Embed Size (px)
Citation preview

Th6rapie g6nique des dystrophies musculaires
N a t h a l i e V i n c e n t - L a c a z e
Au m o m e n t o6 la r4alisation d'essais c l in iques de phase I de transfert de g6ne sur des patients atteints de certains types de dystrophies muscu- laires est envisag6e, en France et aux ]~tats-Unis, cet article se propose de faire le point sur les diff6rents essais qui ont 4t6 r6alis6s sur des mod61es animaux.
Programme de therapie genique-GOn~!hon III, lbis, rue de I'lnternationale, 91000 Evry, France.
L e ,premier essai de faisabilit6 d un traitement par transfert de g6ne sur des patients (mar-
quage des TIL : tumour infiltrating lymphocytes) date de 1989 [74]. Depuis, plusieurs journaux enti6re- merit consacr6s ~ la th6rapie g6nique ont vu le jour, de nom- breuses compagnies priv6es ont 6clos, en majorit6 aux Etats-Unis et plus de 300 essais cliniques ont 6t6 autoris6s dans le monde, essentiel- lement des essais de ~ phase I / II ~ ou c'est-h-dire de tol6rance sur un petit nombre de patients, dans les- quels on ne s'interdit cependant pas de v6rifier l'existence ou non d ' u n effet correcteur sur des param6tres biologiques, histolo- giques, ou m6me cliniques. Pour- tant, il existe relativement peu de rapports sugg6rant un effet th6ra- peutique chez des patients [5, 25, 28]. Cette apparente modestie des r6sultats par rappor t a l 'effort financier et huma in consenti depuis plus de dix ans a conduit l 'opinion, publ ique et sur tout scientifique, d ' u n engouement excessif (~ cela va tout gu6rir ~) vers un scepticisrne tout aussi excessif (<~ cela ne marche pas ~). Au jourd 'hu i pour tant , certaines des causes de cette absence d'effi- cacit6 ont 6t6 identifi6es, et des solutions sont a l '6 tude (par exemple pour l 'am61ioration de l ' importation nucl6aire qui est un des facteurs limitant l'efficacit6 des vecteurs non viraux et celle de cer- tains vecteurs viraux, ou la mattrise de la r6ponse immunitaire). I1 exis-
te une autre raison d 'etre optimiste : la th6rapie g6nique int& resse des grands groupes pharma- ceutiques, dont on sait en g6n6ral qu'ils ont peu l 'habitude d'investir
perte. Les sceptiques se r6fugie- ront alors sur un autre terrain : une th6rapie g6nique pour les cancers ou les maladies cardiovasculaires oui, ou pour certains d6ficits en prot6ines circulantes, a la rigueur, mais pour corriger l 'absence d 'une prot6ine structurale dans l 'en- semble de la musculature, non ! Pourtant deux essais cliniques de phase I de transfert de g6ne sur des patients atteints de dys t rophies musculaires, l 'un en France, le second aux Etats-Unis, sont pro- gramm6s pour les mois a venir. Cet article se propose de faire le point sur les diff6rentes strat6gies de th& rapie g6nique qui ont d6ja 6t6 tes- t6es sur les mod61es animaux et sur les probl6mes sp6cifiques qui se surajoutent aux probl6mes clas- siques de toute technique de trans- fert de g6ne, et qui d6coulent de la nature du tissu cible, le muscle squelettique, et de celle des mala- dies en cause, les dystrophies mus- culaires.
1. Les dystrophies muscu- laires • un ensemble clinique- ment et g~n4tiquement het& rogene I1 s'agit d 'un ensemble de maladies se caract6risant par une atteinte progressive des muscles squelet-
ANNALES DE UINSTITUT PASTEUR / actualit6s (1999) 10, 3/4, 327-338 © Elsevier, Paris 327

tiques. Cette faiblesse musculaire r6sulte d 'un processus patholo- gique affectant la viabilit6 des fibres musculaires elles-m6mes. Ce processus s'appr6cie sur quelques caract6ristiques communes [24] : a) une 616vation du taux sanguin d'enzymes musculaires, et b) des anomalies histologiques associant des degr6s variables, selon la pathologie e t /ou le moment o6 l'on se situe dans l'6volution de la maladie, une variation de la taille des fibres, des foyers de n6crose avec des infiltrats de cellules inflammatoires, des foyers de r6g6- n6ration musculaire, une fibrose se traduisant par un 6paississement du tissu conjonctif, une infiltration adipocytaire. On observe une assez grande varia- bilit6 des manifestations cliniques partir d 'un patron commun : dans l'age de survenue des premiers signes, dans la gravit6 de l'atteinte (de la perte de fonction dans les muscles locomoteurs ou respira- toires a l 'existence de simples crampes a l'effort), dans la topogra- phie de l'atteinte (g6n6ralis6e ou limit6e aux 6paules et aux hanches), dans l 'existence d'at-
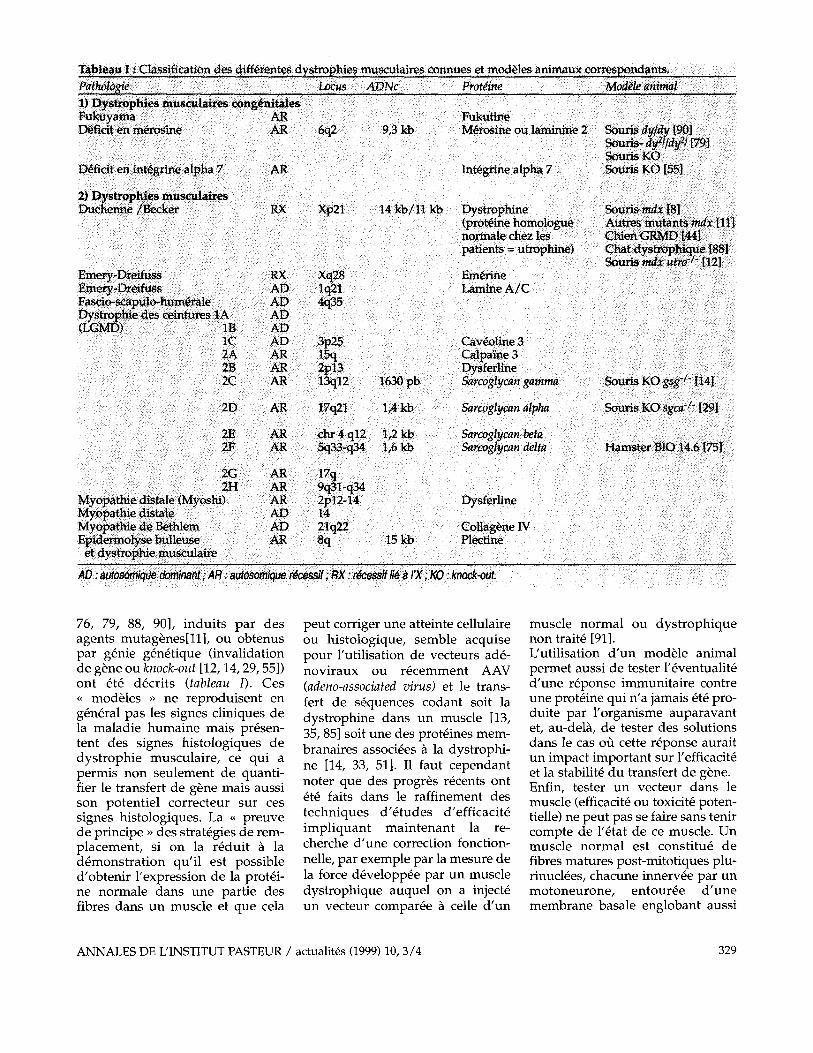
teintes associ6es des syst~mes ner- veux central et p6riph6rique, du muscle cardiaque ou lisse, ou de la peau, dans le mode de transmis- sion et le g6ne impliqu6. Le tableau I reprend les diff6rents groupes de compl6mentations qui ont 6t6 identifi6s ~ ce jour. Pour la majorit6 d'entre eux le g6ne corres- pondant a 6t6 identifi6 ouvrant, dans le cas au moins des dystro- phies transmises de fa~on r6cessi- ve, la voie a une th6rapie par r6in- troduction des s6quences codant la prot6ine d6fectueuse. De faqon int6ressante, plusieurs dystrophies musculaires r6sultent de mutations qui ont un point d'impact commun : la perturbation d 'un 6difice plurimol6culaire qui lie le cytosquelette a la matrice extracellulaire, le complexe dystro- phine/prot6ines associ6es [52] (figure 1). Ont 6t6 d6crites aussi, des mutations perturbant d'autres sys- t6mes membranaires (int6grine ~7 [30], cav6oline 3 [59]) mais aussi des composants de la matrice extracellulaire (m6rosine [31]), des prot6ines cytoplasmiques (calpai- ne-3 [73]) ou nucl6aires (6merine [63], lamine A / C [6]).
D6terminer l'origine du d6ficit pri- maire chez un patient donn6 est un pr6-requis essentiel dans le cas d 'une th6rapie de remplacement mais se r6v61e tr6s difficile dans bien des cas sur le seul examen cli- nique ou parfois m6me apr6s l'ana- lyse immunohistochimique de biopsies musculaires. Dans le cas des pathologies du complexe dys- trophine/prot6ines associ6es, par exemple, la gravit6 de l'atteinte est variable, y compris pour les patients d 'une m6me famille, et l 'absence d 'un des membres du complexe peut entrainer secondai- rement une diminution voire l'ab- sence de certaines prot6ines parte- naires a l'examen des biopsies mus- culaires [82], rendant d61icat le typage exact des patients.
2. Quels modeles animaux et pour quel usage ? D~s lors que l'on cherche ~ 6valuer l'efficacit6 (preuve de principe) ou la toxicit6 potentielle d 'une approche th6rapeutique, se pose le probl6me de la pertinence du module animal utilis6 [69]. Diff6- rents mutants spontan6s [8, 44, 75,
M6rosine "~"
MATRICE EXTRA-CELLULAIRE~ ~
. Dystrophie Musculaire congEnitale
Dystroglycan t~
Dystroglycan Sarcoglycans
a,~,~,,~,~ Dystrophies des ceintures de type 2D, 2E, 2C, 2F
Dystrophine Dystrophies musculaires de Duchenne , de Becker
S
Figure 1. Le complexe des proteines associ6es ~ la dystrophine.
328 ANNALES DE L'INSTITUT PASTEUR / actualit6s (1999) 10, 3/4
i!ii! i~iiiii ii ~!i iiiiii~iiiii~
9,3 kb
AR Int4grine alpha 7
RX Xp21
Em6rine Lamine A/C
AR 17q21 1,4 kb Sarcoglycan alpha
~arCOglycan beta rcoglycan delta
Dysferline
Collag6ne IV 15 kb Plectine
!i~?~iii!511:
76, 79, 88, 90], induits par des agents mutag6nes[11], ou obtenus par g6nie g6n6tique (invalidation de g6ne ou knock-out [12, 14, 29, 55]) ont 6t6 d6crits (tableau I). Ces ~ mod61es ~ ne reproduisent en g6n6ral pas les signes cliniques de la maladie humaine mais pr6sen- tent des signes histologiques de dystrophie musculaire, ce qul a permis non seulement de quanti- fier le transfert de g6ne mais aussi son potentiel correcteur sur ces signes histologiques. La - preuve de principe ~, des strat6gies de rem- placement, si on la r6duit a la d6monstration qu'il est possible d'obtenir l'expression de la prot6i- ne normale dans une partie des fibres dans un muscle et que cela
peut corriger une atteinte cellulaire ou histologique, semble acquise pour l'utilisation de vecteurs ad6- noviraux ou r6cemment AAV (adeno-associated virus) et le trans- fert de s6quences codant soit la dystrophine dans un muscle [13, 35, 85] soit une des prot6ines mem- branaires associ6es a la dystrophi- ne [14, 33, 51]. I1 faut cependant noter que des progr6s r6cents ont 6t6 faits dans le raffinement des techniques d'6tudes d'efficacit6 impliquant maintenant la re- cherche d'une correction fonction- nelle, par exemple par la mesure de la force d6velopp6e par un muscle dystrophique auquel on a inject6 un vecteur compar6e a celle d 'un
muscle normal ou dystrophique non trait6 [91]. Uutilisation d 'un mod61e animal permet aussi de tester l'6ventualit6 d 'une r6ponse immunitaire contre une prot6ine qui n'a jamais 6t6 pro- duite par l'organisme auparavant et, au-dela, de tester des solutions dans le cas oh cette r6ponse aurait un impact important sur l'efficacit6 et la stabilit6 du transfert de g6ne. Enfin, tester un vecteur dans le muscle (efficacit6 ou toxicit6 poten- tielle) ne peut passe faire sans tenir compte de l'6tat de ce muscle. Un muscle normal est constitu6 de fibres matures post-mitotiques plu- rinucl6es, chacune innerv6e par un motoneurone, entour6e d 'une membrane basale englobant aussi
ANNALES DE UINSTITUT PASTEUR / actualit6s (1999) 10, 3/4 329

des petites cellules quiescentes, les cellules satellites, et au contact d 'un r6seau de capillaires dont l'endo- th61ium continu constitue un obs- tacle a la diffusion de composants (cellules, prot6ines, virus) du sang vers le tissu musculaire. Un muscle dystrophique contient des fibres matures, des fibres nouvellement form6es, des foyers de r6g6n6ration avec des myoblastes en prolif6ra- tion, des foyers de n6crose avec des macrophages. I1 peut 6tre, surtout en fin d'6volution de la maladie, le si6ge d 'une fibrose qui sera un s6rieux obstacle a la diffusion du vecteur. Dans certaines formes, comme les dystrophies muscu- laires cong6nitales li6es a un d6ficit en m6rosine, la membrane basale sera par contre d6sorganis6e. Uen- vironnement dans lequel va se trouver le vecteur est donc radica- lement diff6rent dans un tel muscle. II est 6vident dans ces conditions qu'on n'utilisera peut- 6tre pas le m6me vecteur dans un muscle normal (par ex. pour obte- nir la s6cr6tion d 'un facteur circu- lant dans le cadre d'une pathologie non musculaire) ou pour traiter un muscle dystrophique. Cette diff6- rence de structure est un argument suppl6mentaire pour utiliser aussi des animaux mutants dans les 6tudes toxicologiques afin d'6va- luer au mieux la toxicit6 li6e a la r6ponse immunitaire ou au risque de diss6mination en dehors du site d'injection (la vascularisation par exemple pourrait aussi ~tre modi- file dans de tels muscles).
3. Place du transfert de gene dans les approches therapeu- tiques des dystrophies mus- culaires Les tentatives pour corriger les effets de l'absence d'une prot6ine dans les muscles squelettiques ont fait appel d'abord a la greffe de cel- lules musculaires, puis au transfert de g6ne in vivo pour remplacer la prot6ine manquante ou pour apporter des facteurs trophiques. R6cemment, des tentatives de
manipulation de l'expression de g6nes homologues au g6ne rout6 (lorsqu'ils existent) par administra- tion de drogues, ont 6t6 propos6es et pourraient, sous condition d'ob- tention de drogues non toxiques, fournir un moyen plus efficace de traiter une atteinte g6n6ralis6e.
• 3.1. Uinjection de myoblastes
La greffe de myoblastes utilise la propri6t6 des cellules satellites entrer a nouveau en cycle pour donner des myoblastes qui prolif6- rent, et fusionnent, formant de nouvelles fibres musculaires. Ce processus se produit aussi bien in vivo, lors du processus de r6para- tion du muscle, qu'ex vivo, o6 des cellules satellites isol6es a partir de tissu musculaire peuvent prolif6rer. Les myoblastes provenant d 'un donneur sain ou du receveur sont cultiv6s, 6ventuellement g6n6tique- ment modifi6s ex vivo, puis r6im- plant6s dans un muscle receveur, o6 ils vont contribuer dans une pro- portion variable a l'61aboration des fibres [66]. Les essais sur la souris mdx ~ l'aide de myoblastes nor- maux ont permis d'observer en moyenne 30 a 40 % de fibres expri- mant la dystrophine. Plusieurs essais ont 6galement 6t6 tent6s chez des patients atteints de dystrophie musculaire de Duchen- ne [36, 49], mais avec moins de suc- cbs. Diff6rents essais qui ont 6t6 ten- t6s chez des chiens GRMD [44], n'ont pas donn6 de r6sultat positif. Une des raisons du peu d'efficacit6 est la mise en 6vidence d'une mor- talit6 importante (plus de 90 % des cellules inject6es) dans les heures qui suivent l'injection. Cette morta- lit6 est partiellement limit6e par la prise d 'un anti-inflamatoire [70].
• 3.2. Remplacement de la prot4i- ne manquante par transfert de g~ne dans les fibres musculaires
Dans le cas de maladies r6cessives, les techniques de transfert de gbne permettent d'envisager la r6intro- duction des s6quences codant pour la prot6ine absente [1, 16, 51, 71]. En
r6alit6, diff6rentes difficult6s li6es la taille de la s6quence codante par- fois impossible a cloner dans le vec- teur (11 kb pour la dystrophine [43] ?), ou a la r6ponse immunitaire contre la prot6ine ainsi r6introduite, ont fait envisager des strat6gies alternatives. Dans le cas de la cor- rection des dystrophies musculaires de Duchenne et de Becker, on a ainsi propos6 l'utilisation de s6quences codant pour des ~ mini- dystrophines ~ [18] voire de ~ microdystrophines ~ [93] o6 des domaines de la prot6ine jug6s non essentiels sont partiellement ou compl6tement absents. Les ADNc correspondants ont une taille qui autorise le clonage dans les diff6- rents vecteurs existant. L'aptitude de ces dystrophines tronqu6es remplacer la prot6ine compl6te peut-6tre v6rifi6e par croisement de souris transg6niques exprimant ces formes tronqu6e dans les muscles avec des souris mdx [68, 87]. Pour pr6venir le rejet immunolo- gique des cellules g6n6tiquement modifi6es, certains groupes pr6co- nisent plut6t l'utilisation de s6quences codant une prot6ine homologue de la dystrophine, l'utrophine [54], voire de versions tronqu6es de cette prot6ine. L'utro- phine est cod6e par un g6ne situ6 sur le chromosome 6. Elle est abon- dante dans le muscle foetal, mais son expression est limit6e ~ la jonction neuromusculaire dans le muscle adulte [53]. Elle interagit avec le m6me complexe membranaire que la dystrophine [38, 46] et pourrait normalement remplacer celle-ci. Cette hypoth6se a jusqu'a pr6sent 6t6 valid6e par croisement de diff6- rentes souris exprimant a haut niveau l'utrophine dans le muscle adulte avec des souris mdx [81].
• 3.3. Transfert de g6nes codant des facteurs diffusibles
Certains r6sultats sugg6rent que le m6canisme de destruction des fibres pourrait, dans certains cas, d6clencher une mort cellulaire pro- gramm6e ou apoptose [80]. Par ailleurs, plusieurs 6quipes ont
330 ANNALES DE UINSTITUT PASTEUR / actualit6s (1999) 10, 3/4

entrepris d'6tudier le r61e de diff6- rents facteurs de croissance, comme le bFGF (basic fibroblast growth factor), sur la multiplication des myoblastes, la maturation des fibres ou leur survie [77]. Certains de ces facteurs comme I'IGFII (insu- lin-like growth factor) auraient d6montr6 une action antiapopto- tique sur des myoblastes en culture (sugg6rant qu'il pourrait ~tre int6- ressant de d61ivrer ces facteurs dans les muscles dystrophiques de faqon a allonger la dur6e de vie des fibres) [77]. Une approche int6ressante et origi- nale a ainsi 6t6 envisag6e dans ce but. Elle utilise le fait que dans le muscle dystrophique, il existe un afflux de macrophages dans les foyers de n6crose. Ainsi, il a 6t6 montr6 [65] que des macrophages marqu6s et inject6s par voie veineu- s e ~ des souris mdx (un mod61e murin de dystrophie musculaire par absence de dystrophine) pou- vaient 8tre retrouv6s dans les muscles, ce qui sugg6re qu'il serait possible de les utiliser comme navettes, apr6s modification g6n6- tique, pour la d61ivrance des fac- teurs trophiques au niveau des foyers de n6crose musculaire. Plusieurs probl6mes demeurent, en particulier celui de la manipulation ex-vivo de ces macrophages ou de cellules souches m6dullaires, et sur- tout le fait que les facteurs intro- duits vont aussi ~tre d61ivr6s en
dehors des muscles, partout off les macrophages seront recrut6s et acti- v6s.
• 3 .4 . A u t r e s v o i e s
t h 4 r a p e u t i q u e s
Des tentatives de correction d'une mutation par manipulation des ARNm ont 6t6 pr6sent6es a un r6cent colloque organis6 par l'As- sociation franqaise contre les myo- pathies a Paris. Le mod61e murin de dystrophie musculaire de Duchenne pr6sente un codon stop dans l'exon 23 du g6ne de la dys- trophine. Plusieurs 6quipes ont essay6 d' induire une exclusion s61ective de cet exon (exon skipping) au cours de la maturation de I'ARNm en ciblant les sites accep- teurs ou donneurs par des oliginu- cl6otides anti-sens, dans le but de r6tablir le cadre de lecture [15]. La proportion de transcrit excluant l'exon 23 par rapport au transcrit non modifi6 augmente avec la dose d'oligonucl6otide administr6e. I1 a aussi 6t6 propos6 d'inverser au niveau g6nomique, a l'aide d'oligo- nucl6otides chim6res ARN/ADN [47], la mutation pr6sente chez le chien GRMD [44], un autre mod61e animal de dystrophie de Duchenne (mutation entrainant l'exclusion de l'exon 7 du g6ne de la dystrophine au cours de l'6pissage aboutissant un d6calage du cadre de lecture [76]).
Cette approche plus pharmacolo- gique (on injecterait directement des oligonucl6otides synth6tiques) est a rapprocher des tentatives : a) de manipuler au moyen de drogues l'expression de g6nes homologues au g6ne rout6 (ex. : augmenter l'expression du g6ne de l'utrophine endog6ne pour tenter de compenser l'absence de dystro- phine) [26], ou b) d'ignorer les codons stop (en administrant des aminoglycosides par exemple) sur les transcrits mut6s [3]. Ces voies de recherche sont d6velopp6es en parall61e avec les approches clas- siques de transfert de g6ne et sont int6ressantes du fait de la nature de ces maladies dans lesquelles les cel- lules a corriger sont nombreuses et localis6es a plusieurs endroits. Uef- ficacit6 et la toxicit6 potentielles de toutes ces nouvelles approches sont en cours d'investigation.
4. Proprietes des vecteurs testes Diff6rents vecteurs de transfert de g6ne ont 6t6 test6s sur des mod61es animaux de dystrophies muscu- laires (tableau II).
• 4 .1 . I n j e c t i o n d ' A D N n u
Les premiers essais de transfert de g6ne ont utilis6 l'injection directe dans le muscle de souris mdx d'un plasmide contenant I'ADNc corn-
Taille < 40 nm - m Infect ion + +
! TM g~n~ration ~ Gutless. - - m
+ +
SV ADN nu
-t- -- q - l - - + +
f ibres + + f - + / -
+ + +
+ + +
+ + + I -
+ - / + - -
8kb 8kb 9kb 35 kb 5 kb 30 kb > 30 kb
- ? + ? ? +
+ / - - - - + + - -
+ / - +/ - + +/ -
AAV : adeno-associated-virus ; HSV : herpes simplex virus.
ANNALES DE UINSTITUT PASTEUR / actualit6s (1999) 10, 3/4 331

plet de la dystrophine Cette injec- tion a permis d'obtenir 2 a 3 % de fibres exprimant la prot6ine, avec une dur6e d'expression prolong6e (18 mois) [1]. Pour des raisons mal connues, les fibres musculaires se r6v61ent en effet capables de capter de I'ADN nu. Dans le noyau, I'ADN persiste sous forme 6piso- mique, mais la grande majorit6 de I'ADN capt6 par les cellules est s6questr6 dans le cytoplasme ce qui explique en partie la faible efficaci- t6 de transduction [19]. De nom- breux laboratoires s'int6ressent donc maintenant au trafic cytoplas- mique et aux m6canismes d'impor- tation vers le noyau. Afin d 'augmenter l'efficacit6 de transduction par I'ADN nu, on peut aussi appliquer au muscle trait6 un champ 61ectrique [60]. Les r6sultats obtenus chez la souris normale avec des plasmides por- rant un g6ne traceur montre une efficacit6 de transduction sup6rieu- re a celle obtenue apr6s une injec- tion simple, mais il n 'y a pour l'ins- tant pas de donn6es publi6es mon- trant une efficacit6 aussi impor- tante dans un modble de dystro- phie musculaire avec un plasmide contenant un ADNc th6rapeutique. En d6pit de ces progrbs r6cents, l'int6r~t d 'un vecteur plasmidique r6side plut6t pour l'instant dans la possibilit6 de le produire en grandes quantit6s, de transf6rer des s6quences de grande taille (la s6quence codante de la dystrophi- ne par exemple) et dans sa moindre immunog6nicit6 par rapport ~ la plupart des vecteurs viraux.
• 4.2. Inject ion d'un vecteur r6troviral
Ce type de vecteur est d6riv6 du virus de la leuc6mie murine de Moloney (MoMuLV) par la d616- tion de la quasi-totalit6 des s6quences virales, a l'exception des LTR (long terminal repeat) et de la s6quence d'encapsidation. Ainsi modifi6, il doit ~tre propag6 sur des cellules trans-compl6mentantes ayant int6gr6 les g6nes viraux gag, pol et env, n6cessaires a la r6plica-
tion virale. Sa capacit6 d'accueil est limit6e a 8 kb. Le principal int6r6t de ces vecteurs est leur int6gration sous forme de provirus dans le g6nome de la cellule h6te permet- tant leur transmission aux cellules filles en cas de mitose. Cette pro- pri6t6 est particuli~rement int6res- sante si l'expression du transg6ne confbre un avantage s61ectif aux cellules transduites. En revanche, les titres de production restent inf6- rieurs a ceux obtenus avec l'ad6no- virus et l'importation vers le noyau n6cessite la rupture de la membra- ne nucl6aire et donc la division de la cellule h6te. Ce type de vecteur a d'abord 6t6 utilis6 pour transduire un minigb- ne de dystrophine ex vivo dans des myoblastes de souris [17]. En fait, le r6trovirus pourrait permettre une transduction directe in vivo, car dans le muscle dystrophique, il existe une r6g6n6ration active. Ainsi, les myoblastes ont pu ~tre infect6s in vivo par injection intra- musculaire directe, chez la souris mdx, d'un r6trovirus recombinant permettant d'obtenir de 4 a 6 % de fibres exprimant la dystrophine [16].
• 4.3. Vecteurs ad4noviraux
Les vecteurs ad6noviraux sont d6riv6s des ad6novirus humain de type 2 ou 5, non transformants. Les vecteurs dits ~ de premi6re g6n6ra- tion ~ d616t6s pour les r6gions EIA, E1B, et E3 peuvent peuvent ~tre produits a haut titre sur des cel- lules transcompl6mentantes 293, et ont une capacit6 d'accueil maxima- le de 8 kb. Ils se sont r6v616s tr6s efficaces pour transduire les muscles squelettiques d'animaux nouveau-n6s. Uinjection intramus- culaire d 'un vecteur ad6noviral v6hiculant un ADNc codant pour une ~ minidystrophine ~ [71] sur des mutants mdx nouveau-n6s a permis d'obtenir jusqu'a 60 % de fibres transduites pour une dur6e d 'au moins six mois. Ce haut niveau d'expression a permis d'ob- server une diminution nette des signes de dystrophie musculaire
dans le muscle trait6, et une aug- mentation de la dur6e de vie des fibres transduites [85] traduisant une plus grande r6sistance m6ca- nique du sarcolemme en pr6sence de dystrophine [13]. Par contre les vecteurs ad6novi- raux Ad2 et Ad5 de premi6re g6n6- ration ont une efficacit6 de trans- duction diminu6e dans le muscle des souris adultes du fait d'une diminution de l'expression des r6cepteurs utilis6s par ces vecteurs [2, 64] d'une diffusion limit6e ~ tra- vers la membrane basale mature [20] et de l'existence d'une r6ponse immunitaire [92]. I1 a 6t6 d6velopp6 des vecteurs ad6- noviraux a grande capacit6 (ou gut- less) dans lesquels la totalit6 des g6nes viraux sont d616t6s [9], [48]. Ces vecteurs sont moins immuno- g6nes et ont une capacit6 d'accueil de 37 kb qui leur permet de v6hicu- ler la s6quence codante de la dys- trophine enti6re, et non les versions raccourcies test6es pr6c6demment, ou bien la s6quence codant la m6ro- sine (absente dans certaines formes de dystrophies musculaires cong6- nitales). Ils sont produits sur des cellules 293 en pr6sence d 'un virus auxiliaire qui fournit en trans les fonctions n6cessaires a la r6plica- tion. Dans le virus auxiliaire la s6quence d'encapsidation est flan- qu6e de s6quences lox qui permet- tent son excision dans des cellules productrices exprimant la recombi- nase Cre de fa~on a limiter la conta- mination du stock final par le virus auxiliaire (de l'ordre de 0,1% dans les pr6parations actuelles).
• 4.4. Vecteurs AAV
Les vecteurs AAV sont apparus r6cemment. Ils se sont r6v616s tr6s vite tr6s int6ressants pour infecter les muscles squelettiques adultes et obtenir une expression prolong6e, y compris chez des animaux immu- nocomp6tents [10, 23, 41, 61, 78, 89]. Ils sont d6riv6s d 'un parvovi- rus humain, AAV2, non pathog6ne,
ADN simple brin et naturelle- ment d6fectif puisqu'il ne peut se r6pliquer qu'en pr6sence d 'un
332 ANNALES DE UINSTITUT PASTEUR / actualit6s (1999) 10, 3/4

virus auxiliaire, par exemple un ad6novirus. En l'absence de ce virus auxiliaire, I'AAV sauvage s'int6gre dans une r6gion localis6e sur le chromosome 19 [45] et per- siste a l'6tat latent jusqu'a une sur- infection 6ventuelle par le virus auxiliaire [4]. Le vecteur construit conserve seu- lement les s6quences terminales invers6es (ITR) du virus sauvage, s6quences jouant un r61e important dans la conversion de I'ADN viral simple brin en mol4cules double brin pouvant 6tre transcrites. Cette 6tape est semble-t-il l'6tape limi- tante pour l'expression du transg6- ne [21, 22] et n6cessite la participa- tion de facteur(s) cellulaire(s). I1 semble que les fibres musculaires constituent un environnement per- missif pour une conversion rapide en forme double brin [86] et un maintien stable a long terme. Le principal inconv6nient de ce vec- teur est sa faible capacit6 d'accueil (4 a 5 kb) qui ne lui permet pas par exemple d'accueillir la s6quence codant la m6rosine, ni celle codant la dystrophine enti6re, mais seule- ment des versions tr6s courtes de cette derni6re, des ~ microdystro- phines ~ [93] dont la fonctionnalit6 n'est pas d6montr6e. Par contre, ce vecteur peut accueillir les s6- quences codant pour des prot6ines impliqu6es dans les dystrophies musculaires des ceintures (voir tableau I). R6cemment, un vecteur AAV portant I'ADNc d 'un des quatre sarcoglycanes (le sarcoglycan 8 impliqu6 dans la dystrophie des ceintures de type 2B) a 6t6 test6 sur le hamster BIO 14-6 [51] n'expri- mant pas cette prot6ine et porteur d 'une cardiomyopathie et d 'une dystrophie musculaire histolo- gique. Les r6sultats obtenus sur ce module ont mis en 6vidence, pour le vecteur AAV, un bon pouvoir infectant du muscle dystrophique, une stabilit6 d'expression et une correction de certains signes histo- logiques.
• 4.5. Autres vecteurs
Un vecteur bas6 sur HSV1 (herpes simplex virus) pourrait, lui, accueillir I'ADNc complet [37], mais sa toxicit6 potentielle n'en fait pas pour l'instant un tr6s bon can- didat pour une utilisation chez l'homme. De plus, il ne semble pas infecter efficacement les fibres musculaires matures [20]. De m~me, les vecteurs lentiviraux, en particulier ceux d6riv6s du VIH, sont potentiellement tr6s int6res- sants car ils sont eux capables d'in- fecter le muscle adulte [40], mais leur utilisation en th6rapeutique humaine ne pourra ~tre valid6e que sur des crit6res plus s6v6res que ceux utilis6s pour d'autres vec- teurs d6riv6s de virus peu ou pas pathog6nes.
5. Quel est le niveau d'expres- sion requis ? Le niveau d'expression requis pour esp6rer un effet th6rapeutique dans un muscle donn6 peut ~tre appr6- ci6 par l '6tude des ph6notypes observ6s chez certains patients, qui expriment la prot6ine a un taux diminu6, par l '6tude de souris transg6niques, ou r6cemment par l'6tude de la correction de la force d6velopp6e par les muscles trait6s, 6valu6e ex-vivo. Dans le cas de la dystrophie musculaire de Duchen- ne, par exemple il existe des patients ayant une mutation dans le promoteur musculaire et qui se pr6sentent avec une atteinte car- diaque tr6s s6v6re mais une attein- te tr6s mod6r6e des muscles sque- lettiques [62]. Cette dissociation dans la gravit6 de l'atteinte r6sulte du fait que, dans les muscles, des promoteurs alternatifs permettent l'expression de la dystrophine 30 % du niveau normal. Des 6tudes de correction fonctionnelle sur des muscles trait6s ont permis de mettre en 6vidence une diff6rence significative par rapport au t6moin non trait6 a partir de 25 % de fibres marqu6es (N. Deconinck, commu- nication personnelle).
6. Quelles sont les obstacles une transduction efficace des fibres ? Pour atteindre un tel niveau un cer- tain nombre d'obstacles vont devoir ~tre franchis. Certains d'entre eux ont 6t6 6voqu6s au cours de la pr6sentation des diff6- rents vecteurs. Ces difficult6s pro- viennent d 'une part de la nature des pathologies a cibler, d'autre part de la structure des muscles [83].
• 6.1. Contraintes r4sultant de la nature des pathologies cibl6es
Elles peuvent affecter l'ensemble de la musculature, notamment des muscles profonds comme le dia- phragme, ou le muscle cardiaque, ce qui n6cessite d'essayer d'admi- nistrer les vecteurs par voie syst6- mique. II existe plusieurs types cellulaires dans un muscle donn6 ~ un moment donn6, fibres matures, fibres nouvellement form6es, myo- blastes en prolif6ration, cellules satellites, et tous ces types ne sont pas tous 6galement r6ceptifs ~ u n vecteur donn6. Dans les pathologies impliquant des prot6ines du sarcolemme, il existe une moindre r6sistance m6canique de la membrane cellu- laire des fibres [56, 57] qui n6cessi- te pour ~tre corrig6e que la prot6i- ne manquante soit exprim6e tout le long des fibres, faute de quoi la protection contre les dommages caus6s par les contractions r6p6t6es n'est pas efficace.
• 6.2. Principaux obstacles structuraux
I1 s'agit : - d 'un endoth61ium capillaire continu qui emp6che la diffusion des vecteurs administr6s par voie syst6mique ; - du tissu conjonctif qui d61imite les muscles [27], et, a l'int6rieur de chaque muscle, des faisceaux de fibres ;
ANNALES DE UINSTITUT PASTEUR / actualit6s (1999) 10, 3/4 333

- de la membrane basale de chaque fibre qui ne laisse passer que des composants d 'une taille inf6rieure /l 40 nm dans un muscle adulte.
• 6.3. Obstacles biologiques
Ce sont : - la diminution de l'expression des r6cepteurs, par exemple ceux utili- s6s par les vecteurs ad6noviraux, dans les fibres matures ; - une importation inefficace dans les noyaux qui est un problbme g6n6ral du transfert de gbne par les vecteurs synth6tiques ou par les vecteurs r6troviraux (non lentivi- raux) ; - la diffusion limit6e des transcrits ou des prot6ines autour des noyaux g6n6tiquement modifi6s est un probl6me sp6cifique au muscle squelettique ; les fibres musculaires sont des cellules pluri- nucl6es pouvant faire plusieurs millimbtres de long ; chaque noyau contr61e une zone de taille variable selon les protbines, appel6 << domai- ne nucl6aire >> [67, 72] ; - la rbponse immunitaire contre les cellules g6n6tiquement modifi6es.
7. Quelles sont les solutions en cours d'evaluation ? La transduction ~ haut niveau des muscles d 'un membre entier apr6s injection intra-art6rielle d 'un vec- teur a 6t6 d6crite par deux groupes de chercheurs. Le groupe d'H. Stedman a test6 la perm6abilisation des capillaires par une perfusion d'histamine et de papav6rine pr6alablement/t l'injec- tion de vecteurs ad6noviraux ou AAV dans l'art6re iliaque sur des rats ou sur des hamsters [27]. Cet article pr6sente notamment une correction histologique dans l'en- semble des muscles d 'une patte arri6re sur le hamster BIO 14-6 apr6s perfusion d 'un vecteur AAV v4hiculant le g6ne du sarcoglycan delta. Cette technique est toutefois assez d61icate, car le passage des agents perm6abilisants dans la cir- culation g6n6rale tue instantan6-
ment les animaux. La vascularisa- tion de la patte trait6e doit donc ~tre parfaitement isol6e de la circu- lation corporelle par un tourniquet plac6 sur l'art6re et la veine iliaques pendant la dur6e de l'exp6rience. L'application de cette technique sur des patients d6j~ fragiles et suscep- tibles de faire des accidents d'anes- th6sie graves, n'est donc pas 6vi- dente actuellement. Le groupe de J. Wolff a obtenu des r6sultats tout a fait remarquables sur des rats en injectant un plasmi- de par voie art6rielle dans un membre entier, la veine 6tant momentan6ment clamp6e de fa~on /t cr6er une hyperpression impor- tante dans l'arbre vasculaire [7]. La diffusion limit6e apr6s une injection directement dans le muscle n6cessiterait de multiplier les sites d'injection (tous les 5-10 mm). Certains sugg6rent d'uti- liser un traitement enzymatique permettant de d6grader la matrice extracellulaire, d'autres, d'induire une r6g6n6ration musculaire, l'in- fection du muscle par certains vec- teurs comme les vecteurs ad6novi- raux se faisant semble-t-il plut6t par l'infection de myoblastes qui sont ensuite secondairement fusionn6s pour donner des fibres [20]. La baisse d'expression des r6cep- teurs utilis6s normalement par les vecteurs ad6noviraux pourrait ~tre contourn6e par une modification des prot6ines de capside de mani6- re h cibler d'autres r6cepteurs [58]° La limitation de la diffusion des transcrits ou des prot6ines a distan- ce des noyaux n6cessite quanta elle d'optimaliser la cassette d'expres- sion par exemple a l'aide d 'un pro- moteur aussi puissant que possible. Une autre voie de recherche explorer est l 'adjonction de s6quences 3' non traduites permet- tant un << adressage >> efficace des transcrits. En effet, il a 6t6 d6crit que les transcrits pouvaient 6tre adress6s, ou retenus, dans certains compartiments sub-cellulaires [32]. Cette forme d'adressage n'a pas 6t6 d6montr6e pour les prot6ines sar- colemmales, et il existe en fait peu de donn6es, mais on sait par
exemple qu'une s6quence de 1 kb environ de la partie 3' non traduite du transcrit de la dystrophine est extr~mement conserv6e entre le transcrit humain et les transcrits d'autres esp6ces comme le poulet [50]. La r6ponse immunitaire qui accom- pagne l'injection de la plupart des vecteurs, en particulier les vecteurs ad6noviraux peut ~tre contr6e par l 'administration d ' immunosup- presseurs [35, 84], par l'utilisation de vecteurs moins immunogbnes (plasmides, a condition d'61iminer au maximum les s6quences de type CpG [42], les vecteurs lentiviraux, les vecteurs AAV [39] ou m6me les ad6novirus gutless).
Conclusion : des modeles ani- maux aux essais cliniques La place de la th6rapie g6nique dans le traitement des dystrophies musculaires est actuellement potentiellement tr6s importante, parce que ces pathologies ne b6n6- ficient actuellement que de mesures palliatives (qui ont cepen- dant, il faut le signaler, permis d'al- longer la dur6e de vie des patients les plus s6v6rement atteints et d'am61iorer leur qualit6 de vie), que les 6tudes sur diff6rents mod61es animaux ont globalement apport6 la preuve que l'introduc- tion locale d 'un g6ne normal per- met une restauration cellulaire, his- tologique et, r6cemment, fonction- nelle, et que la vectorologie est tr6s probablement amen6e a se d6ve- lopper consid6rablement. Toute- fois, il subsiste de nombreuses dif- ficult6s a surmonter pour atteindre un niveau de transduction efficace dans un muscle donn6. Dans ce contexte entreprendre des essais clinique de phase I dans deux types de dystrophies musculaires peut paraitre pr6matur6. Ne vaudrait-il pas mieux attendre de nouveaux vecteurs plus efficaces, mieux tol6r6s par le syst6me immunitaire, ou que le probl6me de l'adminis- tration syst6mique soit r6solu par des techniques r6ellement appli- cables sur des patients ?
334 ANNALES DE UINSTITUT PASTEUR / actualit6s (1999) 10, 3/4

C o n c e r n a n t ce d e r n i e r po in t , u n e 6 t ape i n t e r m 6 d i a i r e d e p l u s en p l u s s o u v e n t 6voqu6e [34] p o u r r a i t ~tre d e t r a i t e r d e s g r o u p e s m u s c u l a i r e s local is6s , d o n t la fonc t ion est tr6s i m p o r t a n t e p o u r la qua l i t6 d e la v ie q u o t i d i e n n e (pa r ex : le de l to ' /de p e r m e t t a n t d e m o b i l i s e r l ' 6pau l e , o u les m u s c l e s a c t i o n n a n t la p i n c e p o u c e / i n d e x ) , p l u t 6 t q u e l ' e n - s e m b l e d e la m u s c u l a t u r e . Les vec- t eu r s ex i s t an t , n o t a m r n e n t le vec- t eu r A A V d a n s le cas d e p a t h o l o - g ies l i6es a u d6 fau t d ' u n e p r o t 6 i n e cod6e p a r u n A D N c d e ta i l le suffi- s a m m e n t p e t i t e, p o u r r a i e n t ~tre s u f f i s a m m e n t efficaces. U n e s sa i c o n d u i t p a r la soci6t6 Transg6ne d e v r a i t d 6 b u t e r d a n s les p r o c h a i n s m o i s p o u r t e s te r la tol6- r ance a u n e in jec t ion d a n s le b i c e p s d ' u n p l a s m i d e p o r t a n t I ' A D N c d e la d y s t r o p h i n e , s u r d e s p a t i e n t s a t t e in t s d e d y s t r o p h i e m u s c u l a i r e d e D u c h e n n e . U n d e u x i 6 m e essa i o 6 il es t p r o p o s 6 d e t e s te r l ' in jec- t ion, d a n s u n pe t i t m u s c l e accesso i - re d u p i e d , d ' u n v e c t e u r A A V v6hi - c u l a n t I ' A D N c d u sarcoglycan alpha su r d e s p a t i e n t s a t t e in t s d e d y s t r o - p h i e d e s ce in tu r e s d e t y p e 2D, es t en cou r s d ' 61abo ra t i on aux Eta ts- Unis . Ces essa i s son t p a r d6 f in i t i on des essa i s d e to l6rance , n o t a m m e n t d ' 6 v a l u a t i o n d e la r 6 p o n s e i m m u - n i t a i r e 6 v e n t u e l l e m e n t d 6 v e l o p p 6 e con t re le p r o d u i t d u g6ne t ransf6r6, en g6n6ra l t o t a l e m e n t a b s e n t chez les pa t i en t s . A u - d e l a des r6su l t a t s a t t e n d u s , ces essa is son t tr6s i m p o r t a n t s car l eu r a c c e p t a t i o n p a r les i n s t ances offi- c ie l l e s c h a r g 6 e s d ' a p p r o u v e r les e s s a i s t h 6 r a p e u t i q u e s p e r m e t t r a , n o t a m m e n t au t r a v e r s des q u e s - t ions pos6es o u d e s i n f o r m a t i o n s s u p p l 6 m e n t a i r e s d e m a n d 6 e s p a r les expe r t s , d ' am61 io re r la fa i sab i l i - t6 p r a t i q u e d ' e s s a i s f u t u r s d e th6ra- p i e g 6 n i q u e i m p l i q u a n t d e s p a t i e n t s a t t e in t s d e m a l a d i e s neu - r o m u s c u l a i r e s .
R4f~rences [1] Acsadi G., Dickson G., Love D.R., Jani
A., Walsh ES., Gurusinghe A., Wolff J.A., Davies K.E., Human dystrophin expression in mdx Mice after injection of
DNA constructs, Nature 352 (1991) 815-818.
[2] Acsadi G., Jani A., Massie B., Simoneau M., Holland P., Blaschuk K., Karpati G., A differential efficiency of adenovirus- mediated in vivo gene transfer into ske- letal muscle cells of different maturity, Hum. Mol. Genet. 3 (1994) 579-584.
[3] Bedwell D., Kaenjak A., Benos D., Bebok Z., Bubien J., Hong J., Tousson A., Clancy J., Sorscher E., Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line., Nat. Med. 3 (1997) 1280-1284.
[4] Berns K. I., Parvoviridae and their repli- cation, in : B.e.a. Fields Virology, Raven Press, New York, 1990, pp. 1743-1764.
[5] Blaese R.M., Culver K.W., Miller A.D., Carter C.S., Fleisher T., Clerici M., Shea- rer G., Chang L., Chiang Y., Tolstoshev P. et al., T lymphocyte-directed gene therapy for ADA-SCID: initial trial results after 4 years., Science 270 (1995) 475-480.
[6] Bonne G., Di Barletta M., Varnous S., Becane H., Hammouda E., Merlini L., Muntoni F., Greenberg C., Gary E, Urtizberea J., Duboc D., Fardeau M., Toniolo D., Schwartz K., Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy., Nat. Genet. 21 (1999) 285-288.
[7] Budker V., Zhang G., Danko I., Williams P., Wolff J., The efficient expression of intravascularly delivered DNA in rat muscle, Gene Ther. 5 (1998) 272-276.
[8] Bulfield G., Siller W.G., Wight P.A.L., Moore K.J., X chromosome-linked mus- cular dystrophy (mdx) in the mouse, Proc. Natl. Acad. Sci. USA 81 (1984) 1189-1192.
[9] Chen H., Mack L.M., Kelly R., Ontell M., Kochanek S., Clemens P.R., Persis- tence in muscle of an adenoviral vector that lacks all viral genes, Proc. Natl. Acad. Sci. USA 94 (1997) 1645-1650.
[10] Clark K., Sferra T., Johnson P., Recombi- nant Adeno-Associated viral vectors mediate long-term transgene expres- sion in muscle, Hum. Gene Then 8 (1997) 659-669.
[11] Cox G.A., Phelps S.E, Chapman V.M., Chamberlain J.S., New mdx mutation disrupts expression of muscle and non- muscle isoforms of dystrophin, Nature Genetics 4 (1994) 87-93.
[12] Deconinck A.E., Rafael J.A., Skinner J.A., Brown S.C., Potter A.C., Metzinger L., Watt D.J., Dickson J.G., Tinsley J.M., Davies K.E., Utrophin-dystrophin-defi-
cient mice as a model for Duchenne muscular dystrophy, Cell. 90 (1997) 717-727.
[13] Deconinck N., Ragot T., Marechal G., Perricaudet M., Gillis J.M., Functional protection of dystrophic mouse (mdx) muscles after adenovirus-mediated transfer of a dystrophin minigene, Procs. Nat. Aca& Sci. USA 93 (1996) 3570-3574.
[14] Duclos F., Straub V., Moore S.A., Venzke D.P., Hrstka R.E, Crosbie R.H., Durbeej M., Lebakken C.S., Ettinger A.J., van der Meulen J., Holt K.H., Lim L.E., Sanes J.R., Davidson B.L., Faulkner J.A., Williamso R., Campbell K.P., Progressi- ve muscular dystrophy in alpha-sarco- glycan-deficient mice, Cell Biol. 142 (1998) 1461-1471.
[15] Dunckley M., Manoharan M., Villiet P., Eperon I., Dickson G., Modification of splicing in the dystrophin gene in cultu- red mdx muscle cells by antisense oli- goribonucleotides., Hum. Mol. Genet. 7 (1998) 1083-1090.
[16] Dunckley M., Wells D., Walsh E, Dick- son G., Direct retroviral-mediated transfer of a dystrophin minigene into mdx mouse muscle in vivo, Hum. Mol. Genet. 2 (1993) 717-723.
[17] Dunckley M.G., Love D.R., Davies K.E., Walsh ES., Morris G.E., Dickson G., Retroviral-mediated transfer of a dys- trophin minigene into mdx mouse myo- blasts in vitro, FEBS 296 (1992) 128-134.
[18] England S., Nicholson L., Johnson M., Forrest S., Love D., Zubrycka-Gaarn E., Bulman D., Harris J., Davies K., Very mild muscular dystrophy associated with the deletion of 46 % of dystrophin, Nature 343 (1990) 180-182.
[19] Escriou V., Ciolina C., Helbling-Leclerc A., Wils P., Scherman D., Cationic lipid- mediated gene transfer: analysis of cel- lular uptake and nuclear import of plas- mid DNA., Cell Biol. Toxicol. 14 (1998) 95-104.
[20] Feero W., Rosenblatt J., Huard J., Wat- kins S., Epperly M., Clemens P., Kocha- nek S., Glorioso J., Partridge, T., and EP, H., Viral gene delivery to skeletal muscle: insights on maturation-depen- dent loss of fiber infectivity for adeno- virus and herpes simplex type 1 viral vectors, Hum. Gene Ther. 8 (1997) 371-380.
[21] Ferrari F.K., Samulski T., Shenk T., Samulski R.J., Second-strand synthesis is a rate-limiting step for efficient trans- duction by recombinant adeno-associa-
ANNALES DE UINSTITUT PASTEUR / actualit6s (1999) 10, 3 /4 335

ted virus vectors, J. Virol. 70 (1996) 3227-3234.
[22] Fisher K.J., Gao G.P., Weitzman M.D., De Matteo R., Burda J.F., Wilson J.M., Transduction with recombinant adeno- associated virus for gene therapy is limited by leading-strand synthesis, J. Virol. 70 (1996) 520-532.
[23] Fisher K.J., Jooss K., Alston J., Yang Y., Haecker S.E., High K., Pathak R., Raper S.E., Wilson J.M., Recombinant adeno- associated virus for muscle directed gene therapy, Nat. Med. 3 (1997) 306-312.
[24] Gardner-Medwin D., Walton J., The muscular dystrophies, in : Walton J., Karpati G., Hilton-Jones D. (6d.), Disor- ders of volontary muscle, Churchill- Livingstone, Edinburgh, London, Madrid, Melbourne, New York, Tokyo, 1994, 543-594.
[25] Gill D.R., Southern K.W., Mofford K.A., Seddon T., Huang L., Sorgi E, Thomson A., MacVinish L.J., Ratcliff R., Bilton D., Lane D.J., Littlewood J.M., Webb A.K., Middleton P.G., Colledge W.H., Cuth- bert A.W., Evans M.J., Higgins C.E, Hyde S.C., A placebo-controlled study of liposome-mediated gene transfer to the nasal epithelium of patients with cystic fibrosis, Gene Ther. 4 (1997) 199-209.
[26] Gramolini A., Angus L., Schaeffer L., Burton E., Tinsley J., Davies K., Chan- geux J., Jasmin B., Induction of utrophin gene expression by heregulin in skeletal muscle ceils: role of the N-box motif and GA binding protein, Proc. Natl. Acad. Sci USA 96 (1999) 3223-3227.
[27] Greelish J.P., Su L.T., Lankford E.B., Burkman J.M., Chen H., Konig S.K., Mercier I.M., Desjardins P.R., Mitchell M.A., Zheng X.G., Leferovich J., Gao G.P., Balice-Gordon R.J., Wilson J.M., Stedman H.H., Stable restoration of the sarcoglycan complex in dystrophic muscle perfused with histamine and a recombinant adeno-associated viral vector, Nat. Med. 5 (1999) 439-443.
[28] Grossman M., Raper S.E., Kozarsky K., Stein E.A., Engelhardt J.E, Muller D., Lupien P.J., Wilson J.M., Successful ex vivo gene therapy directed to liver in a patient with familial hypercholestero- laemia, Nat. Genet. 6 (1994) 335-341.
[29] Hack A.A., Ly C.T., Jiang E, Clendenin C.J., Sigrist K.S., Wollman R.L., McNal- ly E.M., Gamma-sarcoglycan deficiency leads to muscle membrane defects and apoptosis independant of dystrophin, J. Cell. Biol. 142 (1998) 1279-1287.
[30] Hayashi Y.K., Chou EL., Engvall E., Ogawa M., Matsuda C., Hirabayashi S., Yokochi K., Ziober B.L., Kramer R.H., Kaufman S.J., Ozawa E., Goto Y., Nona- ka I., Tsukahara T., Wang J.Z., Hoffman E.P., Arahata K., Mutations in the inte- grin alpha 7 gene cause congenital myo- pathy., Nat. Genet. 19 (1998) 94-97.
[31] Helbling-Leclerc A., Zhang X., Topalo- glu H., Cruaud C., Tesson E, Weissen- bach J., Tome E, Schwartz K., Fardeau M., Tryggvason K. et al., Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy, Nat. Genet 11 (1995) 216-218.
[32] Hesketh J., Translation and the cytoske- leton: a mechanism for targeted protein synthesis, Mol. Biol. Rep. 19 (1994) 233-243.
[33] Holt K.H., Lim L.E., Straub V., Venzke D.P., Duclos E, Anderson R.D., David- son B.L., Campbell K.P., Functional rescue of the sarcoglycan complex in the BIO 14.6 hamster using delta-sarco- glycan gene transfer, Mol. Cell 1 (1998) 841-848.
[34] Howell J., Is there a future for gene the- rapy?, Neuromuscul. Disord. 9 (1999) 102-107.
[35] Howell J., Lochmuller H., O'Hara A., Fletcher S., Kakulas B., Massie B., Nal- bantoglu J., Karpati G., High-level dys- trophin expression after adenovirus- mediated dystrophin minigene transfer to skeletal muscle of dystrophic dogs: prolongation of expression with immu- nosuppression, Hum. Gene Ther. 9 (1998) 629~34.
[36] Huard J., Bouchard J.P., Roy R., Malouin E, Dansereau G., Labrecque C., Albert N., Richards C.L., Lemieux B., Tremblay J.P., Human Myoblast Transplantation - Preliminary Results of 4 Cases, Muscle & Nerve 15 (1992) 550-560.
[37] Huard J., Goins W.E, Glorioso J.C., Herpes simplex virus type I vector mediated gene transfer to muscle, Gene Therapy 2 (1995) 385-392.
[38] James M., Man N.T., Wise G.J., Jones G.E., Morris G.E., Utrophin-dystrogly- can complex in membranes of adherent cultured cells, Cell. Motil. Cytoskeleton 33 (1996) 163-174.
[39] Jooss K., Yang Y., Fisher K.J., Wilson J.M., Transduction of dendritic cells by DNA viral vectors directs the immune response to transgene products in muscle fibers., J. Virol. 72 (1998) 4212-4223.
[40] Kafri T., Blomer U., Peterson D., Gage E, Verma I., Sustained expression of genes delivered directly into liver and muscle by lentiviral vectors, Nat. Genet. 17 (1997) 314-317.
[41] Kessler P.D., Podsakoff G.M., Chen X., McQuiston S.A., Colosi P.C., Matelis L.A., Kurtzman G.J., Byrne B.J., Gene delivery to skeletal muscle results in sustained expression and systemic deli- very of a therapeutic protein, Proc. Natl. Acad. Sci. USA 93 (1996) 14082-14087.
[42] Klinman D., Yamshchikov G., Ishigatsu- bo Y., Contribution of CpG motifs to the immunogenicity of DNA vaccines, Immunol. 158 (1997) 3635-3639.
[43] Koenig M., Monaco A.P., Kunkel L.M., The complete sequence of dystrophin predicts a rod-shaped cytoskeletal pro- tein, Cell 53 (1988) 219-226.
[44] Kornegay J., Tuler S., Miller D., Levesque D., Muscular dystrophy in a litter of golden retriever dogs, Geno- mics 11 (1988) 1056-1064.
[45] Kotin R.M., Siniscalco M., Samulski R.J., Zhu X.D., Hunter L., Laughlin C.A., McLaughlin S., Muzyczka N., Rocchi M., Berns K.I., Site-specific integration by adeno-associated virus, Proc. Natl. Acad. Sci USA 87 (1990) 2211-2215.
[46] Kramarcy N.R., Vidal A., Froehner S.C., Sealock R., Association of Utrophin and Multiple dystrophin short forms with the mammalian-M(r) 58,000 dystro- phin-associated protein (syntrophin), J. Biol. Chem. 269 (1994) 2870-2876.
[47] Kren B., Bandyopadhyay P., Steer C., In vivo site-directed mutagenesis of the factor IX gene by chimeric RNA/DNA oligonucleotides, Nat. Med. 4 (1998) 285-290.
[48] Kumar-Singh R., Chamberlain J.S., Encapsidated adenovirus minichromo- somes allow delivery and expression of a 14 kb dystrophin cDNA to muscle cells., Hum. Mol. Genet. 5 (1998) 913-921.
[49] Law P.K., Bertorini T.E., Goodwin T.G., Chen M., Fang Q.W., Li H.J., Kirby D.S., Florendo J.A., Herrod H.G., Golden G.S., Dystrophin production induced by myoblast transfer therapy in Duchenne muscular dystrophy, Lancet 336 (1990) 114-115.
[50] Lemaire C., Heilig R., Mandel J., The chicken dystrophin cDNA: striking conservation of the C-terminal coding and 3'untranslated regions between man and chicken, EMBO J. 7 (1988) 4157-4162.
336 A N N A L E S DE L'INSTITUT PASTEUR / actualit6s (1999) 10, 3 / 4

[51] Li J., Dressman D., Tsao Y., Sakamoto A., Hoffman E., Xiao X., rAAV vector- mediated sarcogylcan gene transfer in a hamster model for limb-girdle muscular dystrophy, Gene Ther. 6 (1999) 74-82.
[52] Lim L.E., Campbell K.P., The sarcogly- can complex in limb-girdle muscular dystrophy, Curr. Opin. NeuroL 11 (1998) 443-452.
[53] Love D., Morris G., Ellis J., Fairbrother U., Marsden R., Bloomfield J., Edwards Y., Slater C., Parry D., Davies K., Tissue distribution of the dystrophin-related gene product and expression in the mdx and dy mouse, Proceedings of the National Academy of Sciences USA 88 (1991) 3243-3247.
[54] Love D.R., Hill D.E, Dickson G., Spurr N.K., Byth B.C., Marsden R.E, Walsh ES., Edwards Y.H., Davies K.E., An autosomal transcript in skeletal muscle with homology to dystrophin, Nature 339 (1989) 55-58.
[55] Mayer U., Saber G., Fassler R., Borne- mann A., Echtermeyer F., vonder Mark H., Miosge N., Poschl E., von der Mark K., Absence of integrin alpha 7 causes a novel form of muscular dystrophy, Nat. Genet. 17 (1997) 318-323.
[56[ Mcardle A., Edwards R.H.T., Jackson M.J., How does dystrophin deficiency lead to muscle degeneration? Evidence from the mdx mouse, Neuromuscular Disorders 5 (1995) 445-456.
[57] Menke A., Jockusch H., Decreased osmotic stability of dystrophin-less muscle cells from the mdx mouse, Nature 349 (1991) 69-71.
[58] Michael S.I., Hong J.S., Curiel D.T., Engler J.A., Addition of a short peptide ligand to the adenovirus fiber protein., Gene Ther. 2 (1995) 660~68.
[59] Minetti C., Sotgia E, Bruno C., Scartez- zini P., Broda P., Bado M., Masetti E., Mazzocco M., Egeo A., Donati M., Volonte D., Galbiati E, Cordone G., Bri- carelli F., Lisanti M., Zara F., Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystro- phy, Nat. Genet. 18 (1998) 365-368.
[60l Mir L., Bureau M., Gehl J., Rangara R., Rouy D., Caillaud J., Delaere P., Branel- lec D., Schwartz B., Scherman D., High- efficiency gene transfer into skeletal muscle mediated by electric pulses, Proc. Natl. Acad. Sci. USA 96 (1999) 4262-4267.
[61] Monahan P.E., Samulski R.J., Tazelaar J., Xiao X., Nichols T.C, Bellinger D.A., Read M.S., Walsh C.E., Direct intramus- cular injection with recombinant AAV
vectors results in sustained expression in a dog model of hemophilia, Gene Ther. 5 (1998) 40-49.
[62] Muntoni E, Wilson L., Marrosu G., Mar- rosu M.G., Cianchetti C., Mestroni L., Ganau A., Dubowitz V., Sewry C., A mutation in the dystrophin gene selec- tively affecting dystrophin expression in the heart, Journal of Clinical Investi- gation 96 (1995) 693-699.
[63] Nagano A., Koga R., Ogawa M., Kurano Y., Kawada J., Okada R., Hayashi Y., Tsukahara T., Arahata K., Emerin defi- ciency at the nuclear membrane in patients with Emery-Dreifuss muscular dystrophy, Nat. Genet. 12 (1996) 254-259.
[64] Nalbantoglu J., pari G., Karpati G., Hol- land P.C., Expression of the primary coxsackie and adenovirus receptor is downregulated during skeletal muscle maturation and limits the efficacy of adenovirus-mediated gene delivery to muscle cells, Hum. Gene Ther. 10 (1999) 1009-1019.
[65] Parrish E.P., Cifuentesdiaz C., Li Z.L., Vicart P., Paulin D., Dreyfus P.A., Pes- chanski M., Harris A.J., Garcia L., Tar- geting widespread sites of damage in dystrophic muscle: Engrafted macro- phages as potential shuttles, Gene The- rapy 3 (1996) 13-20.
[66] Partridge T., Morgan J., Coulton G., Hoffman E., Kunkel L., Conversion of mdx myofibres from dystrophin-negati- ve to -positive by injection of normal myoblasts, Nature 337 (1989) 176-179.
[67] Pavlath G.K., Rich K., Webster S.G., Blau H.M., Localization of muscle gene products in nuclear domains, Nature 337 (1989) 570-573.
[68] Phelps S.E, Hauser M.A., Cole N.M., Rafael J.A., Hinkle R.T., Faulkner J.A., Chamberlain J.S., Expression of full- length and truncated dystrophin mini- genes in transgenic mdx mice, Human Molecular Genetics 4 (1995) 1251-1258.
[69] Pilaro A., Serabian M., Preclinical deve- lopment strategies for novel gene the- rapeutic products, Toxicol. Pathol. 27 (1999) 4-7.
[70] Qu Z., Balkir L., van Deutekom J., Rob- bins P., Pruchnic R., Huard J., Develop- ment of approaches to improve cell sur- vival in myoblast transfer therapy, Cell Biol. 142 (1998) 1257-1267.
[71] Ragot T., Vincent N., Chaffey P., Vigne E., Gilgenkrantz H., Couton D., Car- taud J., Briand P., Kaplan J.C., Perricau~ det M., Kahn A., Efficient adenovirus- mediated transfer of a human minidy-
strophin gene to skeletal muscle of mdx mice muscle, Nature 361 (1993) 647-650.
[72] Ralston E., McLaren R. S., Horowitz J.A., Nuclear domains in skeletal myo- tubes: the localization of transferrin receptor mRNA is independent of its half-life and restricted by binding to ribosomes, Exp. Cell Res. 236 (1997) 453-462.
[73] Richard I., Broux O., Allamand V., Fou- gerousse E, Chiannilkulchai N., Bourg N., Brenguier L., Devaud C., Pasturaud P., Roudaut C., Hillaire D., Passos- Bueno M.R., Zatz M., Tischfield J.A., Fardeau M., Jackson C.E., Cohen D., Beckman J.S., Mutations in the proteo- lytic enzyme calpain 3 cause limb-gird- le muscular dystrophy type 2A, Cell 81 (1995) 27-40.
[74] Rosenberg S.A., Aebersold P., Cornetta K., Kasid A., Morgan R.A., Moen R., Karson E.M., Lotze M.T., Yang J.C., Topalian S.L. et al., Gene transfer into humans-immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modi- fied by retroviral gene transduction, N. Engl. J. Med. 323 (1990) 570-578.
[75] Sakamoto A., Ono K., Abe M., Jasmin G., Eki T., Murakami Y., Masaki T., Toyo-oka T., Hanaoka F., Both hyper- trophic and dilated cardiomyopathies are caused by mutation of the same gene, delta-sarcoglycan, in hamster: an animal model of disrupted dystrophin- associated glycoprotein complex, Proc. Natl. Acad. Sci. USA 94 (1997) 13873--13878.
[76] Sharp N.J., Kornegay J.N., Van Camp S.D., Herbstreith M.H., Secore S.L., Kettle S., Hung W.Y., Constantinou C.D., Dykstra M.J., Roses A.D. et al., An error in dystrophin mRNA processing in golden retriever muscular dystro- phy, an animal homologue of Duchen- ne muscular dystrophy, Genomics 13 (1992) 115-121.
[77] Smith J., Fowkes G., Schofield P.N., Pro- grammed cell death in dystrophic (mdx) muscle is inhibited by IGF-II, Cell Death and Differentiation 2 (1995) 243-251.
[78] Snyder R., Spratt S., Lagarde C., Bohl D., Kaspar B., Sloan B., Cohen L., Danos O., Efficient and Stable AAV- Mediated Transduction in the Skeletal Muscle of Adult Immunocompetent Mice, Hum. Gene Ther. 8 (1997) 1891-1900.
[79] Sunada Y., Bernier S., Utani A., Yamada Y., Campbell K., Identification of a
A N N A L E S DE UINSTITUT PASTEUR / actualit6s (1999) 10, 3 / 4 337

novel mutant transcript of laminin alpha 2 chain gene responsible for mus- cular dystrophy and dysmyelination in dy2J mice, Hum. Mol. Genet. 4 (1995) 1055-1061.
[80] Tidball J.G., Albrecht D.E., Lokensgard B.E., Spencer M.J., Apoptosis precedes necrosis of dystrophin-deficient muscle, Journal of Cell Science 108 (1995) 2197-2204.
[81] Tinsley J., Deconinck N., Fisher R., Kahn D., Phelps S., Gillis J., Davies K., Expression of full-length utrophin pre- vents muscular dystrophy in mdx mice, Nat. Med. 4 (1998) 1441-1444.
[82] Vainzof M., Passos-Bueno M., Canovas M., Moreira E., Pavanello R., Marie S., Anderson L., Bonnemann C., McNally E., Nigro V., Kunkel L., Zatz M., The sarcoglycan complex in the six autoso- mal recessive limb-girdle muscular dystrophies, Hum. Mol. Genet. 5 (1996) 1963-1969.
[83] van Deutekom J.C., Floyd S.S., Booth D.K., Oligino T., Krisky D., Marconi P., Glorioso J.C., Huard J., Implications of maturation for viral gene delivery to skeletal muscle, Neuromuscul. Disord. 8 (1998) 135-148.
[84] Vilquin J., Gu6rette B., Kinoshita I., Roy B., Goulet M., Gravel C., Roy R., J.P.
Tremblay, FK506 immunosuppression to control the immune reactions trigge- red by first generation adenovirus° mediated gene transfer, Hum. Gene Ther. 6 (1995) 1391-1401.
[85] Vincent N., Ragot T., Gilgenkrantz H., Couton D., Chaffey P., Gr6goire A., Briand P., Kaplan J.C., Kahn A., Perri- caudet M., Long-term correction of mouse dystrophic degeneration by adenovirus-mediated transfer of a minidystrophin gene, Nature Genetics 5 (1993) 130-134.
[86] Vincent-Lacaze N., Snyder R., Gluzman R., Bohl D., Lagarde C., Danos O., Structure of adeno-associated virus vector DNA following transduction of the skeletal muscle, J. Virol. 73 (1999) 1949-1955.
[87] Wells D.J., Wells K.E., Asante E.A., Tur- ner G., Sunada Y., Campbell K.P., Walsh ES., Dickson G., Expression of human fulMength and minidystrophin in transgenic mdx mice: Implications for gene therapy of Duchenne muscular dystrophy, Human Molecular Genetics 4 (1995) 1245-1250.
[88] Winand N.J., Edwards M., Pradhan D., Berian C.A., Cooper B.J., Deletion of the dystrophin muscle promoter in feli-
ne muscular dystrophy, Neuromusc. Disord. 4 (1994) 433-445.
[89] Xiao X., Li J., Samulski R.J., Efficient long-term gene transfer into muscle tis- sue of immunocompetent mice by Adeno-Associated Virus vector, J. Virol. 70 (1996) 8098-8108.
[90] Xu H., Wu X., Wewer U., Engvall E., Murine muscular dystrophy caused by a mutation in the laminin alpha 2 (Lama2) gene, Nat. Genet. 8 (1994) 297-302.
[91] Yang L., Lochmuller H., Luo J., Massie B., Nalbantoglu J., Karpati G., Petro EB., Adenovirus-mediated dystrophin minigene transfer improves muscle strength in adult dystrophic (mdx) mice, Gene Ther. 5 (1998) 369-379.
[92] Yang Y., Jooss K.U., Su Q., Ertl H.C.J., Wilson J.M., Immune responses to viral antigens versus transgene product in the elimination of recombinant adeno- virus-infected hepatocytes in vivo, Gene Therapy 3 (1996) 137-144.
[93] Yuasa K., Miyagoe Y., Yamamoto K., Nabeshima Y., Dickson G., Takeda S., Effective restoration of dystrophin- associated proteins in vivo by adenovi- rus-mediated transfer of truncated dys- trophin cDNAs, FEBS Lett 425 (1998) 329-336.
338 A N N A L E S DE UINSTITUT PASTEUR / actualit6s (1999) 10, 3 / 4