonaires et laprévention des récidives par dépistage des thromboses veineuses.Comme les autres formes d’HTAP, l’hypertensionthromboembolique contre-indique la prescription d’œstro-progestatifs.Le traitement médical repose sur l’anticoagulation au long cours pardes antivitamines K pour maintenir un international normalized ratio(INR) entre 2 et 3. L’interruption systématique de la veine caveinférieure est encore discutée, proposée par certains uniquement encas de thromboses veineuses des membres inférieurs. Cette dernièreest partielle, ne réduisant pas le débit cardiaque. Enfin, deuxpossibilités thérapeutiques chirurgicales s’offrent à certains patients :la thromboendartériectomie pulmonaire et la transplantationpulmonaire. La thromboendartériectomie pulmonaire peut conduire
Cardiologie Affections de l’artère pulmonaire 11-312-A-10
11

à d’importantes améliorations fonctionnelles, avec des survies allantjusqu’à 15 ans. Elle n’est utilisable que chez les patients ayant deslésions de thrombose proximale, parfois sous-estimées à lascintigraphie et même à l’angiographie. Le scanner peut parfoisdétecter la limite proximale des lésions. Les facteurs de mortalité decette intervention chirurgicale semblent être l’insuffisance cardiaquedroite et l’obésité. Une meilleure sélection plus précoce des patientsdevrait permettre de réduire la mortalité de cette intervention[3, 11, 40].Si la thromboendartériectomie pulmonaire n’est pas possible, laseule possibilité thérapeutique curative reste la transplantationpulmonaire ou cardiopulmonaire, avec un taux de survie de 70 % à1 an et 40 % à 4 ans.
AGÉNÉSIES OU HYPOPLASIESDE L’ARTÈRE PULMONAIRE
L’agénésie de l’AP a été décrite pour la première fois par Fraentzelen 1868. Elle correspond à l’absence complète d’une des branchespulmonaires. Elle se différencie des formes où l’AP est présente,mais aberrante car naissant de l’aorte, ou discontinue comme dansl’interruption proximale unilatérale de l’AP. Anatomiquement, l’unedes branches pulmonaires naît normalement du tronc de l’AP, tandisque la vascularisation de l’autre poumon, souvent hypoplasique, estassurée par une circulation collatérale, plus ou moins riche,constituée par les artères bronchiques ou par des vaisseauxsystémiques d’origine aortique.
¶ IncidenceIl s’agit d’une affection rare, puisque 200 observations sont publiéesdans la littérature. Cette pathologie est plus fréquente à droite, oùelle est isolée dans un tiers des cas, et alors plus fréquente dans lesexe masculin. L’agénésie artérielle pulmonaire gauche est plussouvent associée à des malformations cardiaques tels la tétralogiede Fallot, la coarctation aortique, le retour veineux pulmonaireanormal. L’agénésie de l’AP gauche est retrouvée dans 4 % destétralogies de Fallot (fig 5).
¶ Embryologie et pathogénieLa pathogénie de cette malformation reste inconnue et denombreuses études et théories embryologiques tentent d’expliquerla survenue des agénésies des branches artérielles pulmonaires. Pourcertains, l’anomalie serait embryologiquement d’origine vasculaire.Le sixième arc aortique participe à la formation du canal artériel parsa racine dorsale gauche, et des AP par ses racines ventrales.L’involution totale du segment proximal du sixième arc aortiqueprovoquerait l’agénésie de l’AP correspondante, la vascularisationpulmonaire homologue étant assurée par la circulation aortique et,à gauche, l’involution touchant la racine ventrale du sixième arc, lepoumon gauche serait vascularisé par le seul canal artériel né de lapartie distale du sixième arc. Cette théorie n’explique pas tous lestypes d’agénésie artérielle pulmonaire, notamment dans les formesassociées à la tétralogie de Fallot et, dans cette association, uneanomalie primaire du cloisonnement aorticopulmonaire a étéévoquée par Cucci [21].
Une agénésie pulmonaire complète peut accompagner l’absence devaisseaux, mais une atteinte dissociée n’est pas exceptionnelle,l’agénésie artérielle pulmonaire pouvant s’associer à une aplasie trèspartielle ou localisée du poumon homologue. Cette dissociationfréquente apparaît de nature embryologique car l’arbre aérienpulmonaire est achevé au 40e jour de l’embryogenèse, bien avantl’organisation de la circulation sanguine qui se fait vers le 64e jour,expliquant à l’inverse qu’une hypoplasie importante du parenchymepulmonaire ne s’accompagne pas obligatoirement d’une agénésieprofonde de l’AP homologue.Un tiers des agénésies artérielles pulmonaires unilatérales sontasymptomatiques. Les complications font la gravité de l’anomalieavec, dans 10 % des cas, des hémoptysies parfois massives, etl’HTAP, dans un quart des cas, constitue le risque évolutif le plusimportant. Les infections respiratoires sont fréquentes etrécidivantes, parfois compliquées d’accès de cyanose ou d’épisodesdyspnéiques itératifs [14, 71, 85].
¶ DiagnosticLe diagnostic repose sur l’imagerie [6]. La radiographie thoraciquemontre une asymétrie de volume entre les deux champspulmonaires. À la scintigraphie isotopique, l’un des poumons estmuet. Le scanner thoracique avec injection confirme la discontinuitéde l’AP et les explorations invasives précisent l’état des résistancespulmonaires, la discontinuité artérielle pulmonaire et la nature de lavascularisation systémique de remplacement. Le traitement estdiscuté dans ses indications et ses modalités. En l’absence desymptomatologie, l’abstention est habituelle. Dans les formesd’agénésie artérielle pulmonaire unilatérale complète, il n’y a pas depossibilité de reconstruction vasculaire chirurgicale. Dans cesformes, une pneumonectomie peut être indiquée pour prévenir ledéveloppement de l’HTAP, les surinfections répétées et leshémoptysies. Dans les formes hors du cadre de l’agénésie typique,avec interruption de l’AP ou naissance aberrante de celle-ci à partirde l’aorte, une chirurgie précoce est proposée, de reconstructionanatomique de revascularisation. Le facteur pronostique majeur decette chirurgie est le degré d’hypoplasie pulmonaire associée. Plusrécemment, la survenue d’hémoptysies a conduit à l’indicationd’embolisations artérielles systémiques avec succès dans deux caschez des adultes. Le pronostic de l’agénésie unilatérale de l’AP estpeu favorable, avec un taux de mortalité de 60 % dans les formesassociées à d’autres malformations cardiaques et de 15 % dans lesformes isolées.
STÉNOSES DES BRANCHES ARTÉRIELLES PULMONAIRES
Les sténoses des branches artérielles pulmonaires intéressent letronc, la bifurcation et les branches. Elles sont connues depuislongtemps. De leur cadre sont exclues les agénésies ou hypoplasiesmajeures d’une branche, associées ou non à une séquestrationpulmonaire. De nature malformative, un tiers d’entre elles sontisolées et deux tiers sont associées à d’autres malformationscardiaques (tétralogies de Fallot essentiellement et shunts). Ledéveloppement de la chirurgie cardiaque malformative palliative afait apparaître à titre de complications des sténoses des branchesacquises, après un cerclage pulmonaire distal ou une anastomosesystémicopulmonaire. La fréquence exacte des sténoses isolées desbranches pulmonaires est mal connue, probablement du fait de leurhabituelle bonne tolérance.
¶ Sténoses congénitales des branches artériellespulmonairesLa possibilité de développement de sténoses congénitales des APs’explique par la complexité de l’embryogenèse artériellepulmonaire.Le tronc pulmonaire dérive du tronc aortique en avant et de lafusion des sixièmes arcs aortiques droit et gauche en arrière.La partie proximale de l’AP droite dérive du sixième arc droit, lapartie distale appartenant au système postbranchial qui l’unit auplexus pulmonaire.
5 Agénésie de l’artèrepulmonaire gauche dansune tétralogie de Fallot.
11-312-A-10 Affections de l’artère pulmonaire Cardiologie
12

L’AP gauche est entièrement issue du système postbranchiall’unissant au plexus pulmonaire gauche.Les systèmes branchiaux sont une chaîne vasculaire unissant lesixième arc aux plexus pulmonaires.Le canal artériel appartient au sixième arc gauche.Les anomalies de la jonction entre ces différents segments peuventgénérer des sténoses vasculaires telles que celles du diaphragme oufaux fibreuses, au niveau du tronc et de la bifurcation. Plusfréquemment, il s’agit d’hypoplasies tubulaires localisées à lumièreétroite et paroi épaissie, à laquelle fait suite une dilatationpoststénotique. Parfois, la sténose est diffuse, réalisant unehypoplasie globale de l’arbre artériel pulmonaire.L’examen histologique des parois vasculaires met en évidence, surles zones sténosées, une prolifération intimale irrégulière, avec unemédia amincie et des fibres élastiques réduites en nombre.La cause de ces anomalies vasculaires n’est pas connue, mais se voitdans un contexte malformatif ou fœtopathologique de dysplasievasculaire, tels les syndromes de Marfan ou d’Ehlers-Danlos, etl’embryopathie rubéolique. Le syndrome de Noonan, le syndromed’Alagille, peuvent s’accompagner de sténoses des branchespulmonaires, rencontrées également de façon habituelle dans lesyndrome de Williams et Beuren, où ces sténoses sont particulièrespar leur évolutivité soit vers la régression, soit parfois versl’amélioration avec le temps [58, 63].Le pronostic des sténoses congénitales des branches artériellespulmonaires est fonction des anomalies cardiaques associées(tétralogie de Fallot) et de la diffusion des sténoses sur l’arbreartériel pulmonaire. Les sténoses isolées sont longtemps bientolérées.Le diagnostic clinique est difficile, l’anomalie étant évoquée enprésence d’un souffle systolique doux de siège essentiellementaxillaire uni- ou bilatéral, ou parfois de souffle continu axillaire liéau gradient systolodiastolique intra-artériel pulmonaire ou à lacirculation systémique de suppléance. En cas de sténoses multipleset serrées, le retentissement ventriculaire droit barométriques’exprime à l’ECG par une surcharge ventriculaire droite.L’échographie cardiaque, complétée par le doppler, confirme leretentissement ventriculaire droit de l’obstacle, et le codage couleurpeut localiser l’obstacle s’il est proximal au niveau du tronc ou de labifurcation pulmonaire, mais l’analyse distale des branchesartérielles pulmonaires échappe aux ultrasons et justifie, dans cetableau, un complément d’imagerie par scanner thoracique ouimagerie par résonance magnétique (IRM) et explorationangiographique.Le cathétérisme permet de mesurer la pression ventriculaire droiteet la PAP qui, en amont de la (ou des) sténose(s), a une morphologieparticulière à diastolique basse, voire nulle, avec une encoche dicroteau pied de la descente de la courbe artérielle pulmonaire. Lesgradients de pression de part et d’autre de la sténose sont ainsimesurés, mais d’interprétation parfois difficile en présence dessténoses multiples et étagées en « chapelet ». L’angiographiesélective pulmonaire permet une imagerie précise des lésionssténosantes. Celle-ci peut être également obtenue par le scannerthoracique avec injection et l’IRM.
¶ Traitement
L’évolution et le pronostic des sténoses des branches artériellespulmonaires sont fonction du degré d’hypertension ventriculairedroite, mais les sténoses isolées des branches pulmonaires sontlongtemps bien tolérées, malgré parfois une pression ventriculairedroite systémique, voire suprasystémique.Un geste chirurgical n’est proposé qu’en cas de mauvaise toléranceet de sténoses localisées et proximales.Certaines sténoses proximales sont accessibles à une angioplastietransluminale [45, 79, 80]. La technique est longue, parfois dangereuse,faisant appel à des ballons dont le diamètre est trois ou quatre foiscelui de la sténose. Les résultats sont dans l’ensemble inconstants,avec un risque de lésions comportant les effractions artérielles, le
développement d’anévrisme et la survenue d’un œdème pulmonaireunilatéral [2] au cours de l’angioplastie. L’évolution à long terme deszones dilatées n’est pas encore connue [8]. La reconnaissance dessténoses des branches pulmonaires dans la tétralogie de Fallot esttrès importante et constitue, de par leur gravité, une contre-indication à la réparation précoce de ces formes de tétralogie, carelles aggravent le pronostic chirurgical en raison de la difficulté deleur réparation chez le petit enfant (fig 6).
¶ Sténoses acquises des branches artériellespulmonaires
Elles résultent de lésions des branches pulmonaires dues, dans laplupart des cas, aux anastomoses palliatives préalables ou, plusrarement, au cerclage pulmonaire ayant glissé vers la bifurcation ensténosant plus ou moins sévèrement la branche pulmonaire droite.Les plus graves des sténoses acquises des branches pulmonaires ontété décrites avec l’anastomose systémicopulmonaire droite deWaterston, maintenant abandonnée, et plus rarement avecl’anastomose de Blalock terminolatérale (fig 7). Le diagnostic de cessténoses est angiographique et leur réparation chirurgicale estd’autant plus difficile et aléatoire que l’enfant est petit et la sténosedistale. Les sténoses consécutives à une anastomosesystémicopulmonaire semblent peu accessibles à une angioplastieefficace. Quelques cas de mise en place de stent dans les sténosesdes branches artérielles pulmonaires ont été rapportés, avec desrésultats en cours d’évaluation [99].
DILATATION IDIOPATHIQUEDE L’ARTÈRE PULMONAIRE
Syndrome défini par la dilatation congénitale du tronc de l’AP enl’absence de cardiopathie (shunt intracardiaque gauche-droite) oude maladie pulmonaire parenchymateuse ou vasculaire.L’étiopathogénie de cette anomalie est mal connue, ayant faitévoquer un vice de division du sac aortique ou une altérationprimitive des lames élastiques [93].Les patients sont asymptomatiques et le diagnostic est évoqué sur laradiographie pulmonaire montrant une accentuation de la convexitéde l’arc moyen gauche avec un cœur par ailleurs normal.L’auscultation peut percevoir un souffle systolique éjectionnel avec
6 Sténoses congénitalesdes branches pulmonaires.
Cardiologie Affections de l’artère pulmonaire 11-312-A-10
13

un « click » d’éjection pulmonaire. L’ECG n’est pas contributif,même en présence d’un bloc incomplet droit sans spécificité.L’anomalie suspectée radiologiquement ne justifie plusd’explorations invasives depuis l’utilisation en routine del’échographie cardiaque et du doppler, où le diagnostic est retenuen l’absence d’anomalie intracardiaque de sténose pulmonairevalvulaire significative, et en présence d’un rapport du diamètre del’AP à la bifurcation au diamètre de l’aorte à l’anneau supérieur à
1,4, et d’un rapport de l’anneau pulmonaire à l’anneau aortiquesupérieur à 1,5 [8].
L’évolution se fait sans complication, avec un pronostic excellentautorisant une vie sans limitations.
Dans les formes où une anomalie du tissu élastique est évoquée,une surveillance est nécessaire, car peut se développer un anévrismeartériel pulmonaire, mais alors associé à d’autres lésions artérielles.
Références[1] Abenhaim L, Moride Y, Brenot F, Rich S, Benichou J, Kurz X
et al. Appetite suppressant drugs and the risk of primarypulmonary hypertension. N Engl J Med 1996 ; 335 :609-615
[2] Arnold LW, Klane JF, Kan JS, Fellows KE, Lock JE. Transientunilateral edema after successful balloon dilatation ofperipheral pulmonary artery stenosis. Am J Cardiol 1988 ;62 : 327-330
[3] AzarianR,Brenot F, SitbonO,Parent F, PetitpretzP,MussetD et al. Hypertension artérielle pulmonaire d’originethrombo-embolique chronique : indications thérapeuti-ques. Arch Mal Cœur 1994 ; 87 : 1709-1713
[4] Barst RJ, Rubin LJ, Long WA, McGoon MD, Rich S, BadeschDBetal.Acomparisonofcontinuous intravenousepopros-tenol (prostacycline) with conventional therapy forprimary pulmonary hypertension: the primary pulmonaryhypertension study group. N Engl J Med 1996 ; 334 :296-302
[5] Barst RJ, Rubin LJ, McGoon MD et al. Survival in primaryhypertensionwith long-termcontinuous intravenouspros-tacyclin. Ann Intern Med 1994 ; 121 : 409-415
[6] Bonnet D, Rey P, Verrot D, Cotton T, Fourcade L, Teshner Det al. Agénésie unilatérale de l’artère pulmonaire. Diagnos-tic et prise en charge thérapeutique. Rev Pneumol Clin1995 ; 51 : 83-86
[7] Bourdillon PD, Oakley CM. Regression of primary pulmo-nary hypertension. Br HeartJ 1976 ; 38 : 264-270
[8] Boutin C, Davignon A, Fournier A, Houyel L, Van DoesburgN. Dilatation idiopathique de l’artère pulmonaire. Aspectséchocardiographiques. Arch Mal Cœur 1994 ; 87 : 663-666
[9] Brenot F. Primary pulmonary hypertension: case seriesfrom France. Chest 1994 ; 105 (suppl) : 335-365
[10] BrenotF,HerveP,RainB,SimonneauG.Hypertensionarté-rielle pulmonaire primitive : données de la littérature etexpérience personnelle de 125 cas en 10 ans. Rev Prat1991 ; 41 : 1560-1567
[11] Brune J, Perol M, Mornex JF. L’hypertension artérielle pul-monaire thrombo-embolique. Rev Prat 1991 ; 41 :1554-1559
[12] Bruniaux J, Demontoux S, Lecronier G, Pulvirenti A. Hyper-tension artérielle pulmonaire paroxystique après correc-tion chirurgicale sous circulation extracorporelle des car-diopathies congénitales. Aspects cliniques et traitement.Cœur 1988 ; 10 : 305-310
[13] Caldini P, Gensini GG, Hoffman MS. Primary pulmonaryhypertension with death during heart catheterization. AmJ Cardiol 1959 ; 4 : 519
[14] Canver CC. Isolated unilateral pulmonary artery agenesis. JThorac Cardiovasc Surg 1993 ; 105 : 766-767
[15] Chaouat A, Weitzenblum E, Higenbottam T. The role ofthrombosis in severe pulmonary hypertension. Eur Respir J1996 ; 9 : 356-363
[16] Christman BW, McPherson CD, Newman JH, King GA,Bernard GR, Groves BM et al. An imbalance between theexcretion of thromboxane and prostacycline metabolitesinpulmonaryhypertension.NEngl JMed1992;327:70-75
[17] Circulation artérielle pulmonaire. Actualités cardiovascu-laires médico-chirurgicales. Paris : Masson, 1970
[18] Cloez JL, Isaaz K, Pernot C. Évaluation par échocardiogra-phie doppler de la pression artérielle pulmonaire chezl’enfant. Cœur 1987, 18 : 189-195
[19] Cordier JF. Étiologies et physiopathologie des hyperten-sions artérielles pulmonaires (HTAP). Rev Prat 1991 ; 41 :1534-1540
[20] Corone P, Doyon F, Gaudeau S, Guerin F, Vernant P,Ducam H et al. Natural history of ventricular septal defect.Circulation 1977 ; 55 : 908-915
[21] Cucci CE, Doyle EF, Lewis JE. Absence of a primary divisionof the pulmonary frunk. An ontogenic theory. Circulation1964 ; 29 : 124-131
[22] D’Alonso GE, Barst RJ, Ayres SM, Bergofsky EH, BrundageBH, Detre KM et al. Survival in patients with primary pul-monary hypertension: results from a national prospectiveregistry. Ann Intern Med 1991 ; 115 : 343-349
[23] D’Alonso GE, Gianotti LA, Pohil RL, Reagle RR, DuRee SL,Fuentes F et al. Comparison of progressive exercise perfor-mance of normal subjects and patients with pulmonaryhypertension. Chest 1987 ; 92 : 57-62
[24] Dalen JE,Banas JS, BrooksHL, EvansGL,Paraskos JA,DexterL. Resolution rate of acute pulmonary embolism in man. NEngl J Med 1969 ; 280 : 1194-1199
[25] Dresdale DT, Schultz M, Michtom RJ. Primary pulmonaryhypertension: clinical and hemodynamic study. Am J Med1951 ; 11 : 888
[26] DunnilMS.Postnatalgrowthof the lung.Thorax1962;17 :329
[27] Furchgott RF, Zawadski JV. The obligatory role of endothe-lial cells in relaxation of arterial smooth muscle by acetyl-choline. Nature 1980 ; 288 : 373-376
[28] Furster V, Steele PM, Edwards WD, Gersh BJ, McGoon MD,Frye RL. Primary pulmonary hypertension: natural historyand the importance of thrombosis. Circulation 1984 ; 70 :580-587
[29] Gaine S, Rubin L. Medical and surgical treatment optionsfor pulmonary hypertension. Am J Med Sci 1998 ; 315 :179-184
[30] Gaine S, Rubin L. Primary pulmonary hypertension. Lancet1998 ; 352 : 719-725
[31] Garcia-DoradoD,MillerDD,GarciaEJ,Delcan JL,MarotoE,Chaitman BR. An epidemic of pulmonary hypertension oftoxic rapeseedoil ingestion inSpain. JAmCollCardiol1983;1 : 1216-1222
[32] Glanville AR, Burke CM, Theodore J, Robin ED. Primary pul-monaryhypertension lengthof survival inpatients referredfor heart-lung transplantation. Chest 1987 ; 91 : 675-681
[33] Halbower AC, Tuder RM, Franklin WA, Pollock JS, Förster-mann U, Abman SH. Maturation-related changes in endo-thelial nitric oxide synthase immunolocalization in devel-oping ovine lung. Am J Physiol 1994 ; 267 : L585-L591
[34] Haworth SG. Pulmonary vasculative. In : Pediatric cardio-logy. Edinburgh : Churchill Livingstone, 1987 ; 123-127
[35] Haworth SG, Hall SM, Patel M. Peripheral pulmonary vas-cular and airway abnormalities in adolescents with rheu-matic mitral stenosis. Int J Cardiol 1988 ; 18 : 405-416
[36] Heath D, Edwards JE. The pathology of hypertensive pul-monary vascular disease: a description of six grades ofstructural changes in the pulmonary arteries with specialreference to congenital cardiac septal defects. Circulation1958 ; 18 : 533-547
[37] HeatleL,AngelsenB.Pulmonaryhypertension. In :Dopplerultrasound in cardiology. Philadelphia : Lea and Febriger,1985 : 252
[38] Himbert J, Renais J, GarciaMoll M, Scebat L, Lenegre J. His-toire naturelle des communications interauriculaires. ArchMal Cœur 1965 ; 58 : 690-710
[39] Hoffman JE, Rudolph AM, Heymann MA. Pulmonary vas-cular disease with congenital heart lesions: pathologic fea-tures and causes. Circulation 1981 ; 64 : 873-877
[40] Jamieson SW, Auger WR, Fedullo PF, Channick RN, KriettJM, Tarazi RY et al. Experience and results with 150 pulmo-nary thrombo-endariectomyoperationsover a29-monthsperiods. J Thorac Cardiovasc Surg 1993 ; 106 : 116-127
[41] Kilbourne EM, Rigau-Perez JG, Heath CW Jr, Zack MM, FalkH, Martin-Marcos M et al. Clinical epidemiology of toxic-oil syndrome: manifestations of a new illness. N Engl J Med1983 ; 309 : 1408-1414
[42] Lacombe P. Angioscopie des artères pulmonaires et desveines caves. Rev Prat 1993 ; 43 : 1120-1123
[43] Lakier JB, Stanger P, Heymann MA et al. Early onset of pul-monary vascular obstruction in patients with aortopulmo-nary transposition and intact ventricular septum. Circula-tion 1975 ; 51 : 875-880
[44] Levin DL, Heymann MA, Kitterman JA, Gregory GA, BubbsRH. Persistent pulmonary hypertension of the newborninfant. J Pediatr 1976 ; 89 : 626-630
[45] Lock JE, Casteneda-Zuniga WR, Fuhrman BP, Bass JL.Balloon dilatation angioplasty of hypoplastic and stenoticpulmonary arteries. Circulation 1983 ; 67 : 962-967
[46] Loyd J, RK, Newman J. Familial primary pulmonary hyper-tension: clinical patterns. , Am Rev Respir Dis 1984 ; 129 :194-197
[47] Loyd JE, Butler MG, Foroud TM, Conneally PM, Phillips JA3rd, Newman JH. Genetic anticipation and abnormalgender ratio at birth in familial primary pulmonary hyper-tension. Am J Respir Crit Care Med 1995 ; 152 : 93-97
[48] Martin-Duran R, Larman M, Trugeda A, Vazquez de PradaJA, Ruano J, Torres A et al. Comparison of Doppler-determined elevated pulmonary arterial pressure withpressure measured at cardiac catheterization. Am J Cardiol1986 ; 57 : 859-863
7 Sténose acquise sur anastomose systémicopulmo-naire palliative.
11-312-A-10 Affections de l’artère pulmonaire Cardiologie
14

[49] Masur Y, Remberger K, Hoefer M. Pulmonary capillaryhemangiomatosis as a rare cause of pulmonary hyperten-sion. Pathol Res Pract 1996 ; 192 : 290-295
[50] Masuyama T, Kodama K, Kitabatake A, Sato H, Nanto S,Inoue M. Continuous wave doppler echocardiographicdetection of pulmonary regurgitation and its applicationsto non invasive estimation of pulmonary artery pressure.Circulation 1986 ; 74 : 484-492
[51] McLaughlin VV, Genthner DE, Panella MM, Rich S. Reduc-tion in pulmonary vascular resistance with long-term epo-prosterenol (prostacyclin) therapy in pulmonary primaryhypertension. N Engl J Med 1998 ; 338 : 273-277
[52] Mesa RA, Edell ES, Dunn WF, Edwards WD. Human immu-nodeficiency virus infection and pulmonary hypertension:two new cases and a review of 86 reparted cases. Mayo ClinProc 1998 ; 73 : 37-45
[53] MoncadaS,HiggsEA.TheL-arginine-nitricoxidepathway.N Engl J Med 1993 ; 329 : 2002-2012
[54] MoncadaS,PalmerRM,HiggsEA.Nitrit oxide:physiology,physiopathology and pharmacology. Pharmacol Rev1991 ; 43 : 109-142
[55] Morin FC, Stenmark KR. Persistant pulmonary hyperten-sionof thenewborn: stateof theart.AmJRespCritCareMed1995 ; 151 : 2010-2032
[56] Moser KM, Auger WR, Fedullo PF. Chronic major-vesselthromboembolic pulmonary hypertension. Circulation1990 ; 81 : 1735-1743
[57] Moser KM, Auger WR, Fedullo PF, Jamieson SW. Chronicthromboembolicpulmonaryhypertension:clinicalpictureand surgical treatment. Eur Respir J 1992 ; 5 : 334-342
[58] Myamura H, Watanabe H, Tatebe S, Egochi S. Spontane-ous regression of peripherial pulmonary artery stenosis inWilliams syndrome. Jpn CircJ 1996 ; 60 : 311-314
[59] Newfeld EA, Paul MH, Muster AJ et al. Pulmonary vasculardisease in complete transposition of great arteries. A studyof 200 patients. Am J Cardiol 1974 ; 34 : 75-82
[60] Nixon PA, Joswiak ML, Fricker FJ. A six-minute walk test forassessing exercise tolerance in severely ill children. J Pediatr1996 ; 129 : 362-366
[61] Palevsky HI, Fishman AP. Comparison of acute hemody-namicresponses toprostacyclinewithstandardvasodilata-tors in patients with primary pulmonary hypertension.Chest 1988 ; 93 : 1795
[62] Palevsky HI, Schloo BL, Pietra GG. Pulmonary primaryhypertension vascular structure, morphometry andresponsiveness to vasodilatator agents. Circulation 1989 ;80 : 1207-1221
[63] PapadopoulosGS,FolgerGM.Progressivepulmonaryarte-rial stenosis. Am J Cardiol 1983 ; 51 : 1462
[64] Petitpretz P, Brenot F, Azarian R, Parent F, Rain B, Herve Pet al. Pulmonary hypertension in patients with humanimmunodeficiency virus infection. Comparison with pul-monary hypertension. Circulation 1994 ; 89 : 2722-2727
[65] Pietra GG, Edwards WD, Kay JM, Rich S, Kernis J, Schloo Bet al. Histopathology of pulmonary hypertension: a quali-tative and quantitative study of pulmonary blood vesselsfrom 58 patients in national heart, lung and blood instituteprimary pulmonary hypertension registry. Circulation1989 ; 80 : 1198-1206
[66] Pointet P, JebrakG.Hypertensionartériellepulmonairepri-mitive. Presse Méd 1988 ; 17 : 1560-1567
[67] Rabinovitch M. Problèmes liés à l’hypertension artériellepulmonaire chez l’enfant porteur d’une cardiopathiecongénitale. Cœur 1988 ; 19 : 254-265
[68] RabinovitchM,HaworthSG,CastanadaAR,NadasAS,ReidLA. Lung biopsy in congenital heart disease: a morphome-tric approach to pulmonary vascular disease. Circulation1978 ; 58 : 1107
[69] RabinovitchM,Keane JF,NorwoodWI,CastanedaAR,ReidLA. Vascular structure in lung biopsy tissue correlated withpulmonary hemodynamic findings after repair of congeni-tal heart defects. Circulation 1984 ; 69 : 655-667
[70] Raffy O, Azarian R, Brenot F, Parent F, Sitbon O, Petitpretz Pet al. Clinical significance of the pulmonary vasodilatatorresponse during short-term infusion of prostacyclin inprimary pulmonary hypertension. Circulation 1996 ; 93 :484-488
[71] Rance F, Martinez J, Poget CH, Baunin CH, Rittie JL,Bremont F et al. Interruption proximale unilatérale del’artère pulmonaire. À propos d’une observation. Sem HôpParis 1998 ; 74 : 914-917
[72] Rhodes J, Barst RJ, Garofano RP, Thoele DG, Gersony WM.Hemodynamic correlates of exercise fonction in patientswith primary pulmonary hypertension. J Am Coll Cardiol1991 ; 18 : 1738-1744
[73] Rich S. Primary pulmonary hypertension. Prog CardiovasDis 1988 ; 31 : 205-238
[74] Rich S, Bundage BH. High dose calcium channel blockingtherapy for primary pulmonary hypertension evidence forlong-term reduction in pulmonary arterial pressure andregression of right ventricular hypertrophy. Circulation1987 ; 76 : 135-141
[75] RichS,DantzkerDR,AyresSM,BergofskyEH,BrundageBH,Detre KM et al. Primary pulmonary hypertension: anational prospective study. Ann Intern Med 1987 ; 107 :216-223
[76] Rich S, Kauffmann E, Levy PS. The effects of higt doses ofcalcium-channel blockers on survival in primary pulmo-nary hypertension. N Engl J Med 1992 ; 327 : 76-81
[77] RichS,KierasK,HartK,GrovesBM,Stobo JD,BrundageBH.Antinuclear antibodies in primary pulmonary hyperten-sion. J Am Coll Cardiol 1986 ; 8 : 1307-1311
[78] Rich S, Pietra GG, Kieras K, Hart K, Bruwdage BH. Primarypulmonary hypertension: radiographic and scintigraphicpatterns of histologie subtipes. Ann Intern Med 1986 ; 105 :499-502
[79] Ring JC, Bass JL, Marvin W, Fuhrman BP, Kulik TJ, Foker JEet al. Management of congenital stenosis of a branche pul-monary artery with balloon dilatation angioplasty. Reportof52procedures. J ThoracCardiovascSurg1985;90:35-44
[80] Rocchini AP, Kveselis D, Dick M, Crowley D, Snider AR,Rosenthal A. Use of balloon angioplasty to treat peripheralpulmonary stenosis. Am J Cardiol 1984 ; 54 : 1069-1073
[81] Romberg E. Ueber sklerose des lungen arterie. Dtsch ArchivKlin Med 1891 ; 48 : 197-206
[82] Rothman A, Perry SB, Keane JP, Lock JE. Early results andfollow-up of balloon angioplasty for branche pulmonaryartery stenosis. J Am Coll Cardiol 1990 ; 15 : 1109-1117
[83] Rubin LJ. Primary pulmonary hypertension. Chest 1993 ;104 : 236-250
[84] Serraf A. Circulation pulmonaire et hypertension artériellepulmonaire : application et implication lors de l’utilisationde la circulation extracorporelle. [thèse], Université ParisXI, faculté de médecine Paris-Sud, 1998 : 1-144
[85] SiebertR,QuoixE,BurghardG.Miseaupointàproposd’uncas d’agénésie unilatérale de l’artère pulmonaire. PneumolClin 1984 ; 40 : 135-138
[86] Simonneau G, Herve P, Petitpretz P et al. Detection ofreversible composent inprimarypulmonaryhypertension:valueofprostacycline infusion. [abstract].AmRevRespirDis1986 ; 133 : 1223
[87] Sitbon O, Brenot F, Denjean A, Bergeron A, Parent F,Azarian R et al. Inhaled nitric oxide as a screening vasodila-tator agent in primary pulmonary hypertension. A dose-response study and comparison with prostacycline. Am JRespir Crit Care Med 1995 ; 151 : 384-389
[88] SpeichR, JenniR,OprauilM,PfabM,Russi EW.Primarypul-monary hypertension in HIV infection. Chest 1991 ; 100 :1268-71
[89] Teitel DF, Iwamoto HS, Rudolph AM. Effects of birth-related events on central blood flow patterns. Pediatr Res1987 ; 22 : 557-566
[90] Thebaud B, Mercier JC. Le monoxyde d’azote inhalé : untraitement plus physiologique de l’hypertension artériellepulmonairepersistantedunouveau-né.Arch Pédiatr1997 ;4 : 988-1003
[91] ThurlbeckWM,AngusGE.Growthanddevelopmentof thenormal human lung. Chest 1975 ; 67 (suppl) : 35
[92] Vaksmann G, Rey C, Mycinski C, Dupuis C. Communica-tion interauriculaire avec hypertension artérielle pulmo-naire sévère chez l’enfant. Arch Mal Cœur 1987 ; 80 :455-460
[93] Van Buchem FS, Meveen J, Marring W et al. Idiopathic dila-tation of the pulmonary artery. Dis Chest 1975 ; 28 : 326
[94] Vogel JH, McNamara DG, Hauman G, Rosenberg H, Jamie-son G, McRady JD. Effects of mild chronic hypoxia on thecirculation in thecalveswith reactivepulmonaryhyperten-sion. Circ Res 1967 ; 21 : 661-669
[95] Wagenvoort CA. Vasoconstriction and medial hypertro-phy in pulmonary hypertension. Circulation 1960 ; 22 :535-546
[96] Wagenvoort CA, Heath D, Edwards JE. The pathology ofthepulmonaryvasculature.Springfield :CCThomas,1964
[97] Wheller J, George BL, Mulder DG, Jarmakani JM. Diagnosisand management of post operative pulmonary hyperten-sive crisis. Circulation 1979 ; 60 : 1640-1643
[98] WoodP.Pulmonaryhypertensionwith special reference tothe vasoconstrictive factor. Br HeartJ 1958 ; 20 : 557-570
[99] Worms AM, Marçon F, Pernot C. Angioplastie translumi-nale percutanée de sténoses des branches artérielles pul-monaires après correction chirurgicale de tétralogie deFallot. Arch Mal Cœur 1989 ; 82 : 701-706
Cardiologie Affections de l’artère pulmonaire 11-312-A-10
15