Embed Size (px)
Citation preview



CHEZ LE MÊME ÉDITEUR
Aide-mémoire de pharmacologie, Jean-Luc Eighozi, Dominique DuvalChronobiologie et chronothérapeutique, heure optimaled'administration des médicaments, Alain Reinberg, Gaston Labrecque,Michael SmolenskyLes 200 médicaments essentiels, Maurice RapinToxicologie clinique, Chantai Bismuth, Frédéric Joseph Baud, FrançoiseConso, Jean-Pierre Fréjaville, Robert GamierThérapeutique dermatologique, Louis DubertretThérapeutique rhumatologique, Thomas Bardin, Daniel KuntzLes maladies systémiques, Marcel-Francis Kahn, André P. Peltier,Olivier Meyer, Jean-Charles PietteTraité de médecine, Pierre Godeau, Serge Herson, Jean-Charles PietteTraité de médecine interne Cecil, J.C. Bennett, P. PlumLa Petite encyclopédie médicale Hamburger, Michel Leporrier
DANS LA COLLECTION « Atlas de poche »Anatomie, 3 volumes, W. Kahie, H. Leonhardt, W. PlatzerAnatomie en coupes sériées TDM-IRM, 2 volumes, T.B. Môlier, E. ReifPhysiologie, Stefan Silbernagi, Agamemnon DespopoulosHistologie, Wolfgang KùhnelBiochimie, Jan Koolman, Klaus-Henrich RôhmEmbryologie, Ulrich DrewsGénétique, Eberhard PassargeMéthodes d'analyse, Georg Schwedt
Sédition 19912e tirage 19952e édition 19982e tirage 2001
Pour recevoir le catalogue Flammarion Médecine-Sciences,il suffit d'envoyer vos nom et adresse à
Flammarion Médecine-Sciences4, rue Casimir-Delavigne
75006 PARIS

Atlas de pochede pharmacologieHeinz Lùllmann, Klaus Mohr,Albrecht Ziegler
2e édition française
Traduction de l'allemand deDominique DuvalDocteur es SciencesDirecteur de recherches CNRS
'entre Cyceron, Cyclotron biomédical de Caen
161 planches de Jurgen Wirth
Médecine-SciencesFlammarion
4, rue Casimir-Delavigne, 75006 Paris

IV
Pr émérite Heinz Lûllmann Réalisation des illustrationsDépartement de PharmacologieUniversité de Kiel. Allemagne Pr Jiirgen Wirth
Unité d'arts plastiquesDr Klaus Mohr École Supérieure Technique,Institut pharmaceutique, Darmstadt. Allemagne-Service de Pharmacologieet ToxicologieUniversité de Bonn. Allemagne
Pr Albrecht ZieglerDépartement de PharmacologieUniversité de Kiel. Allemagne
Les marques déposées ne sont pas si- Remarque importante ! commegnalées par un signe particulier. En chaque connaissance, la médecine estl'absence d'une telle indication, il ne en développement permanent. La re-faudrait pas conclure que le titre d'Atlas cherche et la pratique clinique élargis-se Poche corresponde à une marque sent nos connaissances, surtout en celibre, qui concerne les traitements et l'utilisa-Tous les droits de reproduction de cet tion des médicaments. Chaque fois queouvrage et de chacune de ses parties sera mentionnée dans cet ouvrage unesont réservés. Toute utilisation en de- concentration ou une application, lehors des limites définies par la loi sur lecteur peut être assuré que les auteurs,les droits d'auteurs est interdite et pas- l'éditeur et l'imprimeur ont consacrésible de sanctions sauf accord de l'édi- beaucoup de soins pour que cette infor-teur. Ceci vaut en particulier pour les mation corresponde rigoureusement àphotocopies, les traductions, la prise de l'état de l'art au moment de l'achève-microfilms, le stockage et le traitement ment de ce livre.dans des systèmes électroniques. L'éditeur ne peut cependant donner au-
cune garantie en ce qui concerne les in-dications de dose ou de forme d'admi-
© 1990,1996 Georg Thieme Veriag. nistration. Chaque utilisateur est doncRûdigerstrape 14, D-70469 Stuttgart, invité à examiner avec soin les noticesGermany des médicaments utilisés pour établir,
sous sa propre responsabilité, si les in-Traduction autorisée de l'édition allé- dications de doses ou si les contre-indi-mande parue sous le titre Taschenatlas cations signalées sont différentes deder Pharmakologie, publiée par Georg celles données dans cet ouvrage. CeciThieme Veriag. s'applique en particulier aux substances
rarement utilisées ou à celles recem-Pour l'édition française, ment mises sur le marché. ChaqueISBN 2-257-12119-8 dosage ou chaque traitement est
effectué aux risques et périls de l'uti-© 1991, 1998 Flammarion lisateur. Les auteurs et l'éditeur de-Printed in France mandent à chaque utilisateur de leur si-
gnaler toute inexactitude qu ' il aurait puremarquer.

v
Nous remercions nos lecteurs de leur vif intérêt pour ce livre. C'est ainsi qu'il estpossible, deux ans seulement après la sortie de la 2e édition, de proposer une 3e édi-tion remise à jour. Cela est très agréable compte tenu du développement et des avan-cées de la pharmacologie.Nous avons pu introduire des principes actifs récemment mis sur le marché, parexemple les bloqueurs des récepteurs de l'angiotensine II ; de nouvelles planchesont été conçues comme par exemple l'inhibition de la synthèse du cholestérol et lamodulation des réactions immunitaires.Le passage du dessin, jusqu'ici toujours effectué à la main, à une réalisation par or-dinateur a été l'occasion de modifier les représentations anatomiques et d'entre-prendre quelques corrections.Nous remercions en particulier pour leur soutien MM. les Pr B.J. Aldenhoff,K. Christian! et K.O. Gundermann (hôpital de l'Université de Kiel), M. lePr Suverkrup (Institut Pharmaceutique de l'Université de Bonn), Mme le DrE. Kostenis (National Institute ofHeaIth, Bethesda, USA). A côté de cela, d'innom-brables remarques et suggestions de collègues de collaborateurs et d'étudiants enmédecine et en pharmacie, qu'il est impossible de tous citer ici, nous ont été d'ungrand secours.L'image sur la page de couverture est censée symboliser les différents aspects d'unmédicament : son origine, sa structure chimique, son application et surtout le pointle plus important parmi les aspects thérapeutiques, son effet sur l'organisme.Mous espérons offrir, avec cette 3e édition, aux étudiants, aux praticiens et aux phar-maciens, ainsi qu'à tous ceux qui appartiennent au monde de la santé, voire aux pro-fanes curieux, un bon instrument pour se donner une vue d'ensemble sur le mondedu médicament.
Heinz Lûllmann, Kiel Jiirgen Wirth. DarmstadtKlaus Mohr, BonnAlbrecht Ziegler, KielAutomne 1996
•s./''i j i ; .

VI
Avant-propos de la 1'® édition
La pharmacologie est, au sens strict, la science des médicaments. L'Atlas de pochede pharmacologie en propose une présentation concise en textes et en images. Lapremière partie. Pharmacologie générale, traite les domaines qui peuvent être étu-diés indépendamment des diverses molécules, par exemple les formes galéniques, laprise de substances médicamenteuses, leur distribution, leur élimination et leurs mé-canismes d'action moléculaires. Dans la seconde partie, Pharmacologie des spécia-lités, sont présentées les différentes catégories de produits pharmaceutiques en met-tant l'accent sur les aspects fonctionnels et thérapeutiques. L'attention est dirigéesur la façon dont les molécules agissent sur les fonctions de l'organisme, ainsi quesur les multiples possibilités d'application thérapeutique qui en découlent plutôt quesur leurs propriétés chimiques.En réalisant les illustrations, on a cherché à expliquer des interactions complexesavec des « modèles visuels ». La présentation sous forme de schémas conduit obli-gatoirement à la simplification de systèmes ou de structures par nature complexes.C'est ainsi, par exemple, que l'on a renoncé à une description complète des détailsanatomiques pour ne pas gêner la compréhension des figures. La présentation gra-phique des molécules, des organes et des systèmes a été conçue pour chaque thèmede façon hiérarchisée. On n'a donc pas tenu compte des dimensions réelles des élé-ments représentés. La taille et la couleur permettent de distinguer dans une figure lesparties importantes de celles qui le sont moins. Le texte en page de gauche et lesillustrations en regard sont complémentaires. Les données et les interactions phar-macologiques sont résumées et explicitées par l'image. La présentation, que nousespérons attrayante et facile à comprendre, doit en outre aider à saisir la somme desinformations concernant les nombreux médicaments existant et à les garder en mé-moire.L'Atlas de poche de pharmacologie est conçu pour différentes catégories de lec-teurs. Il sera utile aux étudiants en médecine, en pharmacie ou en odontologie pourassimiler rapidement les notions de base et bâtir, en quelque sorte, le gros-œuvred'un édifice pharmacologique. C'est le souhait des auteurs qu'ils puissent ensuiteaccéder ainsi à des notions complémentaires issues de leurs cours ou d'ouvragesplus détaillés de façon à parfaire l'édifice.L'Atlas de poche de pharmacologie permettra également aux médecins et aux phar-maciens de raviver des connaissances déjà acquises et de revoir rapidement les in-teractions pharmacothérapeutiques.L'Atlas de poche de pharmacologie sera enfin une source précise d'informationspour tous ceux qui s'intéressent aux médicaments.Nous remercions le Dr Matéfi (Baie), le Dr Liillmann-Rauch, M. J. Mohr et leDr H. J. Pfànder (tous de Kiel) pour leur aide dans la réalisation de certaines figures.Nous remercions la Bibliothèque Nationale Autrichienne pour l'autorisation de re-produire un fragment du Codex de Constantin.
Heinz Lûllmann, Klaus Mohr, Albrecht Ziegler Jiirgen WirthKiel DarmstadtPrintemps 1990

1__________________________________________VH
Sommaire
Pharmacologie générale ................................................................................. 1
Histoire de la pharmacologie .......................................................................... 2
Origine d'un produit actifDrogues et substances actives ...................................................................... 4Développement d'un médicament ............................................................... 6
Formes galéniquesFormes orales, oculaires ou nasales ............................................................. 8Formes parentérales, inhalées, rectales ou vaginales
et formes topiques .................................................................................... 12Administration par inhalation ..................................................................... 14Dermatologie ................................................................................................ 16Du site d'application à la distribution dans l'organisme ............................. 18
| Sites d'action cellulaire| Les sites d'action des médicaments ............................................................. 20L'Distribution dans l'organisme
Barrières externes de l'organisme ................................................................ 22Barrières entre le sang et les tissus .............................................................. 24Passage à travers les membranes ................................................................. 26Différentes possibilités de distribution d'un médicament ........................... ,28Liaison des médicaments aux protéines plasmatiques ................................ 30
Élimination des médicamentsRôle du foie dans la dégradation des médicaments .................................... 32Biotransformation des médicaments ............................................................ 34Cycle entéro-hépatique, reactions de conjugaison ....................................... 38Élimination rénale ........................................................................................ 40Élimination des substances lipophiles et hydrophiles ................................. 42
PharmacocinétiqueConcentration des médicaments dans l'organisme, évolution en
i fonction du temps : la fonction exponentielle ......................................... 44Cinétique plasmatique des médicaments ..................................................... 46Cinétique plasmatique d'un médicament administré de façon
régulière ou irrégulière.............................................................................. 48Accumulation : doses, intervalles entre deux doses
et contrôle des concentrations plasmatiques ............................................ 50Modification des caractéristiques de l'élimination durant le traitement ..... 50
Mesure de l'effet des médicamentsRelation dose-effet (in vivo) ........................................................................ 52Relation dose-effet (in vitro)........................................................................ 54Courbes doses-réponses ............................................................................... 54Courbes de liaison ........................................................................................ 56

VIII Sommaire
Interaction médicament-récepteur 'Types de liaison ........................................................................................... 58
. Agonistes et antagonistes ............................................................................. 60Antagonisme fonctionnel ............................................................................. 60Stéréochimie de l'action des médicaments .................................................. 62Différents récepteurs..................................................................................... 64Modes de fonctionnement des récepteurs couplés à une protéine G........... 66Cinétique plasmatique et effet d'un médicament ........................................ 68
Effets secondaires des médicamentsEffets secondaires des médicaments ............................................................ 70Allergie aux médicaments ............................................................................ 72Effets nocifs pour l'enfant de la prise de médicaments
pendant la grossesse et l'allaitement........................................................ 74
Effets des médicaments indépendants d'une substance activePlacebo, homéopathie ................................................................................... 76
Pharmacologie des spécialités ....................................................................... 79
Influence des médicaments sur le système sympathiqueSystème nerveux sympathique ..................................................................... 80Organisation du système sympathique, médiateurs
du système sympathique .......................................................................... 82Synapse adrénergique ................................................................................... 82Sous-types de récepteurs adrénergiques et action
des catécholamines ................................................................................... 84Effets sur les muscles lisses.......................................................................... 84Relations structure-activité ........................................................................... 86Substances à action sympathomimétique indirecte ..................................... 88a-sympathomimétiques, a-sympatholytiques ............................................... 90P-sympatholytiques ((3-bloquants) ............................................................... 92Différences entre P-bloquants ...................................................................... 94Antisympathotoniques .................................................................................. 96
Influence des médicaments sur le système parasympathiqueSystème nerveux parasympathique .............................................................. 98Synapse cholinergique .................................................................................. 100Parasympathomimétiques.............................................................................. 102Parasympatholytiques ................................................................................... 104
NicotineTransmission ganglionnaire ......................................................................... 108Actions de la nicotine sur les fonctions de l'organisme ............................. 110Conséquences du tabagisme ......................................................................... 112
Aminés biogènesAminés biogènes, actions et rôles pharmacologiques ................................. 114Dopamine, histamine ................................................................................... 114Sérotonine ..................................................................................................... 116
VasodilatateursVasodilatateurs : vue d'ensemble ................................................................ 118

B^ . Sommaire IXP-————————————————————————-—————————————
Nitrates organiques....................................................................................... 120Antagonistes calciques ................................................................................. 122
Inhibiteurs du système rénine-angiotensine-aldostéroneInhibiteurs de l'enzyme de conversion......................................................... 124Antagonistes des récepteurs de l'angiotensine IL......................................... 124
Médicaments actifs sur les muscles lissesSubstances agissant sur les organes musculaires lisses ............................... 126
Médicaments actifs sur le cœurVue d'ensemble sur les possibilités de moduler la fonction cardiaque ...... 128Le cycle cardiaque, contraction et relaxation .............................................. 128Glycosides cardiaques .................................................................................. 130Autres molécules inotropes positives .......................................................... 132Principes de traitement d'une insuffisance cardiaque chronique ................ 132Traitement des arythmies cardiaques ........................................................... 134Propriétés électrophysiologiques des antiarythmiques appartenant
à la famille des Moqueurs de canaux sodiques ........................................ 136
Anti-anémiquesTraitement de l'anémie ................................................................................ 138
• Vitamine B12 ............................................................................................... 138• Acide folique ................................................................................................ 1381 Métabolisme du fer ...................................................................................... 140
P Anti-thrombotiquesProphylaxie et traitement des thromboses ................................................... 142Dérivés coumariniques, héparine ................................................................. 144Fibrinolyse .................................................................................................... 146Inhibition de l'agrégation plaquettaire ......................................................... 148Inhibition de l'agrégation des érythrocytes ................................................. 148
Substituts du plasma ......................................................................................... 150
HypolipidémiantsHypolipidémiants ......................................................................................... 152
Diurétiques 'Diurétiques : vue d'ensemble ...................................................................... 156Réabsorption du sodium au niveau des reins, diurétiques
osmotiques ................................................................................................ 158Diurétiques de type sulfonamide ................................................................. 160Diurétiques antikaliurétiques, ADH et analogues ....................................... 162
Produits contre les ulcères gastriquesTraitement des ulcères de l'estomac et du duodénum ................................ 164
LaxatifsLaxatifs de lest, mucilages et fibres, laxatifs osmotiques ........................... 168Substances irritant l'intestin, abus des laxatifs ............................................ 170Laxatifs irritant l'intestin grêle : huile de ricin ........................................... 172Laxatifs irritant le côlon ............................................................................... 172Laxatifs lubrifiants .............................................................;—........—........... 172

X Sommaire
Anti-diarrhéiquesTraitement d'une diarrhée .....................;...................................................... 176
Autres médicaments du tractus gastro-intestinal.............................................. 178
Produits agissant sur le système moteurSubstances actives sur le système moteur ................................................... 180Myorelaxants ................................................................................................ 182Myorelaxants dépolarisants .......................................................................... 184Antiparkinsoniens ......................................................................................... 186Antiépileptiques............................................................................................ 188
AnalgésiquesOrigine et conduction de la douleur ............................................................ 192
Analgésiques antipyrétiquesEicosanoïdes ................................................................................................. 194Analgésiques antipyrétiques ......................................................................... 196Anti-inflammatoires non stéroïdiens............................................................. 198Régulation thermique du corps et antipyrétiques ........................................ 200
Anesthésiques locaux ............................................................................i.......... 202
OpioïdesAnalgésiques morphiniques : opioïdes ........................................................ 208
AnesthésiquesAnesthésie et anesthésiques.......................................................................... 214Anesthésiques inhalés................................................................................... 216Anesthésiques injectés................................................................................... 218
HypnotiquesSomnifères, hypnotiques............................................................ ................. 220Rythmes d'éveil et de sommeil et somnifères.............................................. 222
Médicaments du psychismeBenzodiazépines............................................................................................ 224Pharmacocinétique des benzodiazépines...................................................... 226Traitement de la cyclothymie ....................................................................... 228Traitement de la dépression endogène,
antidépresseurs tricycliques....................................................................... 228Traitement de la manie, ions lithium,
prévention de la cyclothymie.................................................................... 232Traitement de la schizophrénie, neuroleptiques........................................... 234Psychomimétiques (substances hallucinogènes ou psychédéliques)............ 238
HormonesHormones hypothalamiques et hypophysaires ............................................ 240Traitement par les hormones thyroïdiennes ................................................. 242Hyperthyroïdie et thyréostatiques ................................................................ 244Utilisations thérapeutiques des glucocorticoïdes ......................................... 246Androgènes, anabolisants, antiandrogènes .................................................. 250Maturation des ovules et ovulation, formation des œstrogènes
et des progestogènes................................................................................. 252

Sommaire Xt
Contraceptifs oraux, pilule ........................................................................... 254Traitement par l'insuline .............................................................................. 256Traitement du diabète sucré avec carence en insuline ................................ 258Traitement du diabète de l'adulte ................................................................ 260
i Substances utilisées pour maintenir l'homéostasie du calcium .................. 262
i Substances antibactériennesMédicaments contre les infections bactériennes .......................................... 264Inhibiteurs de synthèse de la paroi bactérienne ........................................... 266Inhibiteurs de synthèse de l'acide tétrahydrofolique ................................... 270Inhibiteurs de la fonction de l'ADN ............................................................ 272Inhibiteurs de la synthèse protéique ............................................................ 274
I Substances contre les infections à mycobactéries ....................................... 278
1 AntifongiquesSubstances contre les infections provoquées par les champignons ............ 280
'VirustatiquesMédicaments antiviraux ............................................................................... 282
Désinfectants .................................................................................................... 286
Médicaments antiparasitairesSubstances antiparasitaires (endo- et ectoparasites) .................................... 288Antimalariens ............................................................................................... 290
;i CytostatiquesSubstances contre les tumeurs malignes ...................................................... 292
i ImmunomodulateursInhibition des réactions immunitaire.......................................................... 296
AntidotesLutte contre les empoisonnements, antidotes............................................... 298
Traitements de maladies particulièresAngine de poitrine ........................................................................................ 302Anti-angineux ............................................................................................... 304Hypertension et antihypertenseurs ............................................................... 306Différentes formes d'hypotension et leur traitement médicamenteux ........ 308La goutte et son traitement .......................................................................... 310Ostéoporose................................................................................................... 312Polyarthrite rhumatoïde et son traitement.................................................... 314La migraine et son traitement ...................................................................... 316
• Traitement des refroidissements .................................................................. 318Traitement anti-allergique ..................................................................—....... 320Asthme..........................................—...................——————————.———— 322Vomissements et anti-émétiques ——.———.——.————.———— 324
Lectures complémentaires —————————————————'—— 326Liste des médicaments .——..————.————————.———.— 327Index ................-...................-..................——.——.——————--—- 355

Pharmacologie généralePharmacologie générale
I

2 Histoire de la pharmacologie
Histoire de la pharmacologie
De mémoire d'homme, on a toujourscherché à soulager les maux del'homme ou des animaux avec des mé-dicaments. La connaissance des vertuscuratives de certaines plantes ou de cer-tains minéraux était déjà inscrite dèsl'antiquité dans les traités de botanique.Cette croyance en la vertu bénéfiquedes plantes ou de certaines substances,transmise exclusivement par tradition,n'avait été soumise à aucun examen cri-tique.
L'idée
L'impulsion
Claude Galien (129-200) cherche lepremier à déterminer les bases théo-riques de l'utilisation des médicaments.A part égale avec la pratique, la théoriequi permet d'interpréter les observa-tions et les résultats expérimentaux doitconduire à une utilisation rationnelledes médicaments.«• Les empiristes disent que tout seratrouvé par l'expérience. Cependant,nous pensons que les découvertes résul-tent en partie de la théorie. En e f fe t , nil'expérience seule, ni la théorie seulene permettent d'aboutir. »
Théophraste von Hohenheini, sur-nommé Paraceisus (1493-1541), com-mença à remettre en question les ensei-gnements transmis depuis l'Antiquité.A partir de la connaissance des sub-stances actives il créa un système deprescription, s'opposant ainsi aux ab-surdes mélanges de la médecine duMoyen Age. En raison du succès de sesordonnances, il fut même accusé d'em-poisonnement et se défendit par unephrase devenue un axiome en pharma-cologie : « Si vous vouliez expliquer defaçon précise l'action de chaquepoison, il faut alors se demanderqu'est-ce qui n'est pas un poison ?Toute substance est un poison et au-cune n'est inoffensive. C'est simple-ment la dose qui fait qu'une substancen'est pas toxique. » ^

Histoire de la pharmacologie 3
Les débuts
Johan Jakob Wepfer (1620-1695) uti-lisa l'expérimentation animale aveccomme objectif premier le contrôle dela véracité des affirmations concernantles effets pharmacologiques ou toxico-logiques de certaines substances.«J'ai beaucoup réfléchi et finalement
, je me suis résolu à expliquer les phéno-, mènes par l'expérience. »
Il s'efforçait de décrire les actions desdiverses substances en se basant surleurs propriétés chimiques.« La pharmacologie est me scienceexacte. Son enseignement doit nousfournir une connaissance des médica-ments qui permette d'étayer notre choixthérapeutique au lit du malade. »
Développement et reconnaissancegénérale
| L'institutionalisation
Rudolf Buchheim (1820-1879) fondaen 1847 à Dorpat le premier institut uni-versitaire de pharmacologie affirmantainsi l'indépendance de la pharmaco-logie en tant que science.
Oswald Schmiedeberg (1838-1921)avec ses élèves, dont douze furentnommés à des chaires de pharmaco-logie, éleva la pharmacologie alle-mande au plus haut niveau. AvecBernard Naunyn (1839-1925) médecinintemiste, il fonda la première revue pé-riodique consacrée à la pharmacologie;qui est encore publiée de nos jours.
Statu quoAprès 1920, se développèrent, dansl'industrie pharmaceutique et à côté desinstituts universitaires déjà existants,des laboratoires de recherche consacrésà la pharmacologie. Après 1960 furentfondés dans de nombreuses universitéset dans l'industrie des départements depharmacologie clinique.

Origine d'un produit actif
Drogues et substances actives
Jusqu'à la fin du siècle dernier, les mé-dicaments utilisés pour le traitementdes maladies étaient des produits natu-rels, dérivés ou non de la matière vi-.vante, le plus souvent des plantes oudes fragments de plantes séchées maisparfois fraîches. Celles-ci peuvent ren-fermer des substances exerçant une ac-tion thérapeutique mais aussi des com-posés toxiques.
Pour pouvoir disposer de sub-stances médicinales, dérivées du règnevégétal, à longueur d'année et non passimplement au moment de la récolte, onsavait déjà depuis l'antiquité conserverles plantes sous forme séchée ou en lestrempant dans l'alcool ou l'huile végé-tale. La dessiccation d'un produit vé-gétal ou animal aboutit à une drogue.En langage usuel, ce terme de droguedésigne principalement un stupéfiant ouune substance présentant un risqued'abus ou de dépendance. En termesscientifiques, la notion de drogue nerenferme cependant aucune informa-tion sur la nature et l'importance des ef-fets. Les feuilles de menthe séchées oules fleurs du tilleul sont des droguescomme le sont les fleurs et les feuillesséchées du chanvre blanc (marijuana)ou sa résine (haschich) ainsi que la pâteséchée du pavot, qui était obtenue aupa-ravant après incision des capsules(opium brut).
En trempant des plantes ou desfragments de plante dans l'alcool(éthanol), on obtient une teinture : lescomposants pharmacologiquement ac-tifs sont extraits par l'alcool. Les tein-tures ne contiennent cependant pasl'ensemble des substances présentesdans la plante ou la drogue mais seule-ment celles solubles dans l'éthanol.Dans le cas de la teinture d'opium, cessubstances solubles dans l'alcool sontprincipalement des alcaloïdes : mor-phine, codéine, noscapine = narcotine,papavérine, narcéine et bien d'autres.
Le choix d'un produit naturel oud'un extrait pour le traitement d'unemaladie implique en général l'adminis-tration d'un ensemble de molécules denature très diverse. Si bien que laconcentration d'une molécule donnéedans un produit naturel peut varier defaçon importante selon son origine (lieude recueil), son mode de culture (mo-ment de la récolte) et ses conditions destockage (durée et conditions). La pro-portion d'un composant donné peutégalement varier de façon importantepour d'autres raisons.
Après la purification de la mor-phine par F.W. Sertumer (1783-1841),les composants des produits naturelsont été isolés sous forme chimiquementpure dans les laboratoires pharmaceu-tiques.
La purification de ces composantsa pour but :
1. L'identification de la (ou des) mo-lécule(s) active(s)2. L'analyse des propriétés biolo-giques (pharmacodynamiques) de cha-cun des composants ; l'analyse de leurdevenir dans l'organisme (pharmacoci-nétique)3. La possibilité, lors de l'utilisationthérapeutique de l'une de ces moléculesde donner une dose précise et renouve-lable4. La possibilité d'une synthèse chi-mique. Celle-ci permet de s'affranchirde l'épuisement des ressources natu-relles mais permet également l'étudedes relations entre structure chimique etactivité. Ces efforts peuvent même dé-boucher sur la synthèse de moléculesdérivées de la molécule initiale, douéesde propriétés pharmacologiques inté-ressantes.
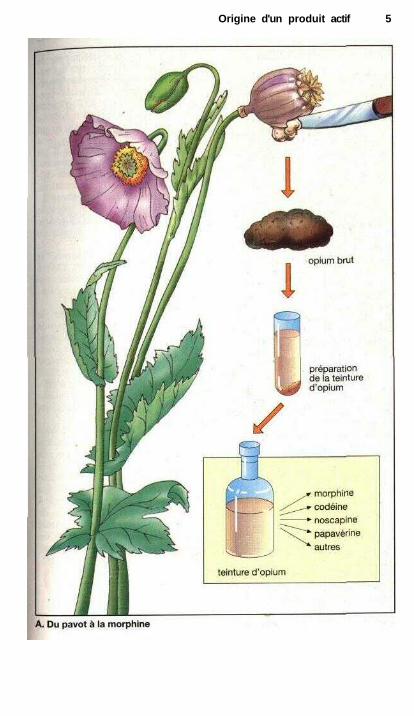
Origine d'un produit actif 5

6 Origine d'un médicament
Développement d'un médicament
Le développement débute par la syn-thèse de nouvelles structures chi-miques. Les substances aux structuresplus complexes peuvent être extraites
•de plantes (glycosides cardiaques), detissus animaux (héparine), de culturesde micro-organismes (pénicilline) ou decellules humaines (urokinase) ou en-core obtenues par recombinaison géné-tique (insuline humaine). Par ailleurs, ilest d'autant plus facile de trouver unemolécule nouvelle que l'on connaît larelation entre structure et activité.
L'investigation préclinique four-nit des informations sur les propriétésdes nouvelles substances. Le premiertri peut être effectué à l'aide d'étudespharmacologiques et biochimiques (parexemple des études de liaison aux ré-cepteurs, p. 56) ou encore d'expé-riences réalisées sur des cellules en cul-ture, des tissus ou des organes isolés.Comme ces modèles ne peuvent jamaisreproduire les événements complexesqui se déroulent dans un organisme vi-vant, les médicaments potentiels de-vront être testés chez l'animal. L'étudechez l'animal indique d'abord si l'effetrecherché a bien lieu et s'il existe deseffets toxiques. Les études de toxicitépermettent de mettre en évidence latoxicité aiguë ou à long terme, les effetsmutagènes, cancérigènes et les éven-tuelles anomalies du développement(effet tératogène). Il faut aussi testerchez l'animal les voies d'administra-tion, la distribution et l'élimination desdiverses molécules (pharmacocinéti-que).
Déjà pendant cette étude precli-nique, on se rend compte que seule unefaible proportion des molécules pourraêtre testée chez l'homme. Les tech-niques galéniques permettent ensuite depréparer les formes d'administration dela substance.
L'étude clinique débute par laphase 1 où l'on détermine chez des vo-lontaires sains si les propriétés obser-vées chez l'animal se manifestent éga-lement chez l'homme, et où l'on établitla relation entre l'effet et les doses. Aucours de la phase 2, le médicamentéventuel est, pour la première fois, testécontre la maladie pour laquelle il estprévu chez un groupe de patients sélec-tionnés. Si la substance montre une effi-cacité réelle et peu d'effets secondaires,on passe alors aux études de phase 3 :l'action thérapeutique de la nouvellesubstance est comparée chez un groupede patients plus important, à celle dumédicament de référence.
Durant ces études cliniques, lamajorité des molécules testées s'avèreinutilisable. Sur 10 000 molécules syn-thétisées, seule une aboutira à un médi-cament.
La décision de mise sur lemarché est prise après une demandeofficielle du laboratoire, par un orga-nisme public (en Allemagne, la com-mission pour la santé). Le demandeurdoit justifier, à l'aide de ses résultatsexpérimentaux, que les critères d'effi-cacité et d'innocuité sont remplis et queles formes galéniques répondent auxnormes de qualité.
Après mise sur le marché, la nou-velle substance reçoit un nom commer-cial (p. 327) et il reste aux médecins à laprescrire et aux pharmaciens à la déli-vrer à leurs malades. Durant l'ensemblede la vie du médicament, on continueraà examiner s'il fait ses preuves (phase 4de l'étude clinique). L'expérience deplusieurs années de prescription permetd'abord d'évaluer les indications et lesrisques et ensuite de définir la valeurthérapeutique du nouveau médicament.
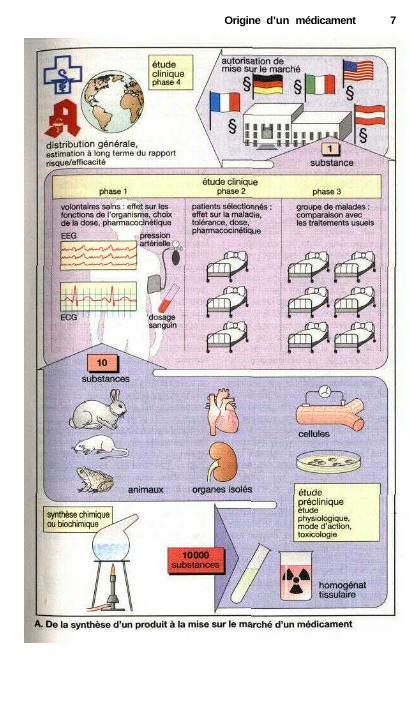
Origine d'un médicament 7

8 Formes galéniques
Formes orales, oculaires ou nasales
La substance active devient un médica-ment après transformation sous uneforme adaptée à l'usage thérapeutique.La forme galénique dépend de la façondont est administrée la substance et doitpermettre au malade et à son médecinune manipulation commode du principeactif (dosage précis, bonne conserva-tion). Lapharmacie galénique s'occupede la fabrication de formes adaptées àl'administration et des contrôles dequalité.
Les formes liquides (A) peuventêtre des solutions, des suspensions(dispersion dans l'eau de petites parti-cules d'une substance insoluble) ou desémulsions (dispersion de fines goutte-lettes d'une solution dans un autre li-quide : par exemple eau dans l'huile).Comme pendant le stockage les suspen-sions peuvent sédimenter et les émul-sions se séparer on aura tendance à pré-férer une solution du principe actif.Dans le cas de substances très peu so-lubles dans l'eau, ce but sera fréquem-ment atteint en utilisant de l'éthanol (oud'autres solvants). On obtient alors desgouttes en solution aqueuse ou alcoo-lique. Ces solutions sont distribuéesdans des flacons spéciaux munis d'uncompte-gouttes. Le malade peut me-surer de façon précise sa dose indivi-duelle en comptant le nombre degouttes (la taille des gouttes dépend dudiamètre de l'orifice du compte-gouttes, de la viscosité et de la tensionsuperficielle de la solution). L'avantagedes gouttes est la possibilité d'ajusteraisément la posologie (selon le nombrede gouttes) aux besoins de chaque ma-lade. Leur inconvénient est parfois ladifficulté de prescrire un nombre précisde gouttes chez des patients âgés ou af-faiblis par la maladie. Si la substanceest en solution dans un volume de li-quide plus important, on parle d'habi-tude d'un sirop ou d'une potion, la doseindividuelle étant mesurée avec une
cuillère doseuse ou encore avec unecuillère à soupe (ss 15 ml) ou une cuil-lère à café (^5 ml). Compte tenu desdifférences de taille des cuillères ducommerce, on ne connaît cependant pasles doses individuelles avec une grandeprécision.
Les gouttes nasales et oculaires(A) sont utilisées pour le traitement desaffections des muqueuses nasales oudes conjonctivites. Dans le cas desgouttes nasales, on augmentera la vis-cosité de la solution pour allonger letemps de contact.
Les formes solides sont les com-primés, les dragées et les gélules (B).Les comprimés sont des objets cylin-driques formés par compression d'unmélange contenant la substance active,un excipient et divers additifs. L'exci-pient (lactose, sulfate de calcium) apour fonction de donner au compriméune taille suffisante pour permettre dele manipuler et de l'avaler facilement. Dfaut se rappeler que la dose unitaire denombreux médicaments est souvent in-férieure à quelques mg. Pour donnerune idée de ce que représentent 10 mg,on a imprimé en bas de la page un carréqui, une fois découpé, pèserait 10 mg.Les additifs tels l'amidon qui gonfle enprésence d'eau ou le bicarbonate quidégage du gaz carbonique au contact del'acidité gastrique, accélèrent la disso-lution du comprimé. Les substancesliantes, facilitent la fabrication descomprimés, leur conservation et leuridentification (couleur).
Les comprimés effervescents nesont pas des formes solides, car ils sedissolvent dans l'eau presque instanta-nément et deviennent alors une solu-tion.
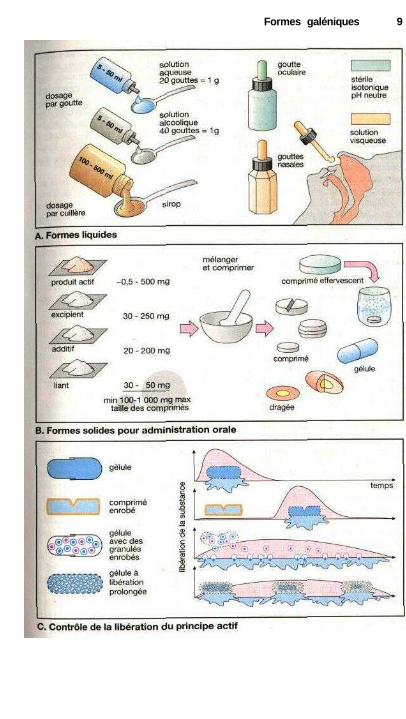
Formes galéniques 9

10 Formes galéniques
Les dragées sont des comprimésrecouverts d'un revêtement. Le noyaude la dragée, le comprimé, est recouvertpar exemple de cire qui protège les mo-lécules fragiles, masque un goût ou uneodeur désagréable, facilite la pnse etpermet d'apposer une marque colorée.Les gélules se composent en générald'une enveloppe de forme ovale, cons-tituée le plus souvent de gélatine, quirenferme la substance active en poudre,sous forme de granulés (p. 9, C) ou plusrarement sous forme d'une solution.
Dans certains comprimés (com-primés à libération prolongée) la sub-stance active est incorporée dans unetrame, permettant ainsi une diffusionlocale au moment de l'imbibition ducomprimé. Dans le cas des solutions, lamolécule active peut être absorbéepresque immédiatement (A, 3e co-lonne) ; au contraire, dans le cas desformes plus solides, il faut d'abord quele comprimé se délite ou que la gélules'ouvre avant que la molécule active nese dissolve, ne traverse la muqueuse del'estomac et de l'intestin et ne passedans le sang (absorption). Comme ledélitement des comprimés et la dissolu-tion de la molécule active réclament dutemps, l'essentiel de l'absorption s'ef-fectuera au niveau de l'intestin (A,2e colonne). Dans le cas d'une solution,le passage dans le sang débute déjà auniveau de l'estomac (A, 3e colonne).
Pour protéger les substances dé-truites en milieu acide, il est possibled'empêcher la désintégration des com-primés dans l'estomac en les recouvrantde cire ou d'un polymère d'acétate decellulose. La désintégration et la disso-lution se produisent alors dans le duo-dénum mais sans que la libération de lasubstance ne soit ralentie en tant quetelle (A, 1" colonne).
La libération de la substance ac-tive, et donc le lieu et la vitesse d'ab-sorption peuvent être contrôlés par lechoix d'un mode de fabrication appro-prié, dragée, gélules à libération pro-longée.
Dans le cas d'un comprimé à libé-ration prolongée, ceci est obtenu en in-corporant la substance active dans unetrame dont elle sera libérée lentement.Au cours du transport, la molécule ac-tive sera libérée dans les différents seg-ments intestinaux traversés et réab-sorbée à ce niveau (A, 4e colonne).Dans ces conditions, la forme exté-rieure du comprimé ne se modifie pasau cours du trajet.
Dans le cas d'une dragée ou d'uncomprimé enrobé, l'épaisseur de l'en-robage peut être choisie de telle façonqu'elle peut se dissoudre soit dans lapartie haute de l'intestin (A, 1" co-lonne), ou bien seulement dans la partiebasse de l'intestin (A, 5e colonne) pourpermettre l'absorption de la substanceactive. En choisissant par exemple untemps de dissolution permettant la tra-versée de l'intestin grêle, on peut ob-tenir une libération dans le côlon.
Pour une gélule, on peut égale-ment allonger la durée de libération duprincipe actif (retardement) en l'utili-sant sous forme de particules recou-vertes d'un revêtement d'épaisseur va-riable, formé par exemple de cire. Leurdissolution dépend de l'épaisseur de lacouche protectrice et aboutit à des vi-tesses de libération et d'absorptiondifférentes. Le principe défini pour lesgélules s'applique également aux com-primés, où des particules de substanceactive enrobées de revêtementsd'épaisseur variable seront compactéesen un comprimé. Les comprimés-retardont l'avantage par rapport aux gélules-retard de pouvoir être facilement sé-cables, ce qui signifie qu'il est possiblede prescrire une dose plus faible quecelle contenue dans le comprimé.
Ce procédé de retardement de lalibération du principe actif sera choisilorsqu'on ne souhaite pas obtenir unpassage rapide de la substance dans lesang, ou bien dans le cas de substancesdont le temps de transit dans l'orga-nisme est très faible et dont l'action doitêtre prolongée grâce à un apportconstant au niveau de l'intestin.
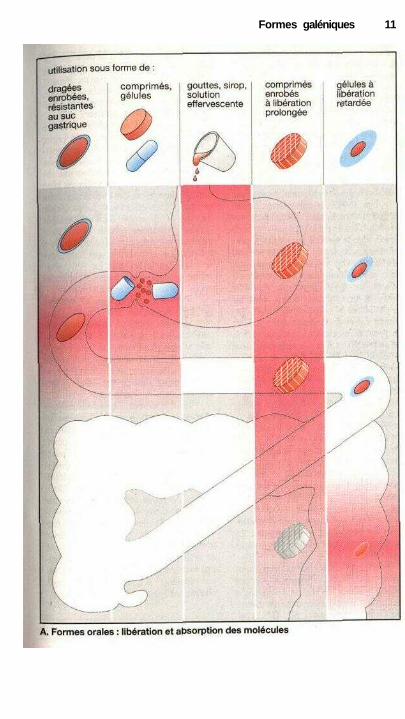
Formes galéniques 11

12 Formes galéniques
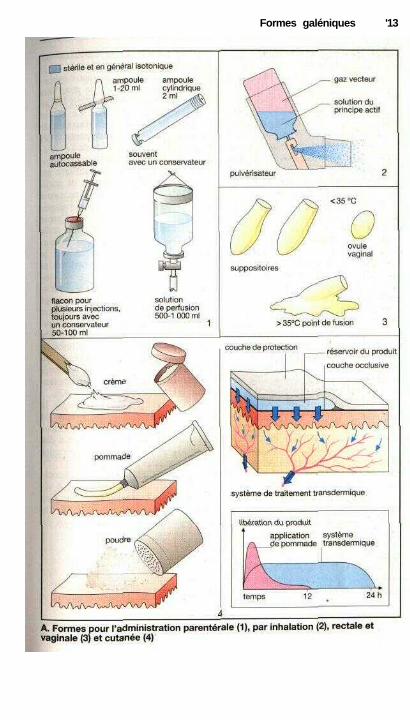
Formes parentérales (1), inhalées (2),rectales ou vaginales (3) et formestopiques (4)
Tous les médicaments ne sont pas obli-gatoirement administrés par la voieorale, c'est-à-dire avalés. Ils peuventêtre aussi donnés par voie parentérale.En parlant de forme parentérale, on dé-signe en général les formes injectablesbien que, en cas d'inhalation ou d'ap-port sur la muqueuse rectale, le sited'absorption soit également parentéral.
Une molécule administrée en in-jection intraveineuse, intramusculaireou sous-cutanée est le plus souvent sousforme liquide (soluté injectable), plusrarement sous forme d'une suspensionadministrée en injection intramuscu-laire, sous-cutanée ou même intra-arti-culaire. Le soluté injectable doit êtrestérile et apyrogène et ne pas contenirde particules en suspension. Il doit éga-lement éviter de provoquer des lésionsau point d'injection et si possible êtreau même pH et à la même pression os-motique que les liquides de l'orga-nisme. Les solutés injectables sontconservés dans des récipients fermés,en verre ou en matière plastique, àl'abri de l'air. La solution contenuedans les ampoules ou les flacons est in-jectée avec une seringue à travers uneaiguille. Il existe un système d'injectiondans lequel on dépose une ampoule cy-lindrique et qui permet d'injecter di-rectement le contenu de l'ampoule àtravers l'aiguille. On parlera d'une per-fusion, lorsque la solution est injectéepar voie intraveineuse pendant untemps plus long. Dans le cas des solu-tions de perfusion, il faut prendre lesmêmes précautions que pour les solutésinjectables.
Les molécules peuvent être vapo-risées sous forme d'aérosols sur lesmuqueuses des cavités de l'organismeen contact avec l'extérieur (par exem-ple l'arbre respiratoire, p. 14). Un aé-
rosol est une dispersion de particules li-quides ou solides dans un gaz, parexemple, l'air. Pour obtenir un aérosol,on pulvérise sous pression la substance,en solution ou en poudre très fine, à tra-vers une buse (pulvérisateur).
Pour déposer une substance activesur la muqueuse du rectum ou du vagin,on utilisera selon le cas des supposi-toires ou des ovules. Dans le cas d'uneprise rectale, on peut rechercher une ab-sorption et un effet systémique ou biencomme dans le cas des ovules vaginauxse limiter à un effet local. La moléculeest en général enrobée dans une matière(graisse, glycérine soluble dans l'eau,gélatine, polyéthylène-glycol), solide àtempérature ambiante et qui fond dansle vagin ou le rectum. Le film ainsiformé se répartit sur la muqueuse et fa-vorise l'absorption des molécules.
Poudres, pommades et crèmes(p. 16) sont étalées sur la peau. Dansbien des cas, elles ne contiennent au-cune molécule active mais assurent unsoin et une protection. On peut cepen-dant y incorporer une substance activesoit pour une action locale, soit plus ra-rement pour obtenir un effet systé-mique.
Les timbres transdermiquessont collés sur la peau. Ils contiennentun réservoir d'où la molécule diffuseraet sera absorbée à travers la peau.L'avantage de ces systèmes transder-miques réside justement dans la possi-bilité de fixer sur l'organisme un dépôtà partir duquel la molécule sera admi-nistrée de façon continue comme parperfusion. Cette voie nécessite cepen-dant des molécules : 1. capables de tra-verser la peau ; 2. agissant à dosefaible, compte tenu de la faible capacitéde réservoir ; 3. dont la fenêtre théra-peutique est assez large puisqu'il n'estpas possible d'ajuster la dose pourchaque malade.
Formes galéniques '13

14 Formes galéniques
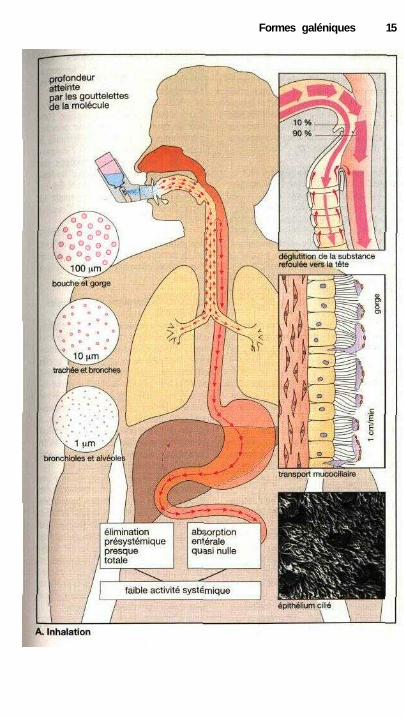
Administration par inhalation
L'inhalation sous forme d'un aérosol(p. 12), d'un gaz ou d'une vapeur,permet d'appliquer une molécule activesur les épithéliums bronchiques et unefaible part de la paroi des alvéoles pul-monaires. Ce mode d'application estchoisi lorsque l'on désire agir sur lamusculature bronchique ou modifier laconsistance du mucus bronchique ouencore lorsque l'on cherche à obtenirpar l'intermédiaire d'une entrée au ni-veau alvéolaire un effet systémique :(anesthésiques inhalés, p. 216).
Les aérosols sont obtenus par pul-vérisation d'une solution ou d'unepoudre très fine. Dans les pulvérisa-teurs classiques propulsés par un gazvecteur, la formation de l'aérosol seradéclenchée en appuyant sur un piston(clapet doseur). Pour une pulvérisationde ce genre, les doses maximales auto-risées seront indiquées en coups depiston / unité de temps. Au momentde l'utilisation, l'embout du pulvérisa-teur sera entouré par les lèvres du pa-tient et l'aérosol sera déclenché lors del'inspiration. L'efficacité de cette for-me d'administration dépend de la tailledes particules émises et de la coordina-tion entre la pulvérisation et l'inspira-tion. La taille des gouttes conditionne lavitesse avec laquelle elles sont entraî-nées dans l'air inspire et par là même laprofondeur atteinte dans l'arbre res-piratoire. Les particules d'un diamètresupérieur à 100 |Jim seront déjà arrêtéesau niveau de la bouche et du pharynx.Si la pulvérisation est d'abord effectuéedans une chambre avant d'être inhalée,on réduit de façon importante la prisede ces grosses particules. Les goutte-lettes ou les poudres d'un diamètre in-férieur à 2 |im atteignent les alvéolesmais seront à nouveau expirées si ellesne sédimentent pas.
Une partie de la substance dé-posée au niveau des bronches sur lacouche de mucus recouvrant l'épithé-lium sera absorbée mais le reste seratransporté en même temps que le mucusbronchique vers la gorge. Le mucusbronchique se déplace en direction ducou sous l'effet des battements coor-donnés des cellules ciliées de l'épithé-lium bronchique. La fonction physiolo-gique de ce courant mucociliaire estl'élimination des poussières et parti-cules inspirées avec l'air. Une partieseulement de la substance pulvériséeparvient en général jusqu'à l'arbrebronchique. Et de cette fraction seuleune faible part pénètre dans la mu-queuse, le reste étant ramené vers lagorge par le transport mucociliaire etavalé. Dans des conditions défavo-rables, 90 % de la dose inhalée aboutis-sent dans le tube digestif. L'avantagedes inhalations, c'est-à-dire le caractèrelocal de l'application, sera particulière-ment utilisé pour des molécules mal ab-sorbées au niveau de l'intestin (cromo-glycate, isoprénaline, ipratropium) ousubissant une élimination présysté-mique (dipropionate de béclométha-sone, budesonide, flunisolide, p. 42).
Lorsque la fraction avalée de lamolécule est absorbée au niveau del'intestin sans être transformée, l'inha-lation permet d'atteindre au niveau desbronches une concentration supérieureà celle des autres organes.
L'efficacité du transport mucoci-liaire dépend du mouvement des cils vi-bratiles et de la viscosité du mucus. Cesparamètres peuvent être modifiés defaçon pathologique (bronchite, toux durumeur).
Formes galéniques 15

16 Formes galéniques
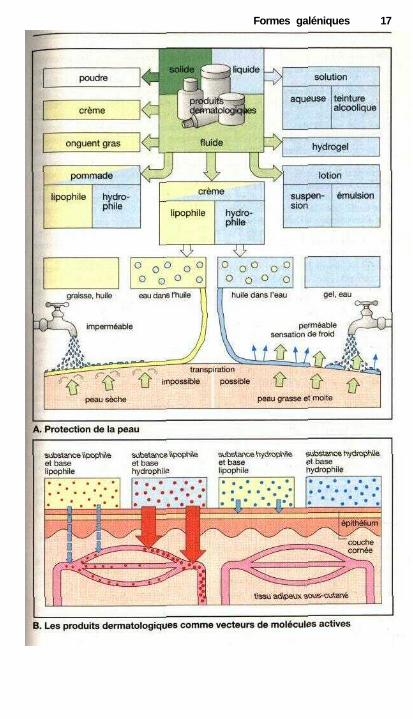
Dermatologie
Des préparations pharmaceutiques(dermatologiques) peuvent être utili-sées sur la peau en guise de soins oupour la protéger des agressions (A) ou
• encore pour laisser diffuser dans lacouche cutanée voire dans l'organismeune molécule active (B).
Protection et soins de la peau (A)Selon l'état de la peau (sèche, fendillée,moite, grasse, élastique) et les typesd'agressions subies (par exemple longsséjours dans l'eau, utilisation régulièrede solutions de désinfection à base d'al-cool (p. 286), bains de soleil prolongés)on pourra utiliser une grande variété demoyens de protection. Ils se différen-cient en fonction de leur consistance, deleurs propriétés physico-chimiques (hy-drophiles, hydrophobes) et éventuelle-ment de leur composition.
Poudres. Elles sont répandues surla peau intacte et contiennent du talc, dustéarate de magnésium, de la silice, oude l'amidon. Elles collent à la peau enformant un film glissant qui peut atté-nuer une irritation mécanique. Lespoudres ont également un effet assé-chant (la surface importante accélèrel'évaporation).
Pommade, crème grasse. Ellessont composées d'une base lipophile(huile de paraffine, de vaseline, graisseanimale) renfermant jusqu'à 1 % depoudre : oxyde de zinc ou de titane,amidon seul ou mélangés.
Pâte, onguent. Il s'agit d'unepommade contenant plus de 10 % depoudre.
Crème. Elle est formée d'uneémulsion aqueuse dans une matièregrasse et s'étale plus facilement qu'unepommade.
Gels et pommades hydrophiles.Leur consistance est due à un agentde structure (gélatine, méthylcellulose,polyéthylène glycol). Une lotion parcontre est une suspension en milieuaqueux de composants solides et inso-lubles.
Les crèmes hydrophiles sontconstituées d'une émulsion d'un corpsgras dans l'eau, stabilisée par un agentémulsifîant.
Tous les composés dermatolo-giques dont la base est lipophile for-ment au contact de la peau une couchehydrophobe. Cette couche résistante àl'eau va également empêcher l'évapo-ration de l'eau. La peau protégée dudessèchement voit son degré d'hydra-tation et son élasticité augmenter. La di-minution de l'évaporation augmente latempérature de la peau sous l'occlu-sion.
Les produits hydrophiles, facile-ment éliminés par lavage, n'empêchentpas la perte d'eau transcutanée. Cetteévaporation provoque une impressionde froid.
Application dermique de moléculesactives (B)Pour parvenir au site d'action, la molé-cule doit pénétrer dans la couche cu-tanée si l'on désire une action topique(par exemple une crème aux corti-coïdes), ou la traverser si l'on souhaiteun effet systémique (application trans-dermique, voir p. 120 par exemple pourl'administration de nitrates organi-ques). La tendance d'une molécule àquitter le support est d'autant plusgrande que son caractère et celui de labase sont différents (par exemple unemolécule hydrophile dans une base hy-drophobe ou l'inverse). Comme la peauconstitue une barrière hydrophobe(p. 22) seules les molécules lipophilespeuvent être absorbées. Lorsque la basede la préparation est hydrophobe, lesmolécules hydrophiles ne pourront pastraverser l'épidémie. Cette forme gale-nique est très utile lorsque l'on cherchepar exemple à obtenir une concentra-tion élevée d'une substance à la surfacede la peau (pommade à la néomycinepour traiter une infection cutanée).
Formes galéniques 17

18 Formes galéniques
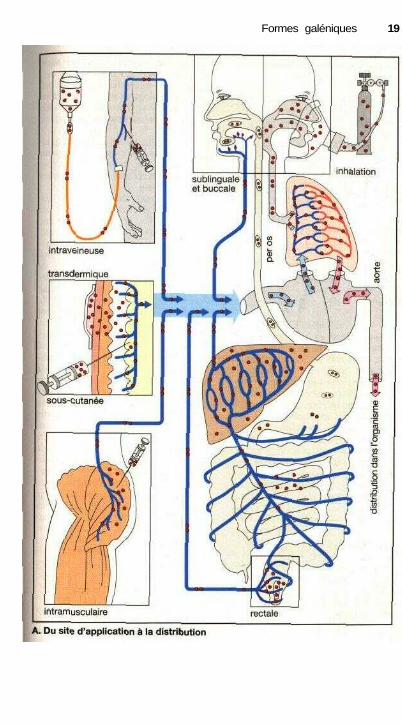
Du site d'application à ladistribution dans l'organisme
En général, les médicaments atteignentleur cible par l'intermédiaire de la cir-culation sanguine. Ceci signifie que lesmolécules doivent d'abord parvenir jus-qu'au sang. Ceci se produit au niveaude la circulation veineuse. Différentssites d'entrée sont possibles. La sub-stance peut être injectée ou perfusée parvoie intraveineuse. Dans ce cas la mo-lécule passe immédiatement dans lesang tandis que dans le cas d'une injec-tion sous-cutanée ou intramusculaire,elle devra d'abord diffuser du site d'in-jection vers le sang. Ces modes d'admi-nistration impliquent une lésion de lapeau et réclament donc des précautionsparticulières. Dans ces conditions, l'ad-ministration par la bouche (per os)beaucoup plus simple sera plus fré-quemment employée ; le passage de lasubstance dans le sang s'effectuant en-suite au niveau de la muqueuse stoma-cale ou intestinale. Cette voie a pourinconvénient de voir la substance tra-verser obligatoirement le foie (systèmeporte) avant d'aboutir dans la circula-tion générale. Il faut se souvenir de cephénomène pour les substances rapide-ment métabolisées au niveau du foie oumême inactivées (élimination presysté-mique, effet de « premier passage »,p. 42). Par voie rectale une partie aumoins des molécules passera égalementpar le système porte avant d'aboutir à lacirculation générale, car seules lesveines de l'extrémité du rectum abou-tissent directement à la veine cave. Lepassage par le foie sera évité dans le casd'une absorption buccale ou sublin-guale car le système veineux drainant lamuqueuse buccale aboutit directementdans la partie supérieure de la veinecave. Le phénomène sera identiquepour une substance inhalée (p. 14).Mais dans ce mode d'administration,on recherche principalement un effet
local et seulement exceptionnellementun effet systémique. Dans certainesconditions, une substance peut égale-ment être appliquée sur la peau : sys-tème thérapeutique transdermique(p. 12). Dans ce cas, la substance dif-fuse lentement à partir d'un réservoir,traverse la peau et aboutit finalementdans la circulation sanguine. Seul unpetit nombre de molécules peut être dé-livré par voie transdermique. La possi-bilité d'utiliser cette voie dépend despropriétés physicochimiques du médi-cament et des nécessités thérapeutiques(effet immédiat ou de longue durée).
La rapidité avec laquelle une sub-stance se répand dans l'organisme serafonction du mode et du site d'applica-tion. La distribution d'une substanceadministrée par voie intraveineuse estplus rapide que pour une injection in-tramusculaire et encore plus rapideque par voie sous-cutanée. Par voiebuccale ou sublinguale, une substancepasse dans le sang plus rapidement quepar administration classique sous formede dragée per os. En effet, le médica-ment est alors proche du site d'absorp-tion et l'on obtient, par dissolution dansla salive d'une dose individuelle, desconcentrations locales très élevées quiaccélèrent l'absorption au niveau del'épithélium de la cavité buccale. Cecine s'applique pas aux médicaments peusolubles dans l'eau ou à ceux qui se ré-sorbent difficilement. Pour ces sub-stances, l'administration orale est indi-quée car le volume de dissolution et lasurface d'échange sont plus importantsdans l'intestin grêle que dans la cavitébuccale.
Sous le terme de de biodisponibi-lité on désigne la fraction de la sub-stance administrée qui parvient dans lacirculation, c'est-à-dire qui sera dispo-nible par voie systémique. Plus l'élimi-nation présystémique d'une substanceadministrée est importante et plus sabiodisponibilité est faible.
Formes galéniques 19

20 Sites d'action cellulaire
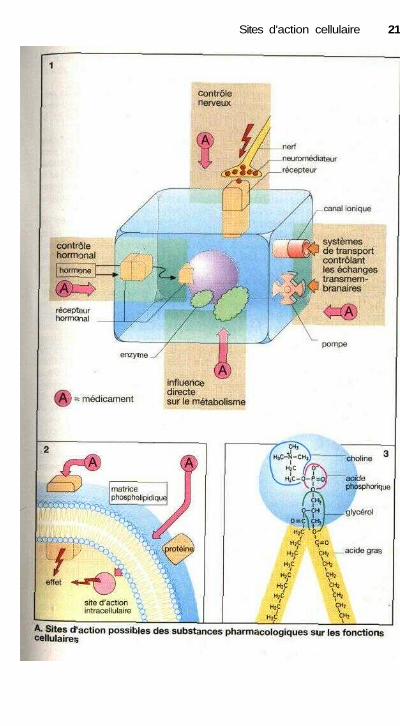
Les sites d'action des médicaments
Le but de l'utilisation des médicamentsest de régler certains événements biolo-giques pour diminuer ou éliminer lesmanifestations de la maladie. La pluspetite des unités vivantes d'un orga-nisme est la cellule. La membrane cel-lulaire ou plasmalemme sépare defaçon efficace la cellule de son environ-nement et permet ainsi le maintiend'une vie intérieure indépendante. Desprotéines de transport dans la mem-brane assurent le contrôle des échangesde matière avec le milieu environnant.Ces protéines peuvent être des pompes,systèmes de transport actif nécessitantde l'énergie (Na+ - K4 ATPase, p. 130),d'autres transporteurs (« carrier », parexemple le co-transport Na/glucose,p. 176) ou des canaux ioniques (canalsodique, p. 136 ou canal calcique,P. 122) (1).
La coordination des fonctions dechaque cellule est indispensable à lasurvie de l'organisme et donc des cel-lules elles-mêmes. Ce contrôle desfonctions cellulaires s'effectue aumoyen de messagers chimiques qui vé-hiculent l'information. Parmi ces mé-diateurs, les neurotransmetteurs libérésau niveau des terminaisons nerveuses,et pour lesquels les cellules possèdentdes sites de liaison spécifiques ou ré-cepteurs, présents sur la membrane.Les hormones sécrétées par les glandesendocrines, qui parviennent aux cel-lules par la circulation sanguine ou lemilieu extracellulaire, servent égale-ment de signaux. Enfin, certains média-teurs peuvent provenir de cellulesproches (par exemple les prostaglan-dines, p. 194) : influence paracrine.
L'action des médicaments estsouvent liée à un effet sur une fonctioncellulaire. Les sites actifs peuvent êtreles récepteurs qui captent spécifique-ment les signaux (agonistes ou antago-nistes des récepteurs, p. 60). La modifi-cation de l'activité d'un système detransport peut également contrôler unefonction cellulaire (ex. : glycosides car-diaques, p. 130, diurétiques de l'anse,p. 160 ou antagonistes calciques,
p. 122). Les molécules peuvent aussiagir directement à l'intérieur des cel-lules, sur le métabolisme général, parexemple en bloquant une enzyme (inhi-biteurs des phosphodiestérases, p. 132)ou en la stimulant (nitrates organiques,P. 120) (2).
Au contraire, des molécules agis-sant sur la couche externe de la mem-brane cellulaire, celles qui touchentl'intérieur des cellules doivent traversercette membrane.
La membrane cellulaire estcomposée d'une double couche depho^spholipides (épaisseur d'environ80 A = 8 nm) dans laquelle sont inté-grées des protéines (protéines inté-grales, par exemple récepteurs ou pro-téines de transport). La molécule dephospholipide comporte deux acidesgras à longue chaîne, estérifiés chacunsur une fonction alcool hydrophile duglycérol. La troisième fonction alcooldu glycérol est liée à un acide phospho-rique qui lui-même porte un résidu sup-plémentaire, par exemple un alcool tella choline (pour donner la phosphati-dylcholine ou lécithine), un acideaminé la serine ou un hexa-alcool cy-clique, l'inositol. En ce qui concerneleur solubilité, les phospholipides sontdes molécules amphiphiles : la partiequi contient les acides gras est lipo-phile, l'autre partie de la molécule (têtepolaire) est hydrophile. Compte tenu dece caractère amphiphile, les phospholi-pides vont s'arranger presque automati-quement en double couche dans un mi-lieu aqueux ; les têtes polaires versl'extérieur, dirigées vers le milieuaqueux, les chaînes d'acides gras tour-nées vers l'intérieur de la membrane,serrées les unes contre les autres (3).
L'intérieur hydrophobe de lamembrane phospholipidique constituepour les molécules polaires et en parti-culier les molécules chargées une bar-rière de diffusion presque infranchis-sable. Les groupes apolaires aucontraire passent facilement à travers lamembrane. Ce phénomène a une in-fluence considérable sur l'entrée, la dis-tribution et l'élimination des médica-ments.
Sites d'action cellulaire 21

22 Distribution dans l'organisme
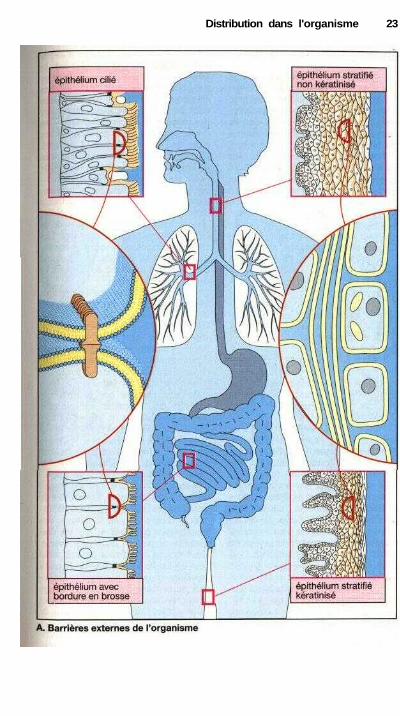
Barrières externes de l'organisme
Avant d'entrer dans la circulation san-guine (absorption), une substance doitfranchir les barrières qui séparent l'or-ganisme de son environnement et déli-mitent le milieu intérieur. Ces limitessont constituées par la peau et les mu-queuses.
Lorsque l'absorption a lieu dansl'intestin, la barrière est alors consti-tuée par l'épithélium intestinal. Cet épi-thélium est formé d'une couche uni-cel-lulaire d'entérocytes et de cellules àmucus. Du côté de la lumière intesti-nale, ces cellules sont liées les unes auxautres formant la zonula occludens (re-présentée par des points noirs dans leschéma du bas à gauche).
Une zonula occludens (ou encoretight junction) est une région dans la-quelle les membranes phospholipi-diques de deux cellules voisines sonttrès proches l'une de l'autre et sontmême reliées par l'intermédiaire deprotéines incluses dans les membranes.Cette zone entoure complètement lescellules comme un anneau, de sorte quechacune d'elle est reliée aux cellulesvoisines, formant une barrière continueentre les deux espaces séparés par lacouche cellulaire, dans le cas de l'in-testin entre la lumière intestinale et l'es-pace intercellulaire. L'efficacité aveclaquelle cette barrière empêche l'é-change de substances peut être ren-forcée par l'alignement d'un grandnombre de ces interactions, comme parexemple dans le cas de l'endothéliumdes capillaires cérébraux. De plus, cesprotéines de liaison semblent égalementservir à contrecarrer un ensemble deprotéines fonctionnelles (pompes, ca-naux ioniques), qui sont caractéris-tiques des domaines membranaires sé-parés.
Seules les molécules dont les pro-priétés chimiques permettent un pas-sage à travers la phase interne lipophilede la double couche (jaune), ou celles
pour lesquelles existe un mécanisme detransport particulier peuvent être absor-bées par voie entérale.
La capacité d'absorption d'un mé-dicament sera caractérisée par le quo-tient entre la quantité absorbée et laquantité présente dans l'intestin.
Dans l'arbre respiratoire, lescellules ciliées de l'épithélium sontégalement liées du côté luminal par deszonulae occludens, de façon à ce que lacavité bronchiale soit séparée des tissuspulmonaires par une double couchephospholipidique continue.
Si l'administration est orale ousublinguale, la molécule se heurte à unebarrière (muqueuse buccale) consti-tuée d'un épithélium stratifié non kéra-tinisé. Les cellules établissent entreelles des contacts ponctuels (desmo-somes, non figurés sur les schémas),mais ces interactions ne ferment pascomplètement l'espace intercellulaire.Cette obturation est réalisée par l'accu-mulation dans l'espace extracellulairede fragments de membrane sécrétés parles cellules (voir encadré semi-circu-laire à droite et au milieu). De cette ma-nière, il existe également dans l'épithé-lium stratifié une couche continue dephospholipides qui, contrairement à cequi se passe dans l'épithélium intes-tinal, est maintenant déposée à l'exté-rieur des cellules. Le même principe debarrière s'observe dans l'épithéliumstratifié kératinisé de la peau.L'existence d'une couche continue dephospholipides signifie que seules lessubstances lipophiles, capables depasser à travers une membrane phos-pholipidique, peuvent pénétrer dansl'organisme à travers les épithéliumsstratifiés. La vitesse d'absorption dé-pend dans ce cas de l'épaisseur de l'épi-thélium. Au niveau de la peau, l'ab-sorption sera rendue encore plusdifficile par la présence d'une couchecornée (stratum corneum) dont l'épais-seur est très variable d'une zone àl'autre.
Distribution dans l'organisme 23

24 Distribution dans l'organisme
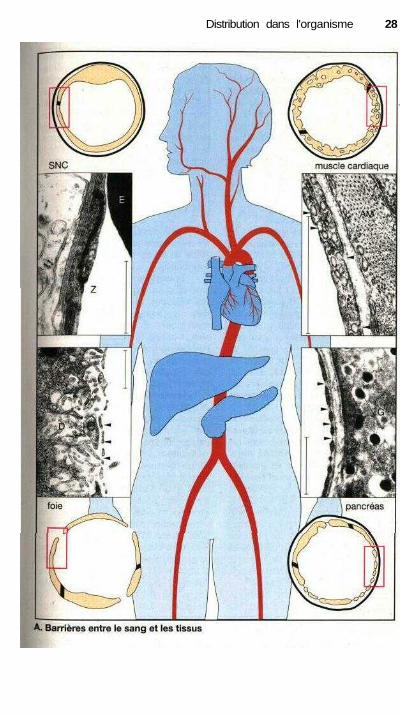
Barrières entre le sang et les tissus
Les substances sont transportées par lesang dans les différents tissus de l'orga-nisme. L'échange de subtances entre lesang et les tissus se déroule principale-ment au niveau des capillaires. Cest eneffet dans le lit capillaire très ramifiéque la surface d'échange est la plus im-portante et la durée d'échange la pluslongue (faible vitesse du flux sanguin).La paroi capillaire constitue égalementune barrière entre le sang et les tissus.Elle est formée d'une couche de cel-lules endothéliales entourée d'unemembrane basale (représentée par untrait noir dans les schémas ci-contre).Les cellules endothéliales sont forte-ment associées entre elles par desjonctions cellulaires (zonula occludensdésignée par Z dans le cliché de micro-scopie électronique en haut à gauche)de telle sorte qu'il n'existe aucun es-pace ni aucune lacune permettant unpassage des molécules du sang versl'espace interstitiel (E : coupe d'un éry-throcyte).
Cette barrière entre le sang et lestissus a une structure variable selon lesrégions du corps et la perméabilité ca-pillaire aux médicaments dépendradonc des fonctions propres de chaquecellule endothéliale.
Dans la majeure partie du réseaucapillaire, par exemple dans lemuscle cardiaque, les cellules endo-théliales sont caractérisées par uneactivité d'endocytose importante.Ceci se manifeste par les nombreuxreplis et les vacuoles visibles dans lescellules endothéliales (indiqués parles flèches dans le cliché de micro-scopie électronique en haut à droite).Cette activité d'endocytose permet untransport de liquide du sang vers l'es-pace interstitiel et en sens inverse.Les molécules dissoutes et les médi-caments peuvent ainsi franchir la bar-rière séparant le sang des tissus (AM :actomyosine d'une cellule cardiaque).Dans ce mode de transport, les pro-priétés physico-chimiques de la sub-stance ne jouent pratiquement aucunrôle.
À côté de cela, il existe d'autresréseaux capillaires (par exemple dans lepancréas) où les cellules endothélialesprésentent une série de fenêtres. Lescellules en effet ne sont pas liées entreelles de façon étroite mais comportentdes pores uniquement recouverts d'undiaphragme (indiqué par les flèchesdans le cliché en bas à droite).Diaphragme et membrane basale sontaisément traversés par les substances defaible masse moléculaire et en particu-lier par la majeure partie des médica-ments. Ce passage est plus difficiledans le cas des macromolécules tellesles protéines et il dépend alors de lataille de la molécule et de sa charge. Lesendothéliums avec des fenestrations in-tracellulaires se trouvent par exempledans le réseau capillaire de l'intestin etdes glandes endocrines.
Dans le cerveau, la moelle épi-nière et le système nerveux central,les cellules endothéliales ont une acti-vité d'endocytose très faible et ne pos-sèdent aucun pore. Pour franchir labarrière hémato-encéphalique, le mé-dicament doit alors traverser la celluleendothéliale et donc franchir les mem-branes luminales et basales. Ce fran-chissement suppose que la moléculepossède des propriétés physicochi-miques particulières (p. 26) ou un mé-canisme de transport propre (voir le casdelaL-DOPAp. 186).
Dans le foie, il n'existe aucun obs-tacle au passage des substances entre lesang et l'espace interstitiel. Les cellulesendothéliales comportent au contact desmilieux extracellulaires des fenêtres degrande taille (100 nm de diamètre, es-pace de Disse : D) où le passage desmolécules n'est gêné ni par un dia-phragme ni par une membrane basale.Des barrières de diffusion peuvent aussiêtre situées de l'autre côté de la paroicapillaire ; barrière placentaire consti-tuée par la fusion des cellules du syncy-tiotrophoblaste, barrière entre le sang etles testicules formée par les cellules deSertoli reliées les unes aux autres.
(Les traits verticaux dans les cli-chés de microscopie électronique cor-respondent à 1 (xm.)
Distribution dans l'organisme 28

26 Distribution dans l'organisme
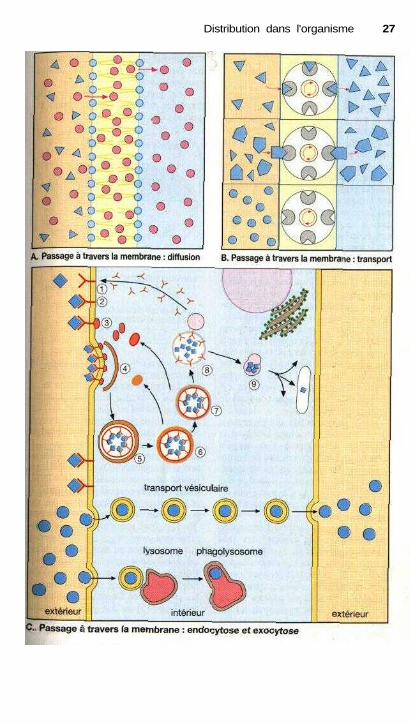
Passage à travers les membranes
La capacité de traverser une doublecouche phospholipidique est indispen-sable à la fois pour l'absorption du mé-dicament, son entrée dans la cellule oules organites subcellulaires et le pas-sage de la barrière hémato-encépha-lique. Nous avons vu que les proprié-tés amphiphiles des phospholipidesconduisent à la formation d'une doublecouche dont l'intérieur est hydrophobeet la surface hydrophile (p.20). Unemolécule pourra traverser cette mem-brane de trois façons distinctes.
Diffusion (A). Les substances li-pophiles (points rouges) peuvent passerde l'espace extracellulaire (coloré enjaune) dans la membrane, s'y accu-muler et de là passer en direction du cy-tosol (coloré en bleu ciel). La directionet la rapidité du transport dépendent durapport des concentrations entre les mi-lieux liquides et la membrane. Plus ladifférence de concentration (gradient)est importante et plus la quantité desubstance transportée par unité detemps sera forte (loi de Fick). Pour lesmolécules hydrophiles (triangles bleus)la membrane lipidique constitue unobstacle infranchissable.
Transport (B). Indépendammentde leurs propriétés physico-chimiqueset de leur caractère lipophile, certainesmolécules peuvent traverser la mem-brane par l'intermédiaire de transpor-teurs. La substance doit alors posséderune forte affinité pour un système detransport qui assurera son transfert àtravers la membrane (les triangles bleuspassent la membrane par l'intermé-diaire d'un transporteur). Le transportactif se produit contre un gradient deconcentration et nécessite de l'énergie.Le transport facilité s'effectue dans lesens du gradient de concentration.
Ce passage peut être inhibé defaçon compétitive par une deuxièmemolécule ayant elle aussi une affinitéélevée pour le même système de trans-port. Si une molécule ne se fixe pas autransporteur (cercle bleu), elle ne serapas transportée. Les médicaments em-
pruntent en fait les systèmes de trans-port propres aux molécules physiolo-giques : par exemple le transporteur desacides aminés dans le cas du passage dela L-DOPA à travers la barrière intesti-nale ou la barrière hémato-encépha-lique (p. 186) ou bien les transporteursdes polypeptides basiques pour le trans-port des aminoglycosides à travers lamembrane luminale des tubules rénaux(p. 276). Seules les substances qui pré-sentent une similitude avec les substratsphysiologiques des systèmes de trans-port ont une bonne affinité pour ceux-ci.
Finalement, le passage à traversles membranes peut s'effectuer sous laforme de petites vésicules entourées demembranes.
Transport vésiculaire / endocy-tose (C). Au moment de la formationdes vésicules, la molécule en solutiondans le liquide extracellulaire s'ytrouve enfermée puis transportée à tra-vers la cellule. Il peut aussi se produireune fusion des vésicules de phagocy-tose, avec les lysosomes aboutissant àune dégradation des substances trans-portées (phagolysosomes).
Internalisation médiée par unrécepteur (C). La molécule se lie à desrécepteurs présents dans la membrane(1, 2) dont les domaines intracellulairesentrent en contact avec des protéinesparticulières (3) (adaptines). Les com-plexes formés se déplacent latéralementdans le plan de la membrane et s'asso-cient avec d'autres complexes. Cetteassociation nécessite la présence d'uneprotéine, la clathrine (4). Le domainemembranaire ainsi affecté s'invaginesous forme d'une vésicule (5). Lacouche de clathrine sera rejetée presqueimmédiatement (6), puis plus tardl'adaptine (7). La vésicule résiduelle vafusionner avec un endosome primaire(8), ce qui aboutit à une augmentationde protons dans la vésicule. Le com-plexe avec le récepteur se dissocie ; lerécepteur est recyclé à nouveau dans lamembrane. Le contenu de l'endosomeprimaire (9) sera ensuite transporté versdifférents organites subcellulaires.
Distribution dans l'organisme 27

28 Distribution dans l'organisme
Différentes possibilitésde distribution d'un médicament
Après l'entrée dans l'organisme, la sub-stance se distribue dans le sang (1), etpeut par son intermédiaire atteindreégalement les tissus. La distributionpeut se limiter à l'espace extracellulaire(volume plasmatique + volume intersti-tiel) (2), ou comprendra également levolume cellulaire (3). Certaines sub-stances peuvent se fixer très fortementaux structures tissulaires de telle sorteque la concentration de la substancedans le sang décroisse fortement maissans entraîner de dissociation (4).
Compte tenu de leur taille, les ma-cromolécules restent confinées à l'inté-rieur des vaisseaux car leur passage àtravers l'endothélium est impossiblemême dans les régions où l'endothé-lium des capillaires est fenestré. Cettepropriété peut avoir des implicationsthérapeutiques. Lorsqu'apres une pertede sang, le lit vasculaire doit être remplià nouveau, on injectera alors une solu-tion remplaçant le plasma (p. 150). Lessubstances fortement liées aux pro-téines plasmatiques se trouveront es-sentiellement dans l'espace vasculaire(p. 30, mesure du volume plasmatique àl'aide de colorants liés aux protéines).Les substances libres (non liées) ont lapossibilité de quitter le flux sanguindans des zones bien précises de l'arbrevasculaire. Ce passage dépendra cepen-dant de la différence de structure desendothéliums (p. 24). Ces variations ré-gionales ne sont pas représentées sur lafigure ci-contre.
La distribution dans l'organismeest fonction de la capacité des sub-stances à traverser les membranes cel-lulaires (p. 20). Les substances hydro-philes (par exemple l'inuline) ne selient pas aux structures externes descellules et ne sont pas captées par lescellules. Il est donc possible de les uti-liser pour évaluer le volume extracellu-laire (2). Les molécules liposolublespassent la membrane cellulaire et il estmême possible d'aboutir à une distribu-tion homogène de la substance dans
l'organisme (3). Le poids du corps peutêtre reparti comme le montre le dia-gramme ci-dessous.
Le graphique ci-contre (p. 29)donne les différents compartiments li-quidions de l'organisme.
eau intracellulaire extracellulaireet érythrocytes
Volumes de distributionpotentiels d'un médicament
La proportion entre le volume duliquide interstitiel et l'eau cellulairevarie selon les périodes de la vie et lepoids du corps. Le pourcentage du li-quide interstitiel est plus important chezle nouveau-né ou le prématuré (environ50 % de l'eau corporelle) et plus faiblechez l'obèse ou le sujet âgé.
La concentration (c) d'une solu-tion est la quantité (D) de la substancedissoute dans un volume (V) : c = D/V.Connaissant la quantité administrée (D)et la concentration plasmatique (c), onpeut évaluer le volume de distributionV = D/c. Il ne s'agit en fait que du vo-lume apparent (V,pp) de distribution,qui serait atteint en supposant une dis-tribution homogène de la molécule dansl'organisme. Cette homogénéité n'estpratiquement jamais observée lorsqueles molécules se fixent aux membranescellulaires (5) ou aux membranes desorganites subcellulaires (6) ou encorese concentrent dans ceux-ci (7). Le vo-lume apparent (V,pp) peut donc être plusimportant que le volume réellement ac-cessible.
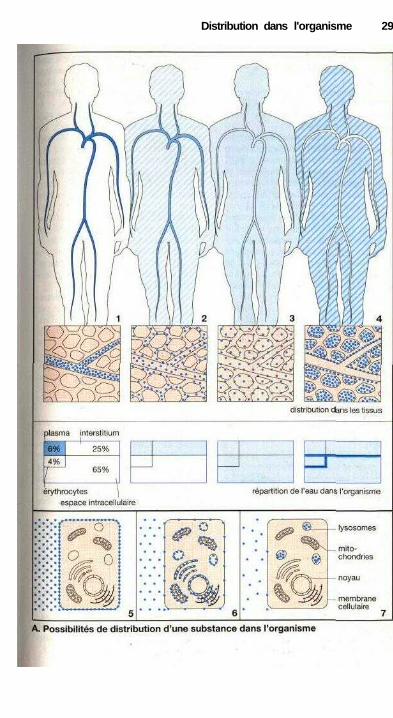
Distribution dans l'organisme 29

30 Distribution dans l'organisme
Liaison des médicamentsaux protéines plasmatiques
Les médicaments peuvent s'associeraux protéines plasmatiques, présentesdans le sang en grande quantité, pourformer des complexes.
Les principales molécules impli-quées dans ce phénomène de liaisonsont en premier lieu l'albumine et dansune moindre mesure les (i-globulines etles glycoprotéines acides. D'autres pro-téines plasmatiques (transcortine, trans-ferrine, globuline de liaison de la thy-roxine) jouent un rôle mais uniquementdans la liaison de molécules spéci-fiques. L'importance de la liaison dé-pend des concentrations respectives dechacun des membres de la réaction et del'affinité de la substance pour les pro-téines. La concentration d'albuminedans le plasma est d'environ 4,6 g/100 ml soit 0,6 mM ce qui représenteune capacité de liaison considérable.L'affinité des substances pour les pro-téines plasmatiques est de l'ordre de10-1 à 105 M (Kd), nettement plus faibleque leur affinité pour des structures deliaison spécifique (récepteurs). Dansces conditions, la liaison de la plupartdes médicaments aux protéines plasma-tiques est pratiquement proportionnelleà la concentration (à l'exception del'acide salicylique ou de certains sulfa-mides).
La molécule d'albumine possèdedes sites de liaison différents pour lesmolécules anioniques et cationiques. Laformation des complexes peut être dueà des liaisons ioniques, bien qu'inter-viennent également des liaisons de Vander Waals (p. 58). L'importance de laliaison est corrélée avec le caractère hy-drophobe de la molécule (propriétéd'une molécule d'être repoussée par lesmolécules d'eau).
La liaison aux protéines plasma-tiques se produit très rapidement et estréversible : ceci signifie qu'à chaquemodification de la concentration de laforme non liée correspond immédiate-ment un changement proportionnel dela concentration de la forme liée. Cette
liaison aux protéines du plasma a unesignification physiologique importantecar la concentration de la forme libreconditionne 1. l'importance de l'effet et2. la vitesse d'élimination.
Pour une même concentration glo-bale (par exemple 100 ng/ml), laconcentration efficace sera de 90 ng/1pour une substance dont 10 % sont liésaux protéines du plasma et seulementde 1 ng/ml pour une substance dont99 % sont liés aux protéines. La dimi-nution de la fraction libre d'une sub-stance par suite de sa liaison auxprotéines affecte aussi sa biotransfor-mation, par exemple hépatique, ou sonélimination rénale : en effet, seule lafraction libre du médicament pénétreradans les cellules hépatiques respon-sables de cette transformation ou serafiltrée par les glomérules.
Lorsque la concentration plasma-tique libre d'une molécule diminue parsuite d'une bioconversion ou de l'élimi-nation rénale, celle-ci sera libérée deses sites de liaison sur les protéines duplasma. La liaison aux protéines plas-matiques s'apparente à une reserve qui,certes, diminue l'intensité de l'actionmais prolonge également la durée del'action en ralentissant la dégradation etl'élimination.
Lorsque deux substances ont uneaffinité élevée pour les mêmes sites deliaison de l'albumine, on pourra ob-server des phénomènes de compétitionau niveau de ces sites : une moléculepeut déplacer une deuxième substancede ses sites de liaison à l'albumine etdonc augmenter la concentration libreet active de cette deuxième molécule(forme d'interaction médicamen-teuse). L'augmentation de la concentra-tion libre de la substance déplacée en-traîne une augmentation de son activitémais également une accélération de sonélimination.
Une diminution de la concentra-tion d'albumine (maladie de foie, syn-drome néphrétique, mauvais état gé-néral) provoque une modification de lapharmacocinétique des substances for-tement liées à l'albumine.
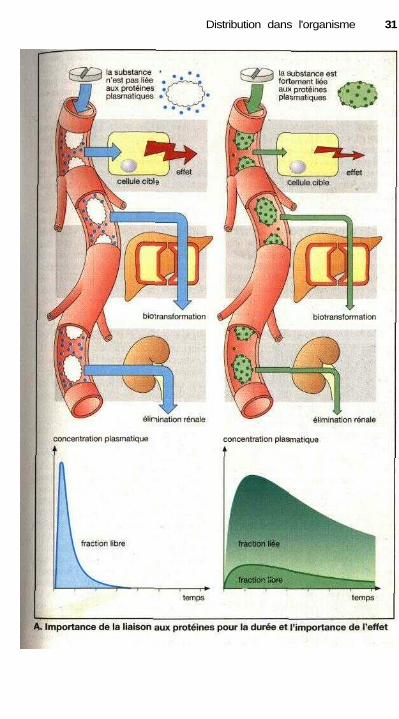
Distribution dans l'organisme 31

32 Élimination des médicaments
Rôle du foie dans la dégradationdes médicaments
Le foie est l'organe principal du méta-bolisme des médicaments, il reçoit parla veine porte 1,1 1 de sang par minuteet environ 350 ml/minute de l'artère hé-patique. Dans le foie coule égalementpresque un tiers du volume sanguinéjecté par le cœur. Enfin le foie contientdans ses vaisseaux et ses sinus 500 mlde sang. Compte-tenu de l'élargisse-ment de la section des vaisseaux au ni-veau du foie, le flux sanguin y sera ra-lenti (A). Par ailleurs, l'organisationparticulière de l'endothélium des sinushépatiques (p. 24) permet même auxprotéines de quitter rapidement le fluxsanguin. L'endothélium perfore auto-rise un contact étroit, inhabituel, entrele sang et la cellule du parenchyme hé-patique et un échange rapide des sub-stances. Ce phénomène est encore favo-risé par la présence de microvillositéssur la surface des hépatocytes tournésvers ces sinus.
L'hépatocyte déverse la bile dansun canalicule biliaire complètement sé-paré de l'espace vasculaire. Cette acti-vité sécrétoire entraîne dans la cellulehépatique un mouvement de liquide di-rigé vers le pôle biliaire (A).
Les hépatocytes contiennent dansles mitochondries ou les membranesdes réticulums lisses (RE1) et rugueux(REr) un grand nombre d'enzymes im-pliquées dans le métabolisme des médi-caments. Les enzymes du réticulumlisse jouent le rôle le plus important carc'est à ce niveau qu'ont lieu les reac-tions d'oxydo-réduction et l'utilisationdirecte d'oxygène moléculaire. Commeces enzymes peuvent également cata-lyser des hydroxylations ou la ruptureoxydaxive de liaisons N-C ou 0-C, onles appelle hydroxylases ou oxydasesà fonctions mixtes. L'élément fonda-mental de ce système enzymatique estle cytochrome P-450.
Sous forme oxydée (Fe" / P450) illie son substrat (R-H). Le complexe
Fe'"/ P450-RH est ensuite réduit par leNADPH. Il lie 0, : 0; - Fe" / P-450-RH. Après capture d'un électron sup-plémentaire, le complexe se dissocie enFe" / P-450, ILO et la substance hy-droxylée R-OH.
Les médicaments lipophiles sontextraits du sang par les cellules du foieplus rapidement que les molécules hy-drophiles et atteignent plus facilementles oxydases mixtes intégrées dans lamembrane du réticulum. Par exemple(B) une substance rendue hydrophobepar la présence d'un substituant aroma-tique (phényï) pourra être hydroxylée etacquérir ainsi un caractère hydrophile(reactions de phase I, p. 34). A côté desoxydases, on trouve également dans leréticulum lisse des réductases et des glu-curonyï transférases. En présence deNAD, ces dernières couplent l'acideglucuronique à un groupe hydroxyle,carbonyle, aminé ou amide (p. 38), parexemple, sur le phénol provenant de lareaction de phase I. Cette réaction decouplage est dite reaction de phase II.Les métabolites de phase 1 et de phase IIpeuvent être à nouveau déversés dans lesang (sans doute par un phénomène detransport passif, fonction des gradients)ou sécrétés dans la bile.
Lors d'une stimulation prolongéed'une des enzymes de la membrane duréticulum, par exemple par un médica-ment tel le phénobarbital, on observeune augmentation du réticulum lisse (Cvs D). Cette induction enzymatique,hypertrophie liée à l'utilisation, touchede la même manière la plupart des en-zymes localisées dans la membrane duréticulum lisse. Ce phénomène entraînenaturellement l'accélération de la dé-gradation de la molécule inductricemais aussi de celle d'autres médica-ments (une autre des formes d'interac-tion médicamenteuse). Cette inductionse développe en quelques jours après ledébut des traitements, multiplie par unfacteur 2-3 la vitesse de transformationet décroît de nouveau après arrêt de lastimulation.
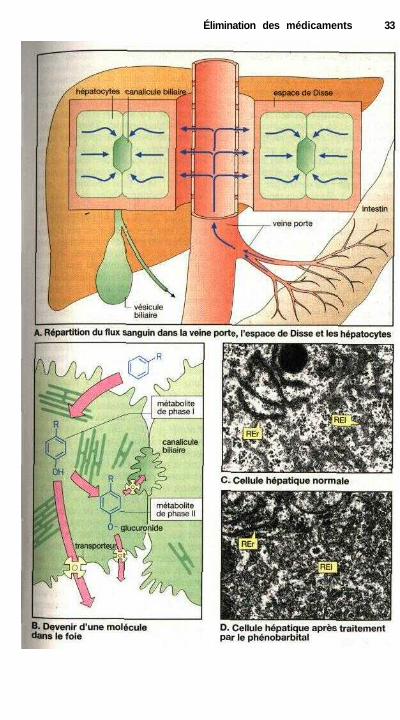
Élimination des médicaments 33

34 Élimination des médicaments
Biotransformation des médicaments
Beaucoup de substances ayant une utili-sation thérapeutique subissent dansl'organisme une transformation chi-mique (biotransformation). Cette mo-dification est en général associée à uneperte d'activité et à une augmentationdu caractère hydrophile, ce qui favorisel'élimination rénale (p. 40). Comme unbon contrôle de la concentration desmédicaments n'est obtenu que pour dessubstances à élimination rapide, beau-coup de médicaments possèdent un sitepréférentiel de dégradation.
La liaison ester constitue l'un deces sites préférentiels d'attaque et seraclivée (hydrolysée) sous l'action d'en-zymes. L'hydrolyse d'un médicament,comme les reactions d'oxydation, deréduction et d'alkylation ou de désalky-lation, appartient aux réactions dephase 1 du métabolisme. On regroupesous ce terme toutes les réactions quiimpliquent une modification de la mo-lécule active. Les réactions de phase IIaboutissent à des produits conjuguésformés à partir des médicaments eux-mêmes, ou des métabolites issus de laphase 1 par conjugaison avec l'acideglucuronique ou sulfurique (p. 38).
Comme exemple de la rapiditéavec laquelle la liaison ester est hydro-lysée, on peut citer le cas d'un neuro-médiateur endogène, l'acétylcholine.Cette molécule est détruite si rapide-ment par l'acétylcholine estérase spéci-fique et les cholinestérases sériques nonSpécifiques (p. 100, p. 102) que son uti-lisation thérapeutique est impossible.L'hydrolyse d'autres esters par les esté-rases se produit plus lentement maistoujours très rapidement par compa-raison avec les autres reactions de bio-transformation. Ceci est mis en évi-dence par l'exemple de la procaïne, unanesthésique local qui ne présente entemps normal aucun effet secondairedans les autres tissus de l'organisme. Lamolécule est en effet inactivée dès sonpassage dans le sang.
La rupture d'une liaison estern'aboutit pas obligatoirement à des mé-tabolites totalement inactifs comme lemontre l'exemple de l'acide acétylsali-cylique. L'acide salicylique, produit del'hydrolyse, possède en effet une acti-vité pharmacologique. Dans certainscas, les substances sont préparées sousforme d'ester, soit pour faciliter l'ab-sorption (énalapriVforme acide, undé-canoate de testostérone/testostérone,p. 250), soit pour permettre unemeilleure biodisponibilité au niveau del'estomac ou de la muqueuse intestinale(succinate d'érythromycine/érythromy-cine). Dans ce cas, ce n'est pas l'esterlui-même qui est actif mais son produitd'hydrolyse. On peut également utiliserune prodrogue inactive qui sera clivéedans le sang en une molécule active.
Quelques médicaments qui possè-dent une liaison amide comme la pnlo-caine (et naturellement les peptides)pourront être hydrolyses et inactivéspar des peptidases.
Les peptidases ont cependant despropriétés pharmacologiques intéres-santes car elles peuvent libérer des pro-duits de dégradation très réactifs àpartir de molécules inactives (fibrine,p. 124) ou des peptides actifs (angio-tensine II, p. 124, bradykinine et enké-phalines, p. 208). Les enzymes impli-quées dans l'hydrolyse de ces peptidesmontrent une étroite spécificité de sub-strat et peuvent être bloquées sélecti-vement. Prenons l'exemple de l'angio-tensine II, un agent vasoconstricteur :l'angiotensine II est issue de l'angio-tensine 1 par élimination des deux aci-des aminés C terminaux leucine ethistidine. Cette réaction est catalyséepar une dipeptidase appelée « angio-tensin converting enzyme » (ACE), quipourra être bloquée par un analoguepeptidique tel le captopril (p. 124).L'angiotensine II sera dégradée parl'angiotensinase A qui coupe l'aspara-gine N terminale de l'angiotensine II.L'angiotensine III ainsi formée n'a au-cune activité vasoconstrictrice.
Élimination des médicaments 35

36 Élimination des médicaments
Les réactions d'oxydation sont dedeux types : celles où un oxygène estajouté à la molécule, et celles où unepartie de la molécule sera éliminée à lasuite d'une oxydation primaire. Les ré-actions d'hydroxylation ou de forma-tion d'époxydes ou de sulfoxides ap-partiennent à la première de ces deuxcatégories. Un substituant alkyl (parexemple le pentobarbital) ou un cyclearomatique (propranolol) pourront êtrehydroxylés. Dans les deux cas, les pro-duits formés seront ensuite conjuguésdans une réaction de phase II, parexemple avec l'acide glucuronique.Une hydroxylation peut également seproduire sur un azote pour former unehydroxylamine (par exemple le paracé-tamol). Le benzène, les composés aro-matiques polycycliques (benzopyrènes)et les hydrocarbures cycliques insaturéspeuvent être transformés en époxydespar des mono-oxygénases. Compte tenude leur caractère électrophile, ces com-posés sont très réactifs et par la mêmetoxiques pour le foie, et vraisemblable-ment cancérigènes.
Le deuxième type de réactiond'oxydation du métabolisme comprendles réactions de désalkylation. Dans lecas des aminés la désalkylation surl'azote débute par l'hydroxylation d'ungroupement alkyl, sur le carboneproche de l'azote. Le produit intermé-diaire n'est pas stable et se dissociepour donner l'aminé désalkylée et l'al-déhyde du substituant éliminé. Ladésalkylation sur l'oxygène (par expour la phénacétine) ou la désarylationsur le soufre (par exemple pour l'aza-thioprine) ont lieu de la même façon.
Une désamination oxydativec'est-à-dire l'élimination d'un groupe-ment NH;, correspond à la désalkyla-tion d'une aminé primaire (R' = H, R2 =H). Le produit intermédiaire hydroxylése dissocie en ammoniaque et en l'al-déhyde correspondant. Ce dernier seraensuite partiellement réduit en alcoolet partiellement oxydé pour donnerl'acide carboxylique homologue.
Les réactions de réduction peu-vent avoir lieu sur un atome d'oxygèneou d'azote. Dans le cas de la réductionde la cortisone en hydrocortisone (cor-tisol) ou de la prednisone en predniso-lone, un groupement céto est trans-formé en groupement hydroxylé. Ceciest d'ailleurs un exemple de la transfor-mation d'un médicament en sa formeactive (bioactivation). Sur l'azote seproduit une réduction du groupementazo ou nitro (par exemple le nitra-zépam). Les groupements nitro serontfinalement réduits en aminé après pas-sage par des groupements nitroso et hy-droxylamine. La déshalogénation estégalement un phénomène de réductiontouchant le carbone (par exemple l'ha-lothane, p. 216).
Les groupements méthyl peuventêtre transférés par une succession deméthyltransférases spécifiques sur lesgroupements hydroxylés (0-méthyla-tion, par exemple la noradrénaline) etsur les groupements aminés (N-méthy-lation, par exemple sérotonine, hista-mine, noradrénaline).
Au niveau des liaisons thio peut seproduire une désulfuration avec rem-placement d'un soufre par un oxygène(par exemple parathion). Cette reactionmontre une fois de plus qu'une réactionde biotransformation ne conduit pasobligatoirement à une inactivation. Leparaoxon (E 600) formé dans l'orga-nisme à partir du parathion (E 605)est la véritable molécule active (p. 102).
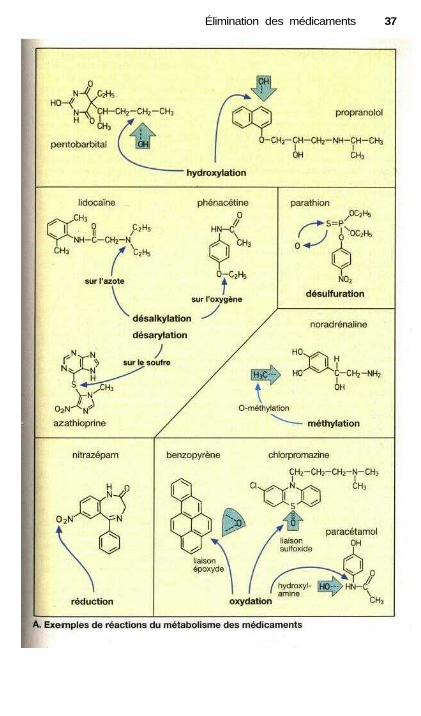
Élimination des médicaments 37

38 Élimination des médicaments
Cycle entéro-hépatique (A)
Les molécules, qui après prise oralesont absorbées au niveau de l'intestin,parviennent au foie par la veine porte etpeuvent être immédiatement couplées àl'acide glucuronique (Figure B, dans lecas de l'acide salicylique), à l'acide sul-furique (Figure B, cas du bisacodylaprès désacétylation) ou à d'autres mo-lécules. Les produits conjugués hydro-philes peuvent aussi être éliminés parvoie biliaire, ils sont alors sécrétés parles hépatocytes dans le liquide biliaire,par l'intermédiaire de mécanismes detransport et aboutissent de nouveau àl'intestin. Les molécules conjuguéeshydrophiles ne peuvent pas traverserl'épithélium intestinal. Les 0-glucuro-nides sont cependant attaqués par lesP-glucuronidases des bactéries ducôlon et la molécule libre peut être ànouveau absorbée. Il se constitue ainsiun cycle entéro-hépatique dans lequelles molécules semblent retenues prison-nières. Les produits de conjugaisonpassent non seulement des cellules hé-patiques dans la bile mais égalementdans le sang. Les glucuronides dont lamasse moléculaire est inférieure à 300passent de façon préférentielle dans lesang, ceux dont la masse est supérieureà 300 passent surtout dans la bile. Lesglucuronides déversés dans le sang parles cellules hépatiques seront filtrés auniveau des glomérules mais comptetenu de leur faible lipophilie, ils ne se-ront pas réabsorbés comme d'autressubstances mais éliminés dans l'urine.
Les médicaments qui subissent uncycle entéro-hépatique seront égale-ment éliminés lentement. On peut citercomme exemple la digitoxine et cer-tains anti-inflammatoires non stéroï-diens.
Réactions de conjugaison (B)
La plus importante des réactions deconjugaison (réactions de phase II) estl'association d'une molécule ou de sonmétabolite à l'acide glucuronique. Legroupement carboxyle de l'acide glucu-
ronique est presque complètement dis-socié dansia zone de pH du sang ou du li-quide extracellulaire ; cette charge néga-tive confère à la molécule conjuguée unepolarité élevée et donc une faible capa-cité de passage à travers les membranes.Cette réaction de conjugaison ne se pro-duit pas spontanément mais seulementlorsque l'acide glucuronique se trouvesous forme active, lié à l'UDF (uridinediphosphate). Les glucuronyl-transfé-rases microsomiales transfèrent l'acideglucuronique de ce complexe à la molé-cule réceptrice. Lorsque cette moléculeréceptrice est un phénol ou un alcool, onobtient un éther-glucuronide, s'il s'agitd'un groupement carboxyle se forme unester-glucuronide. Dans les deux cas, lesmolécules formées sont des 0-glucuro-nides. Avec les aminés, on peut formerles N-glucuronides qui eux ne sont pasclivés par les P-glucuronidases.
Dans le cytoplasme, les sulfo-transférases solubles transfèrent ungroupement sulfate (sous forme activée3'phosphoadénosine-5'phosphosulfate)sur un alcool ou un phénol. Le produitconjugué est alors un acide, commedans le cas des glucuronides.
Ceci le distingue des produitsconjugués formés sous l'action d'acyl-transférase entre un groupement alcoolou un phénol et un groupement acétateactivé (acétyl-coenzyme A). Ce com-posé conjugué ne possède aucun carac-tère acide.
Les acyltransférases sont enfinutilisées également pour le transfert desacides aminés glycine ou glutamine surdes acides carboxyliques. Il se formealors une liaison amide entre une fonc-tion acide de la molécule réceptrice et legroupement aminé de l'acide aminé.Dans le produit conjugué, la fonctionacide de la glycine ou de la glutaminereste libre.
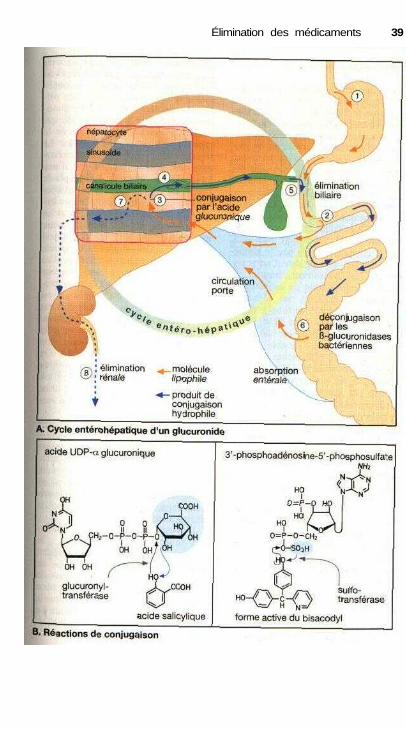
Élimination des médicaments 39

40 Élimination des médicaments
Élimination rénale
La plupart des molécules sont éliminéespar le rein dans l'urine, soit intactes,soit sous forme de produit de dégrada-tion. Cette élimination rénale est liée àla structure particulière de l'endothé-lium au niveau des capillaires du glo-mérule (B). Cette disposition permet eneffet le libre passage des moléculesdont la masse est inférieure à 5 000, etune filtration partielle de celles com-prises entre 5 000 et 50 000. A quelquesrares exceptions près, les molécules àusage thérapeutique et leurs métabolitesont une masse moléculaire bien infé-rieure et seront filtrées par le glomé-ruie, passant du sang dans l'urine pri-mitive. La membrane basale quisépare l'endothélium du capillaire de'l'épithélium contient des glycopro-téines chargées et constitue pour lesmolécules de masse plus élevée, et enfonction de leur charge une barrière defiltration d'étanchéité variable.
En plus de la filtration gloméru-laire (B), certaines molécules plasma-tiques peuvent également aboutir dansl'urine par une sécrétion active (C).Certains cations et certains anions se-ront sécrétés dans la lumière du tubulepar des systèmes de transport spéci-fique, consommant de l'énergie. La ca-pacité de ces transporteurs est cepen-dant limitée. En présence de plusieurssubstrats voisins, on peut observer àleur niveau des phénomènes de compé-tition (p.266).
Au cours du passage à travers lestubules, le volume de l'urine est réduitde plus de 100 fois, aboutissant àconcentrer de façon équivalente la mo-lécule filtrée ou ses métabolites (A). Legradient de concentration ainsi forméentre l'urine et le sang ou le liquide ex-tracellulaire est maintenu pour les mo-lécules qui ne peuvent pas franchirl'épithélium tubulaire. Dans le cas desmolécules lipophiles, ce gradient deconcentration entraînera cependant laréabsorption d'une partie des molé-cules filtrées. Cette réabsorption est duedans la plupart des cas à une diffusion
passive. C'est pourquoi l'importance decette réabsorption est fonction du pH del'urine dans le cas de substances dont ladissociation dépend elle-même du pH.Comme indication du degré de disso-ciation, on utilise la valeur du pK, quiindique le pH auquel la moitié de lasubstance est sous forme protonée. Lareprésentation graphique de ce phéno-mène est donnée dans le cas d'uneaminé dont le pK est de 7. Si le pH del'urine est de 7, la moitié du groupe-ment aminé est sous forme protonée hy-drophile et donc incapable de franchirla membrane (points bleus), l'autremoitié non chargée (points rouges)peut, elle, quitter la lumière du tubuleen suivant le gradient qui se forme. A ceniveau existe encore une réactiond'équilibre entre la base et la forme pro-tonée. Pour une aminé dont le pK estplus élevé (7,5) ou au contraire plus bas(6,5), on obtiendra pour un pH urinairede 7, une quantité plus faible ou plusforte de l'aminé sous forme nonchargée, c'est-à-dire absorbable. Onobtient des variations tout à fait compa-rables avec une substance dont le pK estde 7, en faisant varier le pH de l'urined'une 1/2 unité pH vers le haut ou versle bas.
Le phénomène décrit ci-dessuspour les substances basiques s'appliqueégalement pour les substances acidesmais avec la différence fondamentaleque dans le cas d'un groupementCOOH, c'est la forme chargée qui seforme lorsque l'on augmente le pH uri-naire (alcalinisation) bloquant ainsi lareabsorption.
Il est parfois souhaitable de modi-fier la valeur du pH urinaire, parexemple lors d'un empoisonnementavec des substances protonables, defaçon à accélérer l'élimination dupoison, par exemple une acidificationdans le cas d'un empoisonnement par laméthamphétamine ou une alcalinisationlors d'une intoxication par le phénobar-bital.
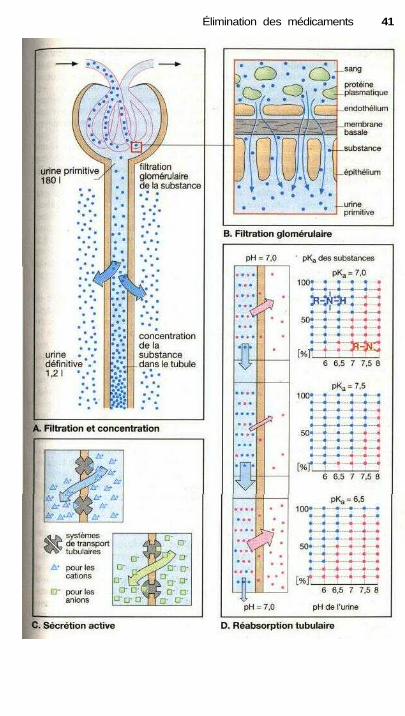
Élimination des médicaments 41

42 Élimination des médicaments
Élimination des substanceslipophiles et hydrophiles
Le caractère lipophile et hydrophile(ou hydrophobe et lipophobe) se définitpar la solubilité des molécules dans desmilieux de faible polarité ou inverse-ment de polarité élevée. Le plasma san-guin, le liquide interstitiel et le cyto-plasme constituent des milieux aqueuxde polarité élevée tandis que les lipides,au moins à l'intérieur d'une bicouchemembranaire (p. 20) et la graisse sontdes milieux apolaires. Les moléculespolaires, hydrophiles, se dissolvent biendans un milieu polaire et les moléculeslipophiles au contraire se dissolventdans des milieux apolaires. Une sub-stance hydrophile qui atteint la circu-lation sanguine ne sera absorbée que defaçon partielle et lentement (non repré-senté) et traversera le foie sans subir demodifications. En effet, ces moléculesqui ne traversent pas, ou seulement len-tement, la membrane des cellules hépa-tiques ne rentrent pas en contract avecles enzymes hépatiques servant à latransformation des molécules. Cettemolécule atteint donc intacte le flux ar-tériel et les reins où elle sera filtrée.Dans le cas des molécules hydrophiles,la liaison aux protéines plasmatiquesest faible (elle augmente en effet avec ledegré de lipophilie), ce qui signifie quela majeure partie de la concentrationplasmatique de telles molécules est dis-ponible pour une filtration gloméru-laire. Une substance hydrophile ne serapas reabsorbée au niveau tubulaire etaboutit donc dans l'urine définitive.Ces molécules subissent donc une éli-mination rénale très rapide.
Une molécule hydrophobe qui,bien qu'elle puisse diffuser dans les cel-lules et entrer en contact avec les en-zymes hépatiques, n'est pas trans-formée en raison de sa nature chimiqueen un composé polaire, persiste dansl'organisme. La fraction filtrée lors dupassage du glomérule sera réabsorbéeau niveau du tabule. Cette réabsorption
est presque totale car la concentrationlibre d'une molécule hydrophobe dansle plasma, est faible (les molécules lipo-philes sont fréquemment liées engrande partie aux protéines). La situa-tion décrite ici d'une molécule hydro-phobe qui ne subit aucune transfor-mation métabolique, n'est passouhaitable pour un médicament. Dansce cas en effet la dose administrée estpratiquement irréversible (difficulté decontrôler le traitement).
Les molécules lipophiles qui sonttransformées dans le foie en métabo-lites polaires permettent un meilleurcontrôle thérapeutique, car cette trans-formation favorise leur élimination. Larapidité de formation des métaboliteshydrophiles conditionne la durée de laprésence du médicament dans l'orga-nisme.
Si la transformation est rapide etles métabolites formés pharmacologi-quement inactifs, seule une fraction dela molécule absorbée atteint intacte lacirculation générale, l'autre partie estéliminée de façon pré-systémique.Lorsque la biotransformation est trèsrapide, l'administration orale n'est paspossible (par exemple la trinitnne,p. 120). La molécule doit être adminis-trée par voie parentérale, buccale outransdermique pour contourner le foie.Indépendamment du mode d'applica-tion, une partie de la substance adminis-trée peut être captée et stockée tempo-rairement au moment du passage àtravers les poumons, avant son passagedans la circulation. Ce processus cor-respond également à une éliminationprésystémique.
Une élimination présystémiquediminue la biodisponibilité d'un médi-cament après absorption orale. La bio-disponibilité absolue est le rapportentre la quantité systémique disponibleet la dose administrée. La biodisponi-bilité relative correspond à la disponi-bilité du médicament dans une formeexpérimentée, comparée à celle d'unepréparation classique.
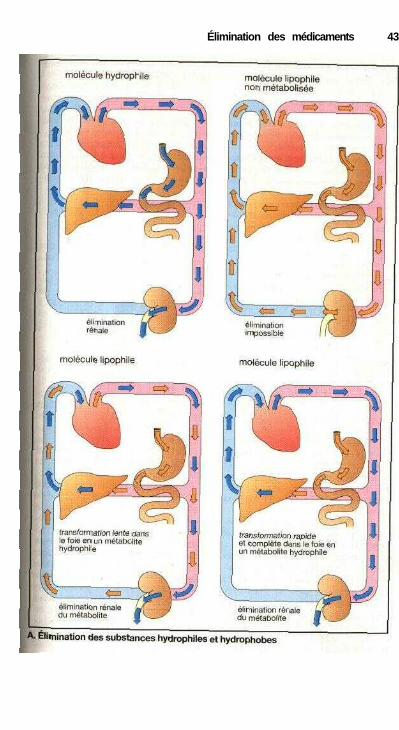
Élimination des médicaments 43

44 Pharmacocinétique
Concentration des médicamentsdans l'organisme, évolutionen fonction du temps :la fonction exponentielle
Divers événements comme l'absorptiondes médicaments et leur éliminationsuivent une loi exponentielle.
En ce qui concerne l'absorption,ceci s'explique essentiellement par lefait que la quantité de substance trans-portée par unité de temps dépend de ladifférence de concentration (gradient)entre les deux compartiments consi-dérés (loi de Fick). Dans le cas d'uneabsorption, le compartiment où laconcentration initiale est la plus élevéeest la lumière intestinale et le comparti-ment avec la concentration la plusfaible est le sang.
Dans le cas de l'élimination ré-nale, l'excrétion dépend à la fois de lafiltration glomérulaire et de la quantitéde substance présente dans l'urine pri-maire. La quantité de substance filtréeau niveau glomérulaire par unité detemps décroît en fonction de la diminu-tion de la concentration de la substancedans le sang. La fonction exponentiellequi rend compte de ce phénomène estprésentée en (A). Dans une fonction ex-ponentielle, le temps nécessaire pourque la concentration plasmatique soitdivisée par deux est constant : cettedurée appelée demi-vie ou période estreliée à la constante de vitesse k par11,2 = In 2/k. Cette valeur et celle de laconcentration initiale c;, permettent decaractériser complètement la fonctionexponentielle.
Compte tenu du caractère expo-nentiel du processus d'élimination onpeut définir le volume du plasma débar-rassé du médicament par unité de temps(dans l'hypothèse où les molécules res-tantes ne se remélangeraient pas defaçon homogène dans la totalité ducompartiment, hypothèse qui n'est ja-mais vérifiée dans la realité). Le vo-lume théorique de plasma débarrassédu médicament par unité de tempsest désigné sous le terme de clearance.Selon que la concentration plasmatiqued'une substance diminue à cause d'une
élimination ou d'une transformationmétabolique on parlera de clearance hé-patique ou rénale. Dans le cas où la mo-lécule est en partie éliminée intacte parles reins et pour l'autre partie dégradée,on additionne les clearances rénale ethépatique en une clearance totale dm,.Cette valeur est la résultante de tous lesévénements participant à l'éliminationet est liée à la demi-vie et au volume dedistribution (V,pp) (p. 28) par la rela-tion :
ti^lnix^'-"lot
La demi-vie est d'autant plus faible quele volume de distribution est petit ou laclearance totale importante.
Dans le cas d'une substance ex-crétée sans modification chimique, onpeut évaluer la demi-vie du produit àpartir de l'élimination cumulée dans lesurines. La quantité totale finalementéliminée correspond à la quantité ab-sorbée.
Dans le cas d'une élimination hé-patique on obtient essentiellement une
. décroissance exponentielle de laconcentration du médicament en fonc-tion du temps parce que les enzymesqui assurent la dégradation travaillentdans le domaine où leur activité est pro-portionnelle à cette concentration. Laquantité de substance transformée parunité de temps diminue ainsi en mêmetemps que la concentration.
L'exception la plus connue à cetteloi exponentielle est l'élimination del'éthanol (alcool éthylique), qui est li-néaire, au moins lorsque la concentra-tion dans le sang dépasse 0,2 %c. Ceciest dû à la faible constante de demi-saturation (Km) de l'enzyme limitantedu métabolisme de l'alcool : l'alcooldéshydrogénase ; cette valeur de Kmest déjà atteinte pour une concentrationen alcool de 80 mg/1 (environ 0,08 %o).Pour une concentration en éthanol su-périeure à 0,2 %c, la quantité métabo-lisée n'augmente plus en fonction de laconcentration et l'élimination par unitéde temps demeure constante.
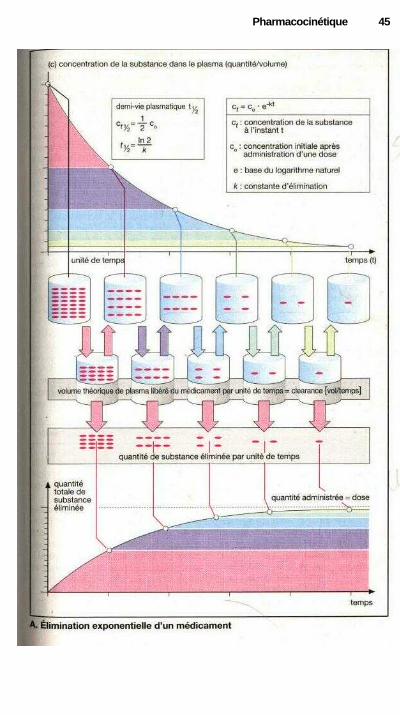
Pharmacocinétique 45

46 Pharmacocinétique
Cinétique plasmatiquedes médicaments
A. Les médicaments sont assimilés parl'organisme puis éliminés par diffé-rentes voies. L'organisme est égale-ment un système ouvert, dans lequel laconcentration du médicament à un ins-tant donné est la résultante à la fois del'influx (entrée) et de l'efflux (élimina-tion). En cas d'administration per os,l'absorption se produit au niveau del'estomac et de l'intestin. La vitesse decette absorption dépend de nombreuxfacteurs parmi lesquels la vitesse dedissolution de la molécule (dans le casd'une forme galénique solide), la vi-tesse du transit stomacal ou intestinal,la capacité de la molécule à traverser lesmembranes, la différence de concentra-tion entre l'intestin et le sang et l'irriga-tion de la muqueuse intestinale. Le pas-sage à travers la muqueuse intestinale(entrée) fait augmenter la concentra-tion sanguine. La substance véhiculéepar le sang atteint les différents organes(distribution) et peut aussi être cap-turée en fonction des propriétés propresde chaque tissu. Les organes bien irri-gués (par exemple le cerveau) reçoiventune fraction plus importante de la sub-stance que les tissus moins bien irri-gués. L'entrée dans les tissus faitbaisser la concentration sanguine. Lepassage de la substance à travers laparoi intestinale diminue lorsque la dif-férence de concentration entre l'intestinet le sang devient plus faible. Le pis.plasmatique atteint un maximumlorsque la quantité éliminée par unité detemps équivaut à celle absorbée,L'influx du médicament dans le foie etles reins représente son entrée dans lesorganes d'élimination. La cinétiqueplasmatique, avec ses phases caractéris-tiques, est la combinaison de trois pro-cessus partiels : entrée, distribution etélimination qui se chevauchent dans letemps. Lorsque l'absorption intestinaleest plus lente que la distribution, la ci-
nétique plasmatique est influencée parl'absorption et l'élimination. Ce phéno-mène peut être décrit de façon mathé-matique simplifiée par la fonction deBateman, où k, et k, sont les constantesde vitesse du processus d'absorption etd'élimination. Lorsque la distributiondans l'organisme est beaucoup plus ra-pide que l'élimination (après une injec-tion intraveineuse), on observe d'abordune décroissance très rapide du picplasmatique suivie d'une décroissancebeaucoup plus lente. La phase de dé-croissance rapide est la phase et (phasede distribution) et le composant pluslent la phase p (phase d'élimination).
B. La vitesse de l'entrée dépenddu mode d'application. Plus l'invasionest rapide, plus court est le temps néces-saire (tn,,,) pour atteindre le pic plasma-tique (Cn,,J ; plus la valeur de €„„ estélevée et plus tôt la concentration plas-matique commence à diminuer de nou-veau.
La surface sous la courbe (AUC,area under curve) est indépendante dumode d'application pour des dosessemblables et une disponibilité totale :loi des surfaces équivalentes. Elle serautilisée pour déterminer la biodisponi-bilité F. Après administration d'unedose équivalente, F est donné par
AUC (voie orale)AUC (voie IV)
La biodisponibilité correspond à lafraction du principe actif qui parvientdans la circulation générale après uneadministration orale.
11 est également possible de com-parer de cette façon plusieurs prépara-tions commerciales contenant desquantités équivalentes de la même mo-lécule : la bioéquivalence correspond àune cinétique plasmatique identique età une même aire sous la courbe.
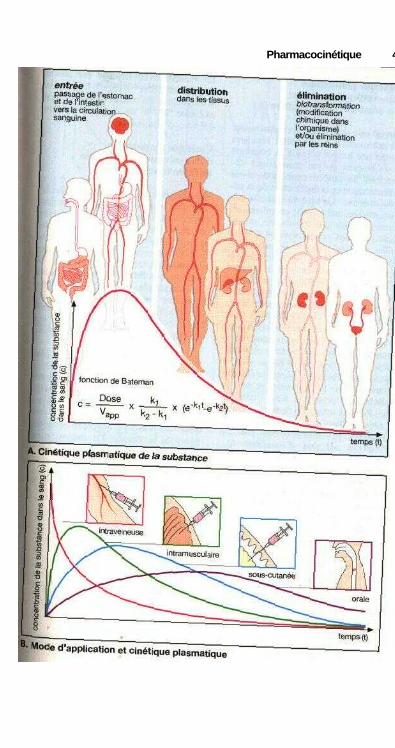
Pharmacocinétique 47

48 Pharmacocinétique
Cinétique plasmatiqued'un médicament durantune administration régulière (A)
Lorsqu'une dose fixe d'un médicamentest administrée à intervalles réguliersdurant une longue période, la cinétiqueet la hauteur du pic plasmatique dé-pendront du rapport entre la demi-vied'élimination et la durée entre deux ad-ministrations. Lorsque la quantité ad-ministrée en une fois est complètementéliminée avant la dose suivante, on ob-tient pour des prises répétées à inter-valle régulier toujours le même picplasmatique. Si une prise de médica-ment se produit avant que la quantitéadministrée lors de la prise précédentene soit complètement éliminée, la dosenouvelle vient s'ajouter au reste de ladose précédente encore présent dansl'organisme : la molécule s'accumule.Plus l'intervalle entre deux administra-tions successives est petit en compa-raison de la demi-vie d'élimination etplus le reliquat auquel vient s'ajouter lanouvelle dose est important et plus lasubstance s'accumule dans l'organis-me. Pour un intervalle de temps donné,l'accumulation du principe actif n'estcependant pas indéfinie, bien plus, onaboutit à un état d'équilibre (c^,steady state). Ceci provient de ce que leprocessus d'élimination est fonction dela concentration. Plus cette concentra-tion augmente et plus les quantités éli-minées par unité de temps sont élevées.Après plusieurs doses, la concentrationest arrivée à un niveau où la quantitééliminée par unité de temps équivaut àla quantité apportée : l'état stationnaireest atteint. C'est autour de ce niveauque la concentration plasmatique os-cille sous l'effet des administrations ré-gulières du médicament. Le niveau del'état stationnaire (cJ est lié à la quan-tité apportée (D) par intervalle d'admi-nistration (r) et à la clearance :c - D
" (TXCl)
La rapidité avec laquelle est atteintl'état d'équilibre indique la vitessed'élimination du produit (la durée pouratteindre 90 % de c,, vaut environ3,3 x 11,2 d'élimination).
Cinétique plasmatiqued'une substance administréede façon irrégulière (B)
Dans la pratique, il s'avère difficile demaintenir une concentration plasma-tique ondulant de façon régulière autourdu niveau thérapeutique désiré. Si parexemple on oublie deux doses consécu-tives (?), la concentration plasmatiquedescend dans une zone inférieure à laconcentration thérapeutique et il faudraune période plus longue de prise régu-lière pour atteindre de nouveau le ni-veau plasmatique souhaité. La possibi-lité et le désir du patient de suivre lesprescriptions du médecin sont désignéssous le terme de compliance.
Ce problème peut également êtrerencontré lorsque la dose journalière aété divisée en trois, de façon à prendreune dose au petite déjeuner, une au dé-jeuner et la troisième au dîner. Dans cesconditions, l'intervalle nocturne estdeux fois plus long que celui entre lesdoses de la journée. La concentrationplasmatique durant les premièresheures de la matinée peut alors des-cendre bien en dessous de la concentra-tion souhaitée et éventuellement de laconcentration absolument nécessaire.
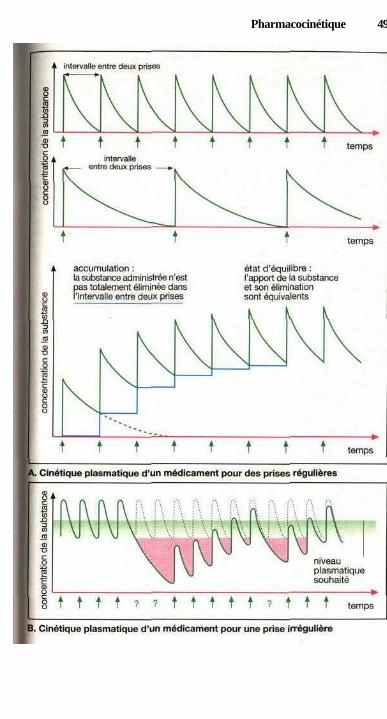
Pharmacocinétique 49

50 Pharmacocinétique
Accumulation : doses, intervallesentre deux doses et contrôle desconcentrations plasmatiques (A)
Dans de nombreuses maladies, l'utilisa-tion d'un médicament n'est couronnéede succès que lorsque sa concentrationplasmatique demeure élevée pendant untemps important. Cette condition peutêtre remplie par une prise régulière sil'on évite soit de laisser chuter laconcentration plasmatique en dessousde la concentration active, soit une ac-cumulation au-dessus du seuil où appa-raissent des symptômes d'empoisonne-ment. Le maintien d'une concentrationplasmatique uniforme n'est pas souhai-table si cela entraîne une réductiond'efficacité (développement d'une tolé-rance), ou lorsque l'utilisation de lasubstance n'est nécessaire que pendantquelques jours.
Il est possible d'obtenir uneconcentration plasmatique constante enutilisant une perfusion, la vitesse deperfusion conditionnant alors la valeurde la concentration atteinte. Cette pos-sibilité est utilisée de façon habituelleen médecine intensive mais pas dans lapratique courante. Pour une prise orale,une solution de compromis est de di-viser la dose journalière en plusieursdoses individuelles (2, 3 ou 4), de sorteque la concentration moyenne dans leplasma ne subisse que des variations defaible amplitude. En fait, il s'avère quela prescription d'un médicament àprendre plusieurs fois par jour serabeaucoup moins suivie (assiduité plusfaible du patient à la prise du médica-ment : faible compliance). L'impor-tance des oscillations plasmatiquesdans l'intervalle entre deux prises peutaussi être réduite en utilisant une formegalénique où la libération du principeactif est ralentie (p. 10) : préparationretard.
La rapidité avec laquelle est at-teint l'état d'équilibre à l'occasion
d'une prise régulière indique la vitessed'élimination. Comme l'indique la for-mule, l'état d'équilibre est presque at-teint après 3 11,2 d'élimination.
Dans le cas d'une substance ac-tive, d'élimination lente, ayant doncune forte tendance à l'accumulation, ilsera plus long d'atteindre le niveauplasmatique requis pour l'action (phen-procoumone, digitoxine, méthadone). Ilest alors possible d'atteindre plus rapi-dement l'état d'équilibre en augmentantla dose initiale (dose d'attaque), cet étatd'équilibre sera ensuite maintenu avecdes doses plus faibles (traitement d'en-tretien). Dans le cas de substances à éli-mination lente, une prise quotidienneunique suffit pour atteindre une concen-tration active presque constante.
Modification des caractéristiquesde l'élimination durantle traitement (B)
Dans tous les cas où l'on utilise desprises médicamenteuses répétées pouratteindre une concentration cumuléeactive, il faut se souvenir que les condi-tions de biotransformation ou d'excré-tion rénale ne restent pas obligatoire-ment constantes au cours du traitement.Il peut se produire une augmentation del'élimination par suite d'une inductionenzymatique (p. 32) ou d'un change-ment du pH urinaire (p. 40). La consé-quence de ce phénomène est une dimi-nution de l'état d'équilibre jusqu'auniveau correspondant à une éliminationplus rapide. L'action initiale du médi-cament peut alors s'atténuer ou mêmedisparaître. Au contraire, une diminu-tion de l'élimination (par exemple dé-veloppement d'une insuffisance rénaledan le cas de médicaments éliminés parle rein) peut entraîner une augmentationdu niveau plasmatique moyen pouvantmême atteindre un seuil toxique.
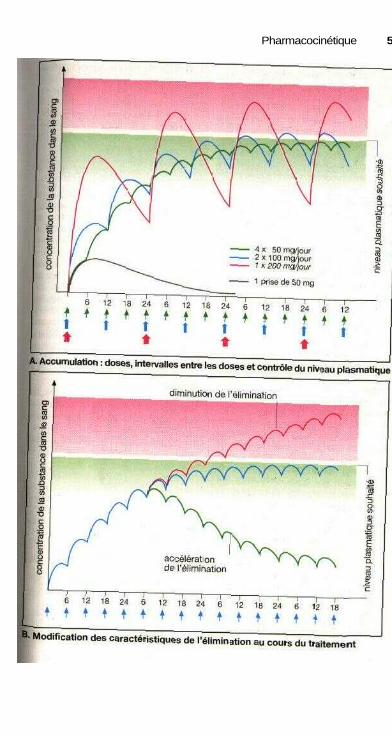
Pharmacocinétique 51

52 Mesure de l'effet des médicaments,
Relation dose-effet (in vivo)
L'action d'un principe actif dépend dela quantité appliquée, c'est-à-dire de ladose. Si l'on choisit une dose inférieureau seuil où se manifeste l'effet, le médi-cament n'aura aucune action. Selon lanature de l'effet attendu, on observera,chez un individu donné, que des dosescroissantes produisent des effets de plusen plus nets, on peut alors déterminerune relation dose-effet. L'action d'unmédicament destiné à faire baisser lafièvre ou à diminuer la pression arté-rielle est ainsi visible, car on peut me-surer la diminution de la température oude la pression artérielle.
La relation dose-effet peut cepen-dant varier d'un individu à l'autre. Chezdes sujets différents, il faudra des dosesdifférentes pour obtenir le même effet.Ceci est particulièrement net dans le casde réactions qui suivent une loi de toutou rien.
L'expérience de hérissement de laqueue présentée en (A) permet d'illus-trer ce phénomène. Les souris blanchesreagissent à la morphine par une pos-ture anormale de la queue et des extré-mités. La relation dose-effet de ce phé-nomène se manifeste sur des groupesd'animaux (groupes de 10) auxquels onadministre des doses croissantes demorphine. Pour des doses faibles, seulsréagissent les animaux les plus sen-sibles, pour des doses croissantes uneproportion de plus en plus importanted'animaux montre une élévation de laqueue tandis que pour des doses élevéestous les animaux du groupe réagissent(B). On peut en déduire une relationentre l'effet (proportion d'animauxavec une réaction) et la dose utilisée. À2 mg/kg, 1 animal sur 10 réagit, pour10 mg/kg, ce sont 5 animaux sur 10 quiréagissent.
La relation entre la dose et lenombre d'animaux qui réagissent estfonction comme nous l'avons déjà dit
de la variabilité des sensibilités indivi-duelles. Celles-ci sont en général distri-buées selon une loi normale commedans l'exemple choisi (C, graphique dedroite). Si l'on porte la fréquence cu-mulée (nombre d'animaux ayant globa-lement réagi pour une dose donnée) enfonction de la dose appliquée (expriméeselon une échelle logarithmique), onobtient une courbe sigmoïde dont lepoint d'inflexion correspond à laconcentration pour laquelle la moitiéd'un groupe a réagi au médicament (C,graphique de gauche). La gamme deconcentration dans laquelle la relationdose-effet s'applique, dépend de la va-riabilité des sensibilités individuelles.
Pour une réaction graduelle, l'éva-luation de la relation dose-effet chez ungroupe de patients sera rendue plus dif-ficile par la variabilité de sensibilitéentre individus. Les mesures seront réa-lisées sur un échantillon pris au hasardet l'on fera la moyenne des résultats.Les doses recommandées pour les trai-tements sont donc adéquates pour lamajorité des patients, mais il existe desexceptions.
L'origine de ces différences desensibilité peut avoir des bases pharma-cocinétiques (les mêmes doses ->• ni-veaux plasmatiques différents) ou phar-macodynamiques (un même niveauplasmadque -> des effets différents).
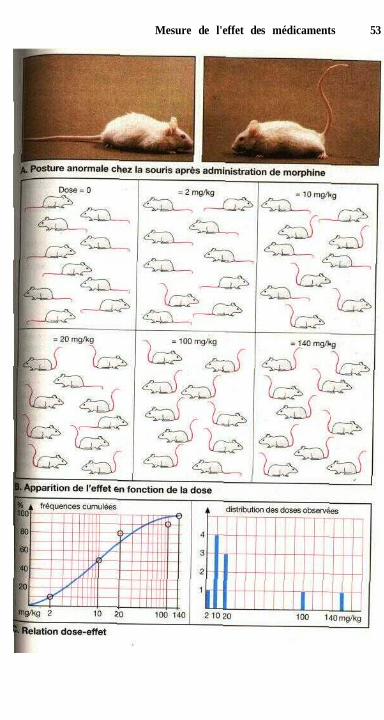
Mesure de l'effet des médicaments 53

54 Mesure de l'effet des médicaments
Relation dose-effet (in vitro) (A)
Dans le cas d'un effet thérapeutique oud'une action toxique (ainsi que pour laphannacodynamie) l'effet se porte engénéral, de façon préférentielle, sur unou quelques organes. Dans le cas de lacirculation par exemple c'est l'actionsur le diamètre des vaisseaux. On adonc suggéré d'isoler l'organe cible dureste des organes, de façon à pouvoirétudier l'action des substances vaso-constrictrices sur différents territoiresde l'arbre vasculaire : veine porte, veinesaphène, artères mésentérique, coro-naire et basilaire. Dans de nombreuxcas, il est possible de maintenir en viependant plusieurs heures dans un étatfonctionnel des organes ou des frag-ments d'organe en utilisant une solutionnutritive appropriée, une oxygénationet une température convenable. La reac-tion de ces préparations à un stimulusphysiologique ou pharmacologiquesera suivie à l'aide d'un appareil de me-sure adapté à la fonction étudiée. Le ré-trécissement d'un vaisseau sera parexemple enregistre en suivant la varia-tion de l'écart entre deux étriers mainte-nant ce vaisseau étiré.
Le travail sur des organes isolésprésente les avantages suivants :1. la connaissance de la concentration
du principe actif qui baigne le tissu ;2. une meilleure possibilité d'observer
et de déterminer l'origine de l'effet ;3. l'élimination des reactions qui peu-
vent, chez l'animal entier, com-penser en partie l'effet propre de lasubstance ; par exemple, l'action dela noradrenaline sur la fréquence
- cardiaque (accélération) peut êtremasquée dans l'organisme entier :l'augmentation de pression artérielleassociée déclenche en effet un méca-nisme de rétrocontrôle dont la résul-tante est une baisse de fréquencecardiaque ;
4. la possibilité de tester l'action dessubstances jusqu'à obtenir un effetmaximum. Il serait par exemple im-possible de suivre sur un organismeintact des effets chronotropes néga-tifs jusqu'à l'arrêt cardiaque.
Les inconvénients de ces systèmessont :1. les lésions inévitables causées au
tissu durant la préparation ;2. la perte du contrôle physiologique
de la fonction de l'organe isolé ;3. le caractère artificiel de l'environne-
ment.
Ces inconvénients jouent un rôle moinsimportant lorsque l'on cherche à com-parer dans un tel système isolé l'actionde différentes substances.
Courbes doses-réponses (B)
En augmentant la concentration par pa-liers égaux, on observe que l'augmen-tation de l ' e f f e t est d'abord constantepuis tend progressivement vers zéro àmesure que l'on se rapproche de laconcentration active maximale. Laconcentration donnant l'effet maximalest difficile à estimer de façon exactetandis que la concentration qui produitla moitié de l'effet maximal possiblepeut être mesurée avec précision (£€50,EC = effective concentration, pointd'inflexion de la courbe sigmoïde ob-tenue en coordonnées semi-logarith-miques). Pour caractériser une courbedose-réponse, il faut donner égalementla valeur de E^ (effet maximal pos-sible) et la pente de la courbe (gammede concentrations dans laquelle la rela-tion s'applique).
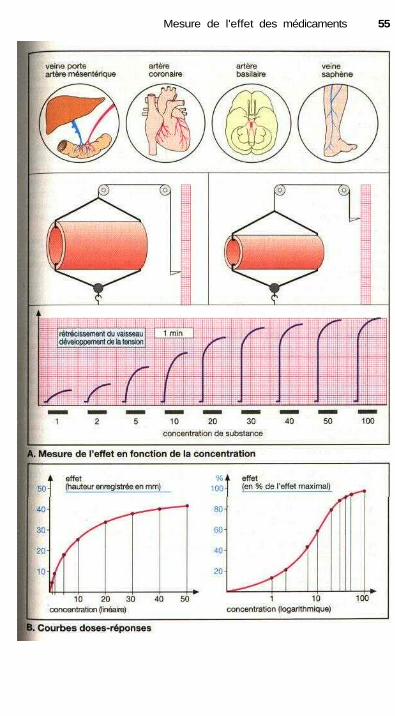
Mesure de l'effet des médicaments 55

56 Mesure de l'effet des médicaments
Courbes de liaison
Pour pouvoir exercer un effet les molé-cules doivent se lier aux cellules del'organe cible. Cette liaison s'effectuele plus souvent sur des structures spé-cialisées ou récepteurs. Dans les étudesde liaison, on détermine l'affinité de lasubstance pour les sites de liaison, la ci-nétique et la localisation cellulaire decette liaison.
Les mesures d'affinité et denombre de sites sont souvent effectuéessur des préparations membranaires dedifférents tissus. La base de cette ap-proche expérimentale est l'hypothèseque les sites de liaison conservent leurspropriétés au cours de l'homogénéisa-tion. Lorsque les sites de liaison sontaccessibles librement, dans le milieu oùsont suspendus les fragments de mem-brane, la concentration au site d'actioncorrespond à celle du milieu. La molé-cule étudiée (marquée par un atome ra-dioactif, de façon à suivre des quantitéstrès faibles) est alors ajoutée au milieu.Lorsque la liaison est complète, lesfragments de membrane et le milieusont séparés, par exemple par filtration,de façon à mesurer la quantité de sub-stance liée aux membranes. La fixationest pratiquement proportionnelle à laconcentration aussi longtemps que ladiminution du nombre de sites libresreste faible (c = 1 et B = 10 % de laliaison totale ; c = 2 et B = 20 %). Avecl'occupation croissante des sites récep-teurs, le nombre de sites libres capablesde lier la substance diminue et l'aug-mentation des sites occupés n'est plusproportionnelle à l'augmentation deconcentration. (Dans l'exemple de lapage 57, pour augmenter la liaison de10 à 20 %, il faut doubler la concentra-tion, pour passer de 70 à 80 %, il fautune augmentation de 20.)
La loi d'action de masse décrit lafonction hyperbolique (B) qui relie laliaison à la concentration du ligand (c).
Cette courbe est caractérisée par l'affi-nité 1/Kp et la liaison maximale Bn,^(qui correspond au nombre total de sitesde liaison par unité de poids de l'homo-génat membranaire).B=B^. —c—C+K.O
Ko est la constante de dissociation àl'équilibre et correspond à la concentra-tion de ligand pour laquelle 50 % dessites de liaison sont occupés. Les va-leurs données en (A), transformées en(B) sous forme d'une courbe de liaisonmontrent un point d'inflexion lorsqueKo =10.
Les expériences de liaisons per-mettent de mettre en évidence de façonélégante l'affinité variable de différentsligands pour un site de liaison.
Ces expériences de liaisons, fa-ciles à réaliser sur le plan expérimental,présentent cependant des inconvénientset en particulier la difficulté d'attribuerprécisément un effet pharmacologiqueà des sites récepteurs caractérisés oul'identification des sites intéressantslorsqu'il y a plusieurs populations desites de liaison. C'est pourquoi on nepeut parler d'un récepteur que lorsqueles critères suivants ont été démontrés :
1. la liaison est saturable (satura-bilité), 2. seules les substances qui pos-sèdent les mêmes propriétés pharmaco-logiques peuvent se lier (spécificité),3. les affinités de différentes sub-stances pour le site de liaison corres-pondent à leur efficacité pharmacolo-gique.
Une expérience de liaison si ellefournit une information sur l'affinitéd'un ligand, ne permet pas de dire sicelui-ci est un agoniste ou un antago-niste (p. 60).
Les sites de liaison, c'est-à-direles récepteurs protéiques peuvent êtremarqués grâce à des molécules radioac-tives puis analysés biochimiquement.
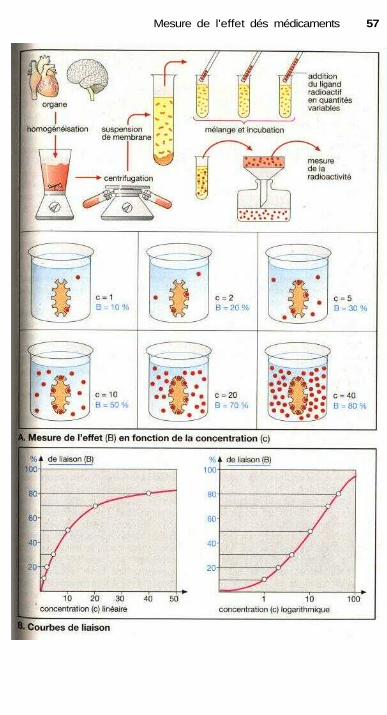
Mesure de l'effet dés médicaments 57

58 Interaction médicament-récepteur
Types de liaison
La condition pour qu'un principe actifpuisse jouer un rôle sur une fonction del'organisme est son interaction avecune structure propre de cet organisme.
Liaison covalente. Deux atomesforment une liaison covalente lorsquechacun d'eux fournit au moins un élec-tron à un nuage électronique commun.L'existence de cette paire d'électronscommune sera représentée dans lesstructures par un trait continu. Laliaison covalente est solide et n'est pas,ou seulement difficilement, réversible.Peu de médicaments se lient de façoncovalente. En effet, la liaison, et doncéventuellement l'effet, persistent long-temps après l'arrêt du traitement si bienque l'action thérapeutique est difficile àcontrôler. Parmi les exemples connus,on trouve des agents anticancéreux al-kylants (p. 294) ou des composés orga-nophosphorés (p. 102). Les réactions decouplage qui se produisent au cours dumétabolisme des médicaments formentdes liaisons covalentes (par exemple unacide glucuronique, p. 38).
Liaison non covalente. Dans cecas, il ne se forme pas de nuage électro-nique commun, la liaison est réversibleet apparaît caractéristique des interac-tions avec les produits pharmaceu-tiques. Un médicament s'associe en gé-néral à plusieurs sites au niveau de lacible, et plusieurs des types de liaisonprésentés ci-dessous peuvent participerà cette interaction.
Interactions électrostatiques (A).Une charge positive et une charge néga-tive s'attirent mutuellement.
Interaction ionique '. un ion estune particule comportant une chargepositive (cation) ou négative (anion),c'est-à-dire qu'un atome a dans sonnuage électronique un électron man-quant ou au contraire un électron excé-dentaire. L'attraction entre deux ions decharge opposée s'exerce à une distanceimportante et constitue la première
force d'attraction vers le site de liaison.Cette liaison ionique est relativementstable.
Interaction ion-dipôle '. lorsque laprobabilité de présence des électrons deliaison n'est pas répartie de façon sy-métrique autour des noyaux des deuxatomes, l'un des atomes porte unecharge négative partielle (5-), l'autreune charge positive partielle (8'1'). Lamolécule qui comporte un pôle négatifet un pôle positif constitue un dipôle.Une charge partielle peut former uneliaison électrostatique faible avec union de signe opposé.
Interaction dipôle - dipôle. C'estl'interaction électrostatique entre deuxcharges partielles de signes opposés. Laliaison hydrogène relie un atome d'oxy-gène porteur d'une charge négative par-tielle à deux atomes comportant unecharge positive partielle.
Interactions de van der Waals(B). Elles se forment entre deux grou-pements apolaires situés à proximitél'un de l'autre. Des altérations sponta-nées et transitoires de la répartitionélectronique d'une molécule (dipôlestransitoires de faible amplitude 68) in-duisent un changement en senscontraire sur la molécule voisine. Laliaison de van der Waals est égalementune forme de liaison électrostatiquemais de faible force.
Interaction hydrophobe (C).L'interaction entre les molécules d'eau(dipôles) est si forte qu'un groupementapolaire, c'est-à-dire non chargé peut àpeine se glisser entre elles ou mêmes'en approcher. Les molécules d'eauserrées les unes contre les autres re-poussent en quelque sorte les groupe-ments apolaires hors de leur milieu. Lesgroupements apolaires ont donc dansl'organisme une forte probabilité deprésence dans un environnement nonaqueux (apolaire) par exemple à proxi-mité des chaînes d'acides gras deimembranes cellulaires ou des zonesapolaires d'un récepteur.
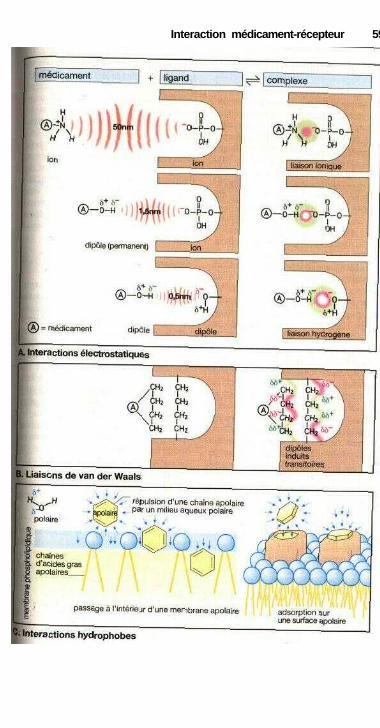
Interaction médicament-récepteur 59

60 Interaction médicament-récepteur
Agonistes et antagonistes (A)
Pour qu'une substance exerce un effetspécifique après interaction avec un siterécepteur, il faut qu'elle puisse non seu-lement s'y fixer (affinité) mais égale-ment posséder la capacité d'agir sur cerécepteur et de déclencher le change-ment d'une fonction cellulaire. Cettepropriété supplémentaire est baptiséeactivité intrinsèque de la substance.Affinité et activité intrinsèque, en-sembles, définissent un agoniste.
Il existe des substances en pré-sence desquelles l'action d'un agonisteest diminuée, ce sont des anti-agomstes,ou antagonistes partiels.
Les antagonistes compétitifspossèdent également une affinité pourle récepteur, mais leur fixation à ce ré-cepteur n'entraîne aucune modificationdes fonctions cellulaires. Les antago-nistes compétitifs n'ont donc aucuneactivité intrinsèque. En présence simul-tanée d'un agoniste et d'un antagonistecompétitif, on aboutit à une compéti-tion des deux molécules pour le récep-teur. Ce sont l'affinité et les concentra-tions respectives des deux compétiteursqui décident si l'un ou l'autre se lie et sil'effet sera ou non déclenché. En aug-mentant la concentration de l'agoniste,on peut surmonter un blocage existant(antagonisme compétitif). En d'autrestermes, la courbe dose-effet d'un ago-niste est déplacée vers des concentra-tions plus élevées (vers la droite) enprésence d'un antagoniste.
Ce type d'antagonisme supposeque la liaison de l'antagoniste au récep-teur est réversible. Si la dissociation del'antagoniste de son récepteur est lentevoire impossible (liaison irréversible) iln'est pas possible de lever l'inhibitionen augmentant la concentration d'ago-niste.
Contrairement aux antagonistescompétitifs, l'antagoniste allostériquese lie en dehors du site récepteur. Cette
liaison provoque une modification de lastructure du récepteur, dont l'affinitépour l'agoniste diminue. La modifica-tion allostérique peut également aug-menter l'affinité de l'agoniste pour lerécepteur et produire un effet de poten-tialisation.
Un agoniste inverse (non repré-senté) en se liant au récepteur influencela fonction cellulaire en sens inverse del'agoniste normal. Dans la nomencla-ture classique, on doit attribuer auxagonistes inverses une activité intrin-sèque négative. L'action d'un agonisteinverse peut être abolie par des antago-nistes.
Antagonisme fonctionnel (B)
Lorsque deux agonistes peuvent agirpar des mécanismes distincts mais ensens opposés sur le même paramètreon aboutit à un antagonisme fonc-tionnel. Si l'on prend comme exemplele diamètre des bronches, l'adrénalineprovoque une dilatation et l'histamineune constnction. Un autre exemple estcelui de l'adrénaline et de l'insulinequi se comportent en antagonistesfonctionnels pour la régulation du glu-cose sanguin. Dans le cas d'antago-nistes fonctionnels, les mécanismesmis enjeu sont totalement différents etles effets maximaux ne sont pas demême amplitude mais seulement de di-rection opposée, il est donc difficile dedéterminer l'état d'équilibre commeon peut le faire pour un antagonismecompétitif.
La dénomination d'antagonismechimique est parfois utilisée lorsqu'unesubstance diminue la concentrationd'un agoniste via la formation d'uncomplexe (par exemple EDTA et cal-cium, protamine et héparine).
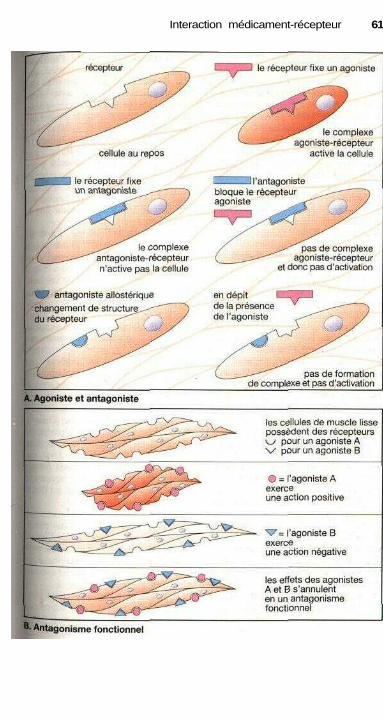
Interaction médicament-récepteur 61

62 Interaction médicament-récepteur
Stéréochimie de l'actiondes médicaments
Beaucoup de médicaments sont des ra-cémiques (par exemple les P-bloquants,les antalgiques acides ou encore l'anti-cholinergique bemétimide (A). Un ra-cémique contient deux molécules, sy-métriques l'une de l'autre dans unmiroir, qui comme la main droite et lamain gauche ne peuvent être superpo-sées : ce sont des structures chiralesou énantiomères. L'origine de ce ca-ractère chiral est dans la plupart des casun atome de carbone portant quatresubstituants différents {centre d'asymé-trie). L'énantiomérie est une forme par-ticulière de stéreo-isomérie. Des stéréo-isomères qui ne sont pas symétriquesl'un de l'autre dans un miroir, sont desdiastéréo-isomères (par exemple qui-nine et quinidine).
Les distances entre atomes sont lesmêmes dans le cas des énantiomèresmais pas dans celui des diastéréo-iso-mères. C'est pourquoi, les énantiomèrespossèdent des propriétés chimiquessemblables (par exemple solubilité,point de fusion), et sont formés en quan-tités égales lors d'une synthèse chi-mique. Dans la nature et sous l'actiondes enzymes l'une des formes estformée de façon préférentielle.
En solution, les énantiomères dé-vient le plan de polarisation de la lu-mière dans des directions opposéesvers la droite d ou (+), forme dextro-gyre, vers la gauche 7 ou (-) forme lé-vogyre. La direction de rotation du plande polarisation de la lumière ne donneaucune indication sur la structure dansl'espace de l'énantiomère. Cette confi--guration est décrite en fonction derègles établies par les préfixes S et R.Certaines molécules sont cependantbaptisées forme D ou L par référence àla structure du D ou L glycéraldéhyde.
Une substance active doit pourexercer son action biologique entrer encontact avec des structures de l'orga-nisme. Cette interaction peut être réa-lisée de façon préférentielle avec l'undes énantiomères : énantiosélectivité.
Énantiosélectivité de l'affinité.Supposons qu'un récepteur possède dessites de reconnaissance pour trois dessubstituants d'un carbone asymétrique(C) (symbolisés en (B) par une sphère,un cône et un cube), dans la plupart descas seul l'un des deux énantiomèresprésentera la configuration optimale. Ilaura donc une tendance plus élevée à selier. C'est ainsi que le déxétimide a uneaffinité 10 000 fois plus élevée pour lesrécepteurs muscariniques que le lévéti-mide (p. 98), le (-) S-propranolol aune affinité 100 fois plus forte que laforme/-!-) R.
Énantiosélectivité de l'activitéintrinsèque. Le mode d'interactionavec le récepteur détermine aussi si uneffet se produira ou non, c'est-à-dire siune substance possède ou non une acti-vité intrinsèque et si elle agit comme unagoniste ou un antagoniste. Un exempleest celui de la dobutamine dont l'énan-tiomère (-) est un agoniste du récepteura-adrénergique et la forme (+) un anta-goniste.
Énantiosélectivité inverse surun récepteur distinct. L'énantiomèredont l'affinité pour un récepteur est laplus faible peut par contre adopter uneconfiguration favorable à son interac-tion avec un autre récepteur. Dans lecas de la dobutamine, l'énantiomère (+)a une affinité pour le récepteur(i-adrenergique 10 fois plus forte quecelle de la forme (—) et les deux formesse comportent comme des agonistes.Par contre, l'effet a-stimulant est limitéà la forme (-) (voir ci-dessus).
Il peut exister de la même manièreque pour l'interaction avec les récep-teurs une énantiosélectivité de l'inter-action avec une enzyme ou une pro-téine de transport. Les énantiomèrespeuvent montrer une affinité et une sus-ceptibilité métabolique distincte.
En conclusion. Les énantiomèrescontenus dans un racémique peuvent sedifférencier par leurs propriétés phar-macocinétiques et pharmacologiques etse comporter comme deux substancesde nature différente.
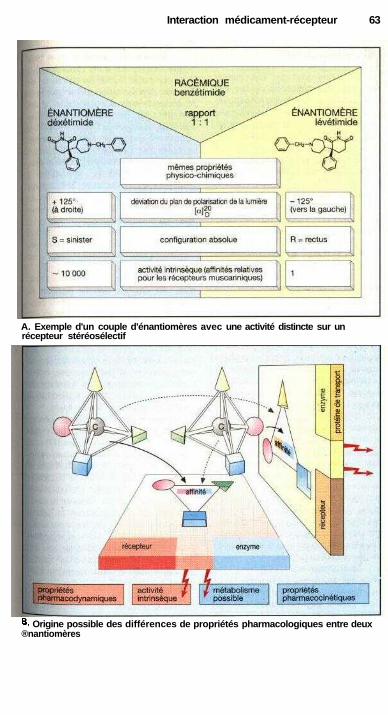
Interaction médicament-récepteur 63
A. Exemple d'un couple d'énantiomères avec une activité distincte sur unrécepteur stéréosélectif
B- Origine possible des différences de propriétés pharmacologiques entre deux®nantiomères

64 Interaction médicament-récepteur
Différents récepteurs
Les récepteurs sont des macromolé-cules dont la fonction est de lier unemolécule signal et de convertir cette in-teraction en un effet c'est-à-dire en unemodification du fonctionnement cellu-laire. Il existe des récepteurs de struc-tures différentes, et la façon dont leuroccupation sera transformée en un effet(transduction du signal) peut égale-ment être très diverse.
Récepteurs couplés à une pro-téine G (A). Ils se composent d'unechaîne d'acides aminés qui traverseplusieurs fois la membrane sous formed'une hélice a. En plusieurs emplace-ments de son domaine extracellulaire,la molécule est glycosylée, c'est-à-direcomporte des résidus sucre. Les septsegments transmembranaires sont vrai-semblablement organisés en un cerclequi contient en son centre une cavité etun site de liaison pour la molécule si-;gnal. L'association du ligand, ou d'unanalogue pharmacologique possédantune activité agoniste, induit un change-ment de conformation du récepteur, etlui permet d'entrer en contact avec uneprotéine G (protéine liant les nucléo-tides guanyliques). Les protéines Gsont situées sur la face interne de lamembrane plasmique et sont forméesde trois sous-unités a, P et -y. Il existedifférentes protéines G qui se différen-cient essentiellement par la structure dela sous-unité a. L'interaction avec le ré-cepteur active la protéine G qui va à sontour moduler l'activité d'une protéine(enzyme, canal ionique). Une propor-tion importante des signaux cellulairesagit par l'intermédiaire de récepteurscouplés à une protéine G. Ceci sera dé-crit plus en détail page 66.
Le récepteur nicotinique de l'acé-tylcholine au niveau de la plaque mo-trice fournit un exemple d'un canal io-nique activé par un ligand (B). Lecomplexe récepteur se compose de5 sous-unités protéiques qui contien-nent chacune quatre domaines trans-membranaires. La fixation simultanéede deux molécules d'acétylcholine
(ACh) sur les deux sous-unités a pro-voque l'ouverture d'un canal ioniqueavec une entrée de Na+ (et une sortie deK4'), une dépolarisation membranaire etle déclenchement d'un potentiel d'ac-tion (p. 180). Les récepteurs ganglio-naires nicotiniques de l'acétylcholine secomposent uniquement de sous-unitésa et P (a;?,). Une partie des récepteursde l'acide "y-aminobutyrique, un neuro-transmetteur (GABA), appartient àcette famille de récepteurs : le récepteurGABA^ contient un canal chlore (et unsite de liaison des benzodiazépinessitué au-dessus du canal, p. 224). Laglycine et le glutamate (dans le cas desrécepteurs ionotropiques) agissent parl'intermédiaire de canaux ioniques ac-tivés par un ligand.
Le récepteur de l'insuline cons-titue une enzyme activée par un li-gand (C). Il s'agit d'un récepteur cata-lytique. Lorsque l'insuline se lie au sitede fixation extracellulaire, une activitétyrosine kinase dans la partie intracellu-laire du récepteur se déclenche. Laphosphorylation de protéines déclencheune modification des fonctions cellu-laires. Les récepteurs des facteurs decroissance appartiennent également àce type de récepteurs catalytiques.
Il existe dans le cytosol des récep-teurs des hormones stéroïdes qui régu-lent la synthèse des protéines (D) (cesrécepteurs migrent dans le noyau cellu-laire après fixation du ligand). Dans lecas de l'hormone thyroïde, le récepteurest présent dans le noyau. La fixation del'hormone dévoile un domaine masquéen temps normal, permettant la liaisondu complexe sur une séquence nucléoti-dique donnée de l'ADN et régulantainsi la transcription du gène en a\al(en général la transcription est initiéeou augmentée et plus rarement blo-quée).
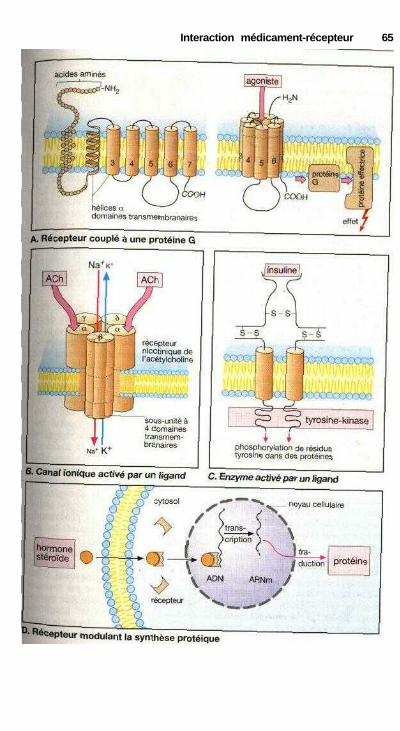
Interaction médicament-récepteur 65

66 Interaction médicament-récepteur
Modes de fonctionnementdes récepteurs couplésà une protéine G
Dans le cas des récepteurs couplés àune protéine G, le mécanisme de trans-duction du signal est en principe iden-tique (A). A la suite de la liaison d'unagoniste sur le récepteur, la conforma-tion de la protéine se modifie. Ce chan-gement se propage jusqu'à laprotéine G : la sous-unité a libère leGDP et fixe le GTP, elle se dissocie desdeux autres sous-unités, entre encontact avec une protéine effectrice etaltère son état fonctionnel. La sous-unité a est capable d'hydrolyser lente-ment le GTP en GDP, le complexe a-GDP ne possède aucune affinité pour laprotéine effectrice et s'associe de nou-veau avec les sous-unités P--y (A). Lesprotéines G peuvent diffuser latérale-ment dans la membrane et ne sont pasassignées à un récepteur unique. Ilexiste cependant une relation entre lestypes de récepteurs et les types de pro-téines G (B). Les sous-unités a des dif-férentes protéines G se distinguent lesunes des autres par leur affinité pourdifférentes protéines effectrices et parl'effet exercé sur ces effecteurs. Lecomplexe G(,-GTP de la protéine Gs sti-mule l'adénylate cyclase, tandis que lecomplexe G.-GTP de G, l'inhibe. Lesrécepteurs muscariniques de l'acétyl-choline, ceux de la noradrénaline, del'adrénaline, de la dopamine, de l'hista-mine, de la morphine, des prostaglan-dines, des leucotriènes et de biend'autres molécules signal et hormonesfont partie des récepteurs couplés auxprotéines G.
En ce qui concerne les protéineseffectrices des récepteurs couplés auxprotéines G, il faut citer principalementl'adénylate cyclase (ATP -* secondmessager intracellulaire, AMPc), laphospholipase C (phosphatidyl inositol-* deux seconds messagers intracellu-laires, inositol triphosphate, ?3 et dia-cylgiycérol, DAG), et les protéinescanal (B).
De nombreuses fonctions cellu-laires peuvent être gouvernées par laconcentration intracellulaire d'AMPc,car l'AMPc augmente l'activité de laprotéine-kinase A, qui à son tour cata-lyse le transfert d'un groupement phos-phate sur une protéine effectrice.L'élévation de la concentration d'AMPcstimule par exemple le tonus desmuscles lisses, la force de contractiondu muscle cardiaque et augmente laglycogénolyse et la lipolyse (p. 84). Laphosphorylation d'un canal calcique fa-vorise son ouverture lors d'une dépola-risation membranaire. Il faut noter quel'AMPc est inactivé par les phospho-diestérases. Les inhibiteurs de ces en-zymes maintiennent élevée la concen-tration d'AMPc et peuvent déclencherdes effets comparables à ceux de l'adré-naline.
Le récepteur lui-même peut êtreégalement phosphorylé, et perdre de cefait sa capacité à activer les protéines G.C'est un des mécanismes qui peuventconduire à la diminution de sensibilitéd'une cellule après stimulation pro-longée par un agoniste.
L'activation de la phospholipase Cconduit à la coupure d'un phospholi-pide membranaire, le phosphatidyl ino-sitol 4,5 biphosphate en inositol tri-phosphate (?3) et en diacylgiycérol(DAG). L'IP3 déclenche la libérationd'ions Ça2* à partir de stocks intracellu-laires, ce qui déclenche par exemple lacontraction des cellules musculaireslisses, la dégradation du glycogène ouune exocytose. Le diacylgiycérol activela protéine kinase C, qui phosphorylécertaines enzymes contenant des ré-sidus serine ou thréonine.
La sous-unité a de certaines pro-téines G, est capable de déclencherl'ouverture d'une protéine canal. C'estde cette façon que seront par exempleactivés des canaux potassiques (actionde l'acétylcholine sur les ganglions,p. 100, action des opioïdes sur la trans-mission de l'excitation nerveuse,p.208).
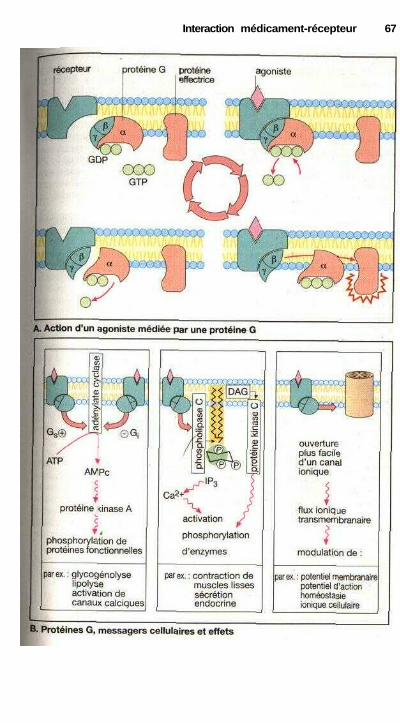
Interaction médicament-récepteur 67

68 Interaction médicament-récepteur
Cinétique plasmatiqueet effet d'un médicament
Après l'administration d'un principeactif, sa concentration dans le plasmaaugmente, atteint un maximum puis dé-croît graduellement sous l'effet de l'éli-mination jusqu'à retourner au niveau dedépart (p. 46). La concentration plas-matique à un instant donné est fonctionde la dose initiale. Dans la zone desconcentrations thérapeutiques, il existe,pour beaucoup de médicaments, une re-lation linéaire entre la hauteur du picplasmatique et la dose (cinétique li-néaire en fonction de la dose (A), notezl'échelle différente sur les ordonnées).Cette relation n'est cependant pas véri-fiée pour certaines molécules, dont lesreactions d'élimination sont déjà acti-vées de façon importante dans lagamme des concentrations thérapeu-tiques de sorte qu'une élévation supplé-mentaire de la concentration plasma-tique n'entraîne pas une augmentationproportionnelle de l'élimination. Dansces conditions, pour des doses élevées,une proportion relativement faible de lasubstance sera éliminée par unité detemps.
La cinétique d'action et celle de laconcentration plasmatique ne sont pasidentiques, car la relation entre laconcentration et l'action est une fonc-tion hyperbole (B, voir aussi p. 54).Ceci signifie que, pour une cinétique li-néaire de la concentration en fonctionde la dose, la cinétique d'action dépendégalement de la dose (C).
Si l'on administre une dose faible(1 dans l'exemple représenté), laconcentration plasmatique varie dansune gamme (0 à 0,9) où le changementde concentration est encore relié defaçon presque linéaire au changementd'activité. La cinétique de la concentra-tion plasmatique et celle de l'effet sonttrès semblables (graphique de gauche Aet C selon le cas). Si par contre on
donne une dose élevée (100), la concen-tration plasmatique demeure longtempsdans une zone (entre 90 et 20) où unchangement de la concentration n'en-traîne aucune variation nette de l'effet.Après des doses élevées (100) on ob-serve donc un plateau dans la courbed'activité en fonction du temps. L'effetne décroît que lorsque le niveau plas-matique a suffisamment diminué (< 20)pour que la variation de la concentra-tion plasmatique se traduise à nouveaupar un changement d'intensité del'effet.
Cette relation entre la cinétique del'effet et la dose peut avoir une applica-tion pratique. Lorsque l'on désire al-longer la durée d'action on administreune dose supérieure à celle strictementnécessaire pour obtenir l'action désirée,c'est le cas par exemple de la pénicil-line G (p. 266) où l'on préconise uneprise toutes les huit heures, en dépitd'une demi-vie d'élimination de 30 mi-nutes. Cette pratique n'est possible na-turellement que lorsque le dépassementde la dose n'entraîne pas d'effettoxique.
Il peut se produire que l'on ob-tienne en cas d'administration régulièreun effet pratiquement constant, bienque le niveau plasmatique oscille defaçon importante dans l'intervalle entreles doses.
La relation hyperbolique reliant laconcentration dans le plasma et l'effetexplique pourquoi la cinétique d'actionne peut être décrite par une loi expo-nentielle. On ne peut calculer une demi-vie que pour l'entrée ou l'éliminationou encore pour la variation du niveauplasmatique mais par pour l'apparitionou la disparition de l'effet.
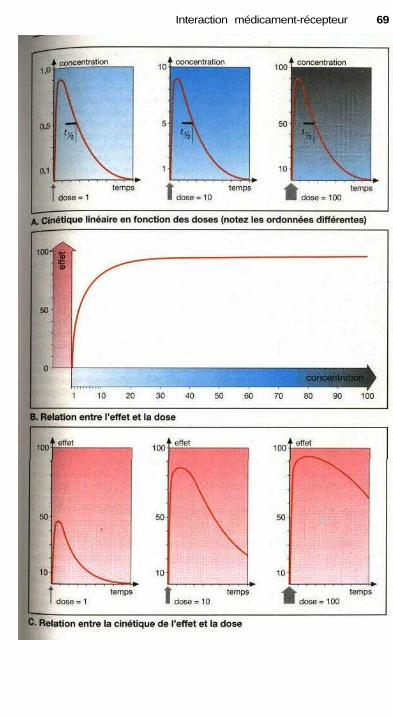
Interaction médicament-récepteur 69

70 Effets secondaires des médicaments
Effets secondaires des médicaments
L'effet souhaité (principal) d'un médi-cament est de modifier les fonctions del'organisme de sorte que les symptômesdu patient s'estompent. Par ailleurs, unmédicament peut également présenterdes effets secondaires indésirables quientraînent leurs symptômes propres,déclenchent des maladies ou sont mor-tels.
Origine des effets secondaires :surdosage (A). La substance est uti-lisée à une dose supérieure à celle né-cessaire pour obtenir l'effet principal :ceci conduit d'autres fonctions de l'or-ganisme à en subir les conséquences.La morphine (p. 208) par exemple àdose optimale agit en apaisant la dou-leur par son action sur les voies sensi-tives aboutissant dans le système ner-veux central. L'administration d'unequantité trop élevée de morphine freineles centres respiratoires avec risque deparalysie respiratoire. L'influence de ladose sur ces deux phénomènes peut êtrereprésentée sous forme de courbesdose-réponse. L'écart entre les deuxcourbes indique la différence entre lesdoses thérapeutiques et toxiques : cetintervalle de sécurité s'appelle la fe-nêtre thérapeutique.
« C'est en premier lieu la dosequi fait le poison » (Paracelse). Cettemaxime s'applique à tous les médica-ments mais aussi aux toxines de l'envi-ronnement. Aucune wbstance en elle-même n'est toxique. L'appréciation dudanger réside dans la connaissance :1. de la dose active, 2. de la dose à la-quelle peuvent apparaître des effets nui-sibles.
Sensibilité accrue (B). Lorsqu'unefonction donnée de l'organisme est par-ticulièrement sensible, on peut obtenirun effet indésirable même pour unedose normale. Une sensibilité accrue ducentre respiratoire à la morphine s'ob-serve chez les patients atteints d'unemaladie pulmonaire chronique, chezdes nouveau-nés ou sous l'influenced'une autre molécule déprimant les
centres respiratoires. La courbe dose-réponse est déplacée vers la gauche,une dose plus faible de morphine suffità provoquer une paralysie respiratoire.Une hypersensibilité peut égalementprovenir d'une anomalie génétique dumétabolisme. C'est ainsi que de nom-breux médicaments (primaquine sulfa-méthoxazol) déclenchent une destruc-tion prématurée des érythrocytes(hémolyse) chez des sujets souffrantd'un déficit en glucose 6-phosphatedéshydrogénase. La pharmacogéné-tique est une branche de la recherchequi s'intéresse à la relation entre le gé-notype de l'individu et sa réaction auxmédicaments.
Il faut distinguer ces formes d'hy-persensibilité de l'allergie qui a traitaux réactions du système immunitaire(P. 72).
Mauvaise spécificité (C). Mêmepour une dose appropriée et une sensi-bilité normale, des effets indésirablespeuvent se produire lorsque le médica-ment n'agit pas de façon totalementspécifique sur l'organe ou le tissu cible(malade). Par exemple l'atropine, unesubstance parasympatholytique ne selie pratiquement qu'aux récepteursmuscamuques de l'acétylcholine, maisceux-ci se trouvent dans différents or-ganes. La prométhazine, antihistami-nique et neuroleptique, est capabled'influencer plusieurs types de récep-teurs (NA = noradrénaline). Son actionn'est donc spécifique ni d'un organe nid'un récepteur. Les conséquencesd'une spécificité imparfaite peuventêtre fréquemment évitées, lorsque lemédicament n'a pas besoin de la circu-lation sanguine pour parvenir à sa cibleet peut être administre par voie locale(utilisation d'un parasympatholytiqueen gouttes oculaires ou en inhalation).
Pour chaque prise d'un médica-ment, on doit tenir compte des effets se-condaires. Avant de prescrire le médi-cament, il faut évaluer le bénéficeattendu et les risques. Ceci supposeune connaissance de l'effet principal etdes effets secondaires.
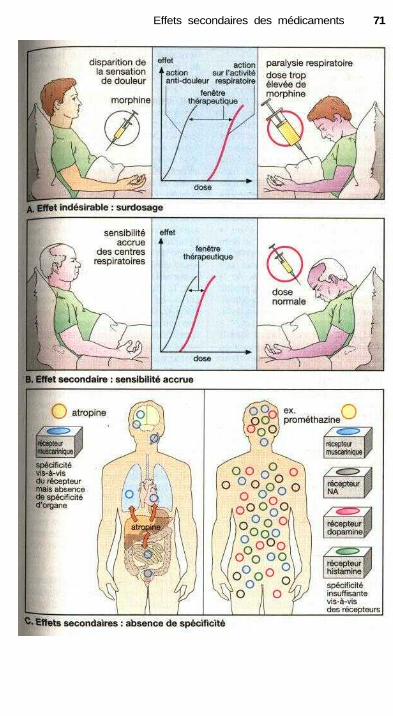
Effets secondaires des médicaments 71

72 Effets secondaires des médicaments
Allergie aux médicaments
Le système immunitaire a normalementla charge d'éliminer les particulesétrangères ayant pénétré dans l'orga-nisme (par exemple les bactéries). Lesreactions immunes peuvent se produirede façon inutile ou exagérée et porteratteinte à l'organisme (par exemple parune réaction allergique contre un médi-cament, contre le principe actif ou l'ex-cipient). Seuls quelques médicaments(par exemple des protéines étrangères àl'organisme) atteignent une taille suffi-sante pour pouvoir à eux seuls consti-tuer un stimulus antigénique. Dans laplupart des cas, la substance (ou hap-tène) doit d'abord se lier à une protéineappartenant à l'organisme, pour agircomme antigène. Dans le cas de lapénicilline G par exemple, un produitd'hydrolyse (groupement penicilloyl)permet la formation d'une liaison cova-lente avec une protéine.
Lors du premier contact avec lasubstance, le système immunitaire estsensibilisé : dans les organes lym-phoïdes se multiplient des cellules B(productrices d'anticorps) et des lym-phocytes T, caractéristiques de l'anti-gène et formant des cellules mémoires.Au deuxième contact, les anticorpssont déjà disponibles, les cellules mé-moires se multiplient rapidement et l'onvoit apparaître une réponse immunolo-gique notable : réaction allergique. Ellepeut être violente même pour desfaibles doses. On distingue quatre typesde réaction :
1. Réaction anaphylactique. Des an-ticorps de type I g E , spécifiques de lasubstance se fixent par leur fragment Fcaux récepteurs situés sur la surface ex-terne des mastocytes. La liaison de lamolécule pharmaceutique constitue lestimulus pour la libération d'histamineet d'autres médiateurs. Dans le pire descas, se déclenche un choc anaphylac-tique, potentiellement morte), avec unehypotension, un bronchospasme (crised'asthme), un œdème dans la région dularynx, l'apparition de démangeaisons
(urticaire), la contraction des musclesde l'intestin accompagnée de diarrhées(p.320).
2. Réaction cytotoxique. Des com-plexes substance-anticorps ( I g G ) se dé-posent à la surface des cellules san-guines. Ces complexes peuvent êtreformés avec des molécules de médica-ment déjà présentes dans le sang ou pri-maires. Au niveau du complexe setrouve un facteur d'activation du com-plément. Le complément est composéde différentes protéines, circulant dansle sang sous forme inactive, et qui sontactivées en cascade sous l'action d'unstimulus donné. Le complément activé(dirigé de manière normale contre lesagents infectieux) peut rompre la mem-brane cellulaire et lyser les cellules, ac-tiver la phagocytose, attirer les neutro-philes et les granulocytes (réactionchimiotactique) et déclencher une réac-tion inflammatoire. L'activation ducomplément peut avoir pour les cellulessanguines les conséquences suivantes :anémie hémolytique, granulocytopénie,thrombocytopénie.
3. Vasculitis à immuns complexes(maladie sérique, reaction d'Arthus).Les complexes entre le médicament etles anticorps se déposent sur la paroides vaisseaux, le complément est alorsactivé et déclenche une réaction inflam-matoire. Les neutrophiles attirés vers lefoyer inflammatoire, libèrent leurs en-zymes lysosomiales en tentant de pha-gocyter ces complexes et ces enzymesvont dégrader la paroi vasculaire (vas-culitis). Les différents symptômes peu-vent être : fièvre, œdème, gonflementdes ganglions, arthrite, névrite et né-phrite.
4. Eczéma de contact. Une substanceappliquée sur la peau, se lie à la surfacede lymphocytes T, dirigés spécifique-ment contre elle. Ces lymphocytes libè-rent dans leur environnement des mes-sagers (lymphokines) qui activent desmacrophages et déclenchent une réac-tion inflammatoire.
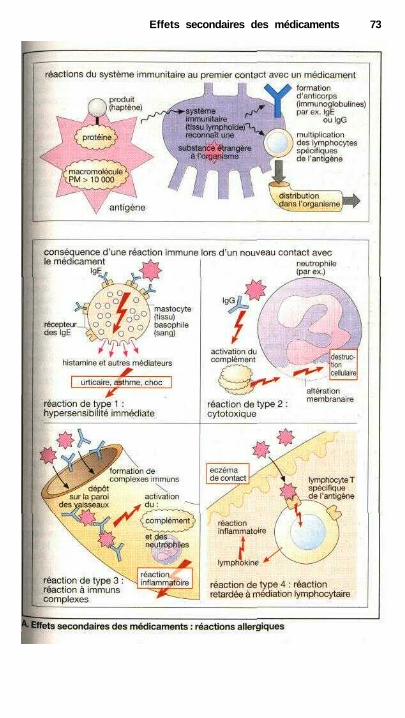
Effets secondaires des médicaments 73

74 Effets secondaires des médicaments
Effets nocifs pour l'enfant de la prisede médicaments pendant la grossesseet l'allaitement
Les substances absorbées par la mèrepeuvent atteindre l'enfant et produiredes effets indésirables.
Grossesse (A). Ce sont surtout lesmalformations des membres provo-quées par un somnifère (la thalidomide)qui ont attiré l'attention sur le risqueque les médicaments peuvent provo-quer des malformations (tératogéni-cité). Les effets provoqués chez lefœtus par les médicaments peuvent êtrede deux types :
1. Les effets qui dérivent des ef-fets typiques des molécules. Parexemple : masculinisation d'un fœtusféminin par les androgènes, hémorragiecérébrale provoquée par les anticoagu-lants oraux, bradycardie en présence deP-bloquants.
2. Les effets propres aux orga-nismes en formation et qui ne peuventêtre prévus à partir des autres propriétéspharmacologiques de la substance.
Pour estimer le risque que peut re-présenter la prise d'un médicament du-rant la grossesse, il faut tenir comptedes points suivants :
a) Moment de l'administration dumédicament. Les conséquences pos-sibles de la prise d'un médicament dé-pendent du stade de développement del'embryon (voir A). Le risque associé àun médicament dont l'effet est spéci-fique est également délimité dans letemps. Les tétracyclines par exempleexercent un effet sur les dents et les osprincipalement après le troisième moisde grossesse lorsque commence la mi-néralisation.
b) Perméabilité placentaire. Laplupart des molécules peuvent passerdu sang de la mère à celui de l'enfant auniveau du placenta. Les cellules acco-
lées du syncytiotrophoblaste consti-tuent une barrière de diffusion. Saperméabilité aux substances médica-menteuses est cependant plus élevéeque ne peut le laisser croire la notion de« barrière placentaire ».
c) Tératogénicité de la moléculeconcernée. Pour des produits connus etutilisés souvent, il existe des estima-tions statistiques du risque. De nom-breux médicaments n'ont aucun effettératogène démontrable. Pour les médi-caments nouvellement introduits, iln'est en général pas encore possible dedisposer d'une évaluation statistiquefiable du risque.
Il existe une action tératogèneavérée par exemple pour les dérivés del'acide rétmoique (étrétinate, isotréti-noïne) administrés per os pour le traite-ment des maladies de peau, dans le casdes anticoagulants oraux ou des tétracy-clines. Une forme particulière d'altéra-tion chez l'enfant peut être induite parle diéthylstilbestrol, une molécule es-trogénique. Lorsque la mère a ététraitée pendant la grossesse, on observechez les filles, vers l'âge de 20 ans, unrisque accru de carcinome du cervix etdu vagin.
Dans l'estimation du rapport effi-cacité-risque, il faut également penser àl'intérêt que peut présenter pour l'en-fant un traitement correct de sa mère.C'est ainsi qu'il ne faut pas arrêter untraitement antiépileptique car une épi-lepsie non soignée est au moins aussidangereuse pour l'enfant que l'éventua-lité de l'administration d'anti-épilep-tique.
Allaitement (B). Il existe une pos-sibilité qu'une substance présente dansl'organisme maternel passe dans le laitet soit ainsi absorbée par l'enfant. Pourapprécier l'importance du danger, ilfaut examiner les points présentés en(B). En cas de doute, il est faciled'éviter de mettre l'enfant en danger enle sevrant.
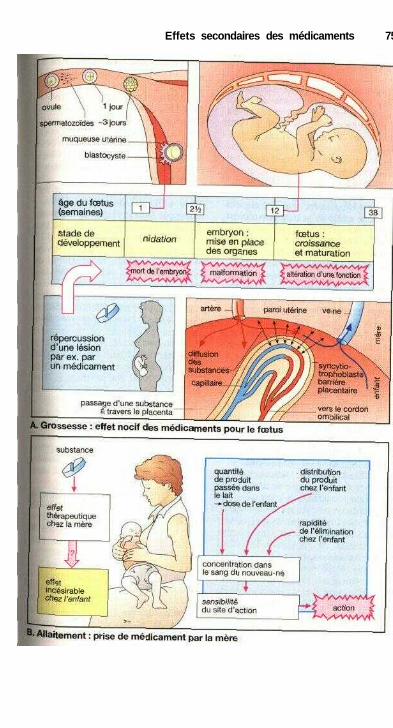
Effets secondaires des médicaments 75

76 Effets des médicaments indépendants d'une substance active
Placebo (A)
Un placebo est une forme médicamen-teuse ne contenant aucune substanceactive, une apparence de médicament.L'administration d'un placebo peutaussi bien déclencher des effets béné-fiques (soulagement des maux) que deseffets néfastes. Ceci dépend d'une mo-dification de l'état psychologique dupatient après une visite chez le mé-decin.
Consciemment ou inconsciem-ment, le médecin peut laisser transpa-raître à quel point il est intéressé par lessouffrances de son malade et combien ilest assuré de son diagnostic et de sonOrdonnance. En face d'un praticien cha-leureux, compétent et plein d'assu-rance, le malade se sentira en de bonnesmains, sera moins angoissé et pourra,plein d'optimisme, entrevoir sa gué-rison.
L'état physique influence l'étatpsychologique mais inversement, celui-ci peutjouer sur les sensations de l'or-ganisme. On cite le cas de blessésgraves qui, pendant la bataille, sentaientà peine leur blessure et commençaient àressentir de violentes douleurs à leur ar-rivée à l'hôpital, en sécurité. Ou encorede patients souffrant d'un ulcère à l'es-tomac par suite de stress psychologiqueet qui se plaignaient que « quelquechose leur était resté sur l'estomac ».
Etude clinique. Dans un cas isolé,il est parfois impossible de décider si laguérison provient de la substance elle-même ou de la situation thérapeutique.Il est alors nécessaire de réaliser uneétude statistique chez un grand nombrede patients en comparant les effetsd'une substance (verum) et ceux d'unplacebo. Étude contrôlée contre pla-cebo. Une étude prospective est plani-fiée à l'avance alors que dans une étuderétrospective, la décision d'analyser estprise après la fin du traitement. Les ma-lades sont répartis au hasard en deuxgroupes (randomisés) : traitement ouplacebo. Dans une étude en double
aveugle, ni le médecin, ni le malade nesait qui reçoit le placebo ou le médica-ment. Il est enfin possible, au coursd'un deuxième cycle de traitementd'effectuer un échange entre les traite-ments (placebo et médicament), étudecross-over. Dans ce cas, on peut com-parer les effets d'une susbtance à ceuxdu placebo non seulement entre deuxgroupes de patients mais également àl'intérieur d'un même groupe.
Homéopathie (B). C'est une mé-thode différente de traitement déve-loppée par Samuel Hahnemann à partirde 1800. Son hypothèse était qu'unedrogue (au sens de médicament), qui àdes concentrations usuelles (médecineallopathique) suscite un ensemble desymptômes précis peut, à dose trèsfaible, et chez un malade dont les symp-tômes sont proches de son « profil d'ac-tion », entraîner une guérison (principede similitude). L'organisme possède enlui-même la capacité de se guérir etcette force est activée par des doses trèsfaibles de la substance, conduisant ainsià une autoguénson. Chez son malade,l'homéopathe ne doit pas diagnostiquerles causes de la maladie mais trouver ladrogue dont le profil symptomatolo-gique se superpose au mieux avec la sé-miologie de la maladie : il faut doncdiagnostiquer un médicament. Il estdonc nécessaire de procéder à un inter-rogatoire approfondi du patient concer-nant ses maux. La substance est alorsutilisée fortement diluée.
L'action directe des médicamentshoméopathiques sur les fonctions del'organisme n'est pas détectable.L'action curative repose sur la force desuggestion de l'homéopathe et sur l'at-tente du malade.
Lorsqu'une maladie peut être for-tement influencée par des paramètrespsychologiques, et qu'il n'existe pas detraitement efficace, il est souhaitabled'utiliser la force de suggestion commemode de traitement. L'homéopathieconstitue alors l'une des solutions pos-sibles.
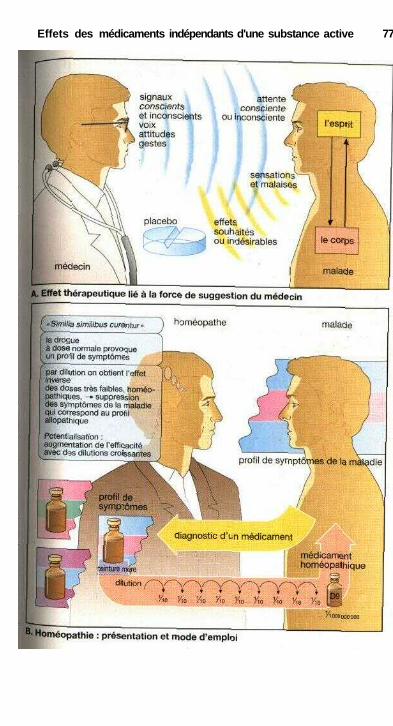
Effets des médicaments indépendants d'une substance active 77

•j Pharmacologie des spécialités

80 Influence des médicaments sur le système sympathique
Système nerveux sympathique
Au cours de l'évolution, il a fallu déve-lopper un système de contrôle efficacepour coordonner chez les individus decomplexité croissante les fonctions dechaque organe et pour pouvoir adapterleur comportement aux changementsdes conditions d'environnement. Cesystème de contrôle se compose du sys-tème nerveux central avec le cerveau etla moelle épinière, ainsi que deux voiesséparées de communication avec les or-ganes périphériques, le système ner-veux somatique et le système nerveuxvégétatif. Le système nerveux soma-tique (nerfs de la sensibilité superfi-cielle et profonde, des organes des senset des muscles squelettiques) sert à per-cevoir l'état du monde environnant et àgouverner les mouvements du corpsadaptés à la situation (perception senso-rielle : menace -* réaction : fuite ouattaque). Le système nerveux végé-tatif associé au système endocriniencontrôle le monde intérieur. Il accordeles fonctions des organes internes auxbesoins de l'organisme. Le contrôle parvoie nerveuse permet une adaptationtrès rapide tandis que le système endo-crinien règle l'état des fonctions à longterme. L'activité du système nerveuxvégétatif est indépendante du contrôlevolontaire et fonctionne de façon auto-nome (d'où son nom de système ner-veux autonome). Ses centres se trou-vent dans l'hypothalamus, la moelleépinière et le tronc cérébral.
Le système nerveux végétatif pré-sente une partie sympathique et unepartie parasympathique (p. 98). Lesreseaux de ces deux systèmes compor-tent, à côté de nerfs efférents (issus dusystème nerveux central), des nerfs af-férents. Dans les organes qui sont in-nervés à la fois par le système sympa-thique et le système parasympathique,l'activation de ces systèmes déclencheen général des réactions opposées.
En cas de maladie (dérangementdes fonctions d'un organe), on cher-chera souvent en utilisant des produitspharmaceutiques qui agissent sur le
système végétatif à ramener à la nor-male le fonctionnement de l'organe.
L'effet biologique de susbtancesqui inhibent ou stimulent le systèmesympathique ou inversement le systèmeparasympathique, peut être aisémentdéduit de l'observation des rôles dusystème sympathique ou parasympa-thique (A : conséquences d'une acti-vation sympathique). L'activation dela partie sympathique du système ner-veux végétatif peut être considérée defaçon simplifiée comme l'ensemble desréactions de l'organisme permettantd'aboutir rapidement à un état d'acti-vité plus élevée, propice à une fuite ouun combat.
Les deux situations réclament uneactivité musculaire intense. L'oxygèneet les substrats énergétiques doiventêtre amenés aux muscles en quantitésuffisante et c'est pourquoi le flux san-guin au niveau des muscles, la fré-quence et la force de contraction ducœur vont augmenter pour pouvoirpomper plus de sang dans la circulation.De plus, le rétrécissement des vais-seaux irriguant les intestins détournerale flux sanguin vers les muscles.Comme dans cette situation, la diges-tion des aliments est superflue et mêmegênante, le transport vers l'avant ducontenu intestinal est freiné, le péristal-tisme décroît et les muscles dusphincter se contractent. Cependant,pour augmenter la fourniture d'élé-ments nutritifs aux muscles et au cœur,le glucose hépatique doit être libérédans le sang ainsi que les acides gras dutissu adipeux. Les bronches s'élargis-sent de façon à accroître le volume res-piratoire et par là même l'apport d'oxy-gène au sang.
Les glandes sudoripares sont aussiinnervées par le système sympathique(mains moites lors d'une émotion),elles constituent une exception en cequi concerne le neurotransmetteur (acé-tylcholine, p. 106).
Les conditions de vie des hommesmodernes sont différentes de celles del'homme des cavernes mais les fonc-tions biologiques n'ont pas changé.
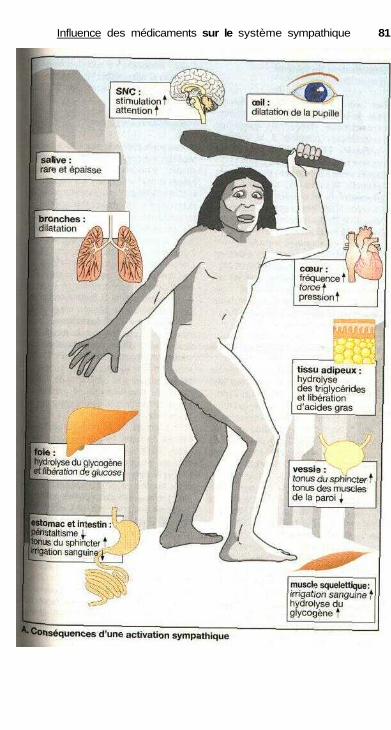
Influence des médicaments sur le système sympathique 81

82 Influence des médicaments sur le système sympathique
Organisation du systèmesympathique
Les neurones sympathiques efférentsvoni de la moelle épinière à la chaîneparavertébrale (rangée de ganglionssympathiques parallèle à la colonnevertébrale). Les ganglions constituentdes ensembles de points de contact (sy-napse) entre les neurones provenant dela mœlle épinière (1, neurone gan-glionnaire) et les cellules nerveuses quienvoient leurs prolongements vers lapériphérie de l'organisme (2, neuronepost-ganglionnaire). A ce niveau, ilsentrent en contact avec les cellules desorganes cibles au niveau des synapsespost-ganglionnaires. A côté de ces neu-rones, il en existe d'autres, dont les in-terconnexions ont lieu d'abord dansl'organe cible ou encore qui aboutissentsans intermédiaire aux glandes surré-nales.
Médiateurs du système sympathique
Tandis que l'acétylcholine joue le rôlede médiateur chimique au niveau dessynapses entre les neurones 1 et 2 (pré-et post-ganglionnaires, voir le principede la transmission cholinergique,p. 98), c'est la noradrénaline qui rem-plit cette fonction pour les synapses desneurones de type 2 (B). Un neuronesympathique de type 2 n'établit pas unesynapse avec une seule cellule de l'or-gane cible, il se ramifie de nombreusesfois et chaque prolongement établit aupassage des contacts avec plusieurscellules. Au voisinage de ces synapsesse trouvent des épaississements desaxones (varicosités) qui se succèdentcomme les perles d'un collier à chaquecontact du nerf avec une cellule cible.De cette façon, lors de la stimulation dunerf un domaine cellulaire plus impor-tant sera activé bien que l'action de lanoradrénaline libérée par un neurone detype 2, reste limitée à la proximité dessynapses.
L'activation d'un neurone detype 1, conduisant aux glandes surré-nales déclenche par l'intermédiaire
d'une libération d'acétylcholine 1sécrétion d'adrénaline (p. 108) qui srépand dans l'organisme par le san(hormone, A).
Synapse adrénergique
La noradrénaline est stockée à proximité des varicosités dans des petites v<sicules entourées d'une membran(grana 0 0,05 - 0,2p.m). La dopaminssynthétisée dans l'axoplasme à partir dla tyrosine et via plusieurs reactions irtermédiaires, sera capturée à l'inténeudes ces vésicules. La dopamine est ensuite convertie en noradrénaline pal'enzyme dopamine p-hydroxylas(Lors d'une stimulation électrique dnerf sympathique, une partie des vésicules déverse son contenu et donc 1noradrénaline dans la fente synaptiqwLa noradrénaline libérée reagit avedes récepteurs adrénergiques posisynaptiques présents sur la membrandes cellules cibles ou présynaptiquesur la membrane des varicosités. La stimulation des récepteurs a; présynaptiques entraîne une inhibition de la libération de noradrénaline et permet u;rétrocontrôle négatif du processus dlibération.
L'action de la noradrénaline déversée disparaît très rapidement : environ 90 % sont recaptés rapidementpar un processus de transport actild'abord dans l'axoplasme puis de 1dans les vésicules (recapture neuronale). Une petite partie de la noradrénaUne sera inactivée par la Catéchol-0Méthyl-Transférase (COMT, enzymdu cytoplasme des cellules cibles) eune autre partie par la Mono-AminéOxydase (MAO, dans les mitochondries des cellules nerveuses ou des ce!Iules cibles).
Le foie est richement pourvu eices enzymes et contribue de façon importante à la dégradation de l'adrénaline ou de la noradrénaline existantedans le sang.
Le produit final de la dégradatioldes catécholamines par la COMT et 1MAO est l'acide vanylmandélique.
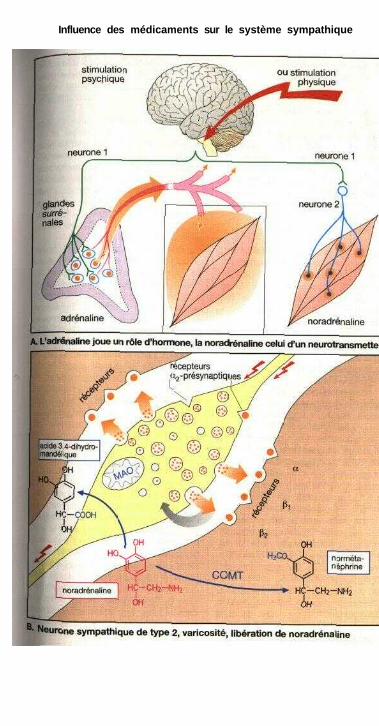
Influence des médicaments sur le système sympathique 83

84 Influence des médicaments sur le système sympathique
Sous-types de récepteursadrénergiques et actionsdes catécholamines
D'un point de vue pharmacologique, onpeut distinguer des récepteurs a, et a,(p. 90), des récepteurs P|, |3; et même(Î3. Les différents récepteurs adréner-giques sont distribués de façon très hé-térogène dans chaque tissu. Lesagonistes adrénergiques (sympatho-mimétiques directs) peuvent être uti-lisés à diverses fins thérapeutiques.
Effets sur les muscles lisses
Les effets opposés d'une stimulationdes récepteurs a et (3 sur le muscle lissereposent sur les différences dans latransduction du signal (p. 66) c'est cequi est représenté en (A) dans le cas desmuscles de la paroi vasculaire. La sti-mulation du récepteur oi| déclenche parl'intermédiaire d'un second messagerintracellulaire (?3) une libération ac-crue d'ions Ca1*. Associé à la calmodu-line, le calcium permet l'activation dela myosine-kinase, ce qui conduit àla phosphorylation d'une protéinecontractile, la myosine, et à l'augmen-tation du tonus (— vasoconstriction).
L'AMPc inhibe l'activation de lamyosine-kinase. Les récepteurs P;aboutissent via une protéine G activa-trice, Gs, à une augmentation de la for-mation d'AMPc (— vasodilatation), lesrécepteurs a; via une protéine inhibi-trice G, provoquent une diminutiond'AMPc (-> vasoconstriction).
La vasoconstriction provoquéepar l'application locale d'a-sympatho-mimétiques sera utilisée dans le casd'une anesthésie locale (p. 204) ou dansdes gouttes nasales décongestionnantes(naphtazoline, tétryzoline, xylométazo-line, p. 90, 318, 320). L'administrationsystémique d'adrénaline joue un rôleimportant pour augmenter la pressionartérielle dans le traitement d'un chocanaphylactique.
Bronchodilatation. La dilatationdes bronches due à une stimulation desrécepteurs (i; (ex. : fénotérol ou salbu-tamol) est un mode de traitement trèsimportant dans l'asthme (p. 322).
Tocolyse. L'effet inhibiteur desPz-sympathomimétiques (par ex. le fé-notérol) sur la contractilité utérine peutêtre utilisé pour calmer des contrac-tions précoces (risque d'accouchementprématuré). Une vasodilatation médiéepar une stimulation p;, associée à unechute de la pression artérielle, conduit àune tachycardie réflexe, à laquelleparticipe également une action stimu-lante (31 de la substance.
Effets cardiaques. Les catéchola-mines augmentent toutes les fonctionsdu cœur par le biais des récepteurs p,et de l'AMPc : force d'éjection (effetinotrope positif), vitesse de raccour-cissement (effet klinotrope), fréquencedes battements (effet chronotrope),propagation de la stimulation (effetdromotrope) et excitabilité (effet bath-motrope). Dans le tissu nodal, la dépo-larisation diastolique est accélérée desorte que le seuil de déclenchement dupotentiel d'action soit atteint plus ra-pidement (effet chronotrope positif, B).L'action des (3-sympathomimétiquessur le cœur peut être utilisée encas d'arrêt cardiaque : administrationd'adrénaline. L'utilisation de (3-mimé-tiques pour traiter une insuffisance car-diaque est associée à un risque d'ar-rythmie.
Effets métaboliques. La stimula-tion des récepteurs p; augmente, vial'AMPc, la dégradation du glycogène(glycogénolyse) en glucose dans le t'oieet les muscles squelettiques. Le glucosehépatique sera déversé dans le sang.Dans le tissu adipeux, les triglycéridesseront dégradés en donnant des acidesgras (lipolyse, médiée par les récep-teurs p3 ?), qui seront ensuite déversésdans le sang. Les effets métaboliquesdes catécholamines n'ont aucune utilitéthérapeutique.
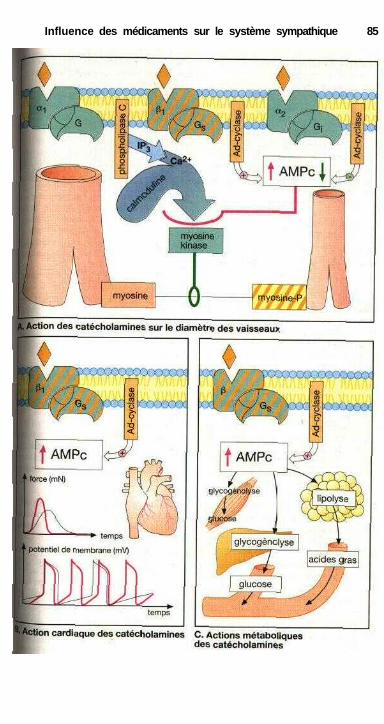
Influence des médicaments sur le système sympathique 85

86 Influence des médicaments sur le système sympathique
Relations structure-activité
II n'est pas possible avec l'adrénalined'exercer un effet spécifique sur l'undes sous-types de récepteurs car ellepossède une affinité importante pourtous les récepteurs a et p. Elle neconvient pas non plus pour une admi-nistration orale car elle est mal ab-sorbée et sera éliminée par voie presys-témique.
La noradrénaline est une catécho-lamine (catéchol est un nom usuel pourun 0-hydroxyphénol), qui se distinguede l'adrénaline par une affinité élevéepour les récepteurs a et une affinitémoindre pour les récepteurs ?;. Dans lecas de l'isoprénaline, la dissociation estpresque totale (A) :noradrénaline -* a, (3iadrénaline -» a, pi, (i,isoprénaline -* P[, P;La connaissance de la relation entrela structure chimique et l'effet (rela-tion structure-activité) permet lasynthèse de sympathomimétiques quiont une affinité préférentielle pour undes sous-types de récepteurs adrener-giques.
L'élément chimique commun àl'élaboration de tous les sympathomi-métiques directs (substances agissantcomme agonistes sur les récepteursadrénergiques) est la structure phényl-éthylamine. Le groupement hydroxylesur la chaîne latérale est importantaussi bien pour l'affinité envers les ré-cepteurs a que p. La substitution surl'azote diminue l'affinité pour les ré-cepteurs a et augmente celle pour lesrécepteurs P, de telle sorte qu'avec unrésidu isopropyl, on atteint déjà une af-finité optimale pour les récepteurs p(isoprénaline = isopropylnoradréna-line). L'allongement ultérieur de cesubstituant favorise l'action sur les ré-cepteurs p, (sélectivité P; par ex. salbu-tamol, fénotérol). Les deux groupe-ments hydroxyle du noyau aromatiquesont indispensables à l'affinité, une af-finité élevée pour les récepteurs a estattachée à la position de ces groupe-ments OH en 3, 4 ; cependant, certainsdérivés qui portent des groupes hy-
droxyle en 3,5 (orciprénaline, terbuta-line, fénotérol), ont une affinité pour lesrécepteurs P.
Les groupements hydroxyle de lamolécule de catécholamine diminuentconsidérablement son caractère lipo-phile. La polarité est augmentée par lefait que l'azote concerné est presqueentièrement protoné dans la zone despH physiologiques. Le remplacementde l'un ou de tous les groupements hy-droxyle se traduit par une améliorationdu passage à travers les barrières mem-branaires (barrière entre l'intestin et lesang : absorption après administrationorale, barrière hémato-encéphalique :action sur le système nerveux central),mais en même temps par une diminu-tion d'affinité.
L'absence de l'un ou des deuxgroupements hydroxyle est lié à uneaugmentation de l'activité sympatho-mimétique indirecte, qui correspond àla capacité d'une substance à libérer lanoradrénaline de ses sites de stockage,sans être elle-même un agoniste adré-nergique (p. 88).
Un changement de la position desgroupements hydroxyle sur le cycle (or-ciprénaline, fénotérol, terbutaline) ouleur substitution (salbutamol) protège lamolécule de la dégradation par laCOMT (p. 82). L'introduction d'un ré-sidu alkyl de petite taille sur l'atome decarbone proche de l'azote, comme lasubstitution sur l'azote du groupementméthyl par un résidu de plus grandetaille, rend plus difficile la dégradationparlaMAO(p.82).
Comme la structure chimique né-cessaire pour une affinité élevée ou lesconditions requises pour permettre uneadministration orale ne coïncident pas,il est nécessaire de faire des compromislors du choix d'une substance. Si l'onveut utiliser l'affinité élevée de l'adré-naline, on n'a pas en même temps unebonne absorption au niveau de l'intestin(adrénaline, isoprénaline) ; si par contreon souhaite également une bonnebiodisponibilité, après administrationorale, il faut accepter des concessionsen ce qui concerne l'affinité pour les ré-cepteurs (éthyléphrine).
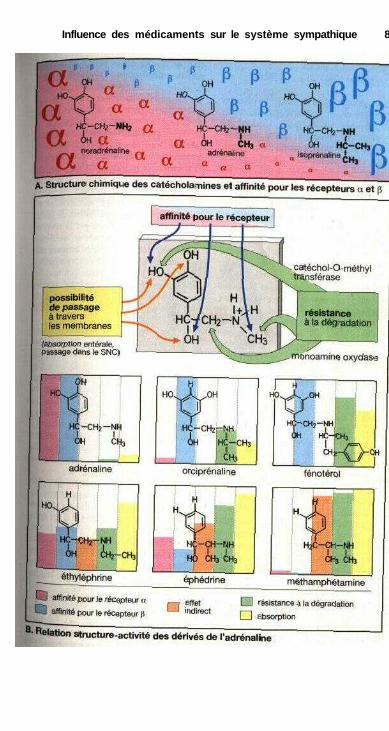
Influence des médicaments sur le système sympathique 87

88 Influence des. médicaments sur le système sympathique
Substances à actionsympathoinimétique indirecte
Plusieurs systèmes participent à côté desrécepteurs au fonctionnement de latransmission adrénergique. Ce sont lessystèmes de recapture active qui trans-portent le médiateur de la fente synap-tique dans le cytosol (axoplasme) àtravers la membrane cellulaire, lessystèmes de transport de l'axoplasmevers les grana, ainsi que l'enzyme dedégradation, la monoamine oxydase(MAO). La noradrénaline possède uneaffinité pour les récepteurs, pour le sys-tème de transport et les enzymes de dé-gradation. Des molécules ayant subi unemodification chimique vont se différen-cier de la noradrénaline par leur affinitérespective pour les différents systèmes(p. 86) et agiront préférentiellement surl'une ou l'autre des fonctions.
Inhibiteurs de la monoamineoxydase (A). Ils touchent la mono-amine oxydase, enzyme essentielle-ment localisée dans les mitochondries,qui maintient la concentration de nora-drénaline dans l'axoplasme à une va-leur faible. L'inhibition de l'enzymeaugmente la concentration de noradré-naline. Comme la dopamine est égale-ment dégradée par la MAO, l'inhibitionde l'enzyme augmente la quantité dedopamine disponible pour la synthèsede noradrénaline. La quantité de nora-drénaline stockée dans les grana et demême celle libérée à chaque excitationaugmentent à cause de l'inhibition del'enzyme.
Dans le système nerveux central,l'inhibition de la MAO influence enplus de l'accumulation de la noradréna-line celle de la dopamine et de la séro-tonine aboutissant ainsi, probablementà cause de l'importance majeure de ceneurotransmetteur, à une activation gé-nérale (effet thymérétique). La tranyl-cypromine sert dans quelques cas parti-culiers d'antidépresseur. Elle bloque defaçon durable grâce à une liaison cova-lente les deux sous-types MAO-A etMAO-B. Le moclobémide est un inhibi-
teur réversible de la MAO-A ; il seraparfois utilisé comme antidépresseur ;la sélégiline est utilisée comme anti-parkinsonien ; on obtient dans ce casune augmentation de la concentrationde dopamine (p. 186).
Sympathomimétiques indirects(B). Ce sont des substances qui aug-mentent la concentration de noradréna-line dans la fente synaptique que ce soitpar inhibition de la recapture (cocaïne,sympathomimétique indirect et anes-thésique local), par une accélération dela libération, par une inhibition de la dé-gradation par la MAO ou par la sommedes trois effets (amphétamine, métham-phétamine encore appelées aminés sti-mulantes). L'efficacité des sympatho-mimétiques indirects peut diminuer etfinalement disparaître (tachyphylaxie)lorsqu'on aboutit à un épuisement dustock de noradrénaline le plus prochedu plasmalemme.
Les Sympathomimétiques indi-rects peuvent traverser la barrière hé-mato-encéphalique et produire au ni-veau central un sentiment de bien êtrecorporel, l'activité est accrue, l'humeureuphorique et la sensation de faim, oude fatigue effacée. Après le déclin del'effet viennent contrariété et abatte-ment. Ces effets secondaires incitent àune nouvelle prise des produits (risqueélevé de dépendance). Pour éviter lesabus, ces substances sont inscrites dansle tableau B (stupéfiants).
L'utilisation abusive de sub-stances amphétaminiques pour une sti-mulation transitoire des capacités(dopage) présente le danger d'un épui-sement de l'organisme. Comme la sen-sation de fatigue est absente, un sportifpar exemple, aura pu mobiliser ses der-nières forces. Dans les cas extrêmespeut se produire une défaillance cardio-vasculaire (B).
Les « coupe-faim » (anorexi-gènes), sont chimiquement très prochesdes amphétamines (par ex. fenflura-mine, mazindol). Leur utilisation peutégalement conduire à la dépendance etleur utilité thérapeutique est douteuse.
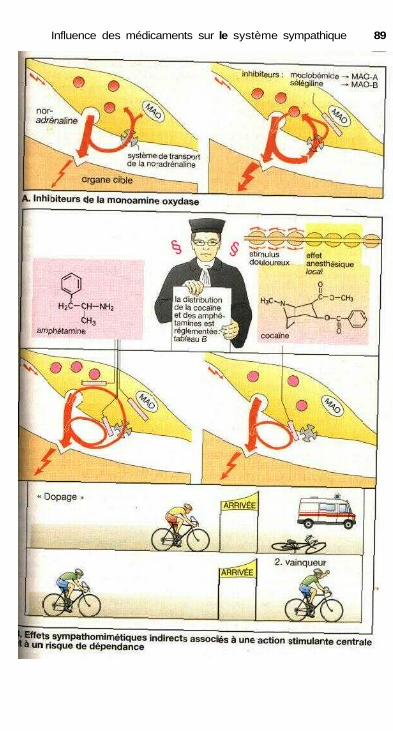
Influence des médicaments sur le système sympathique 89

90 Influence des médicaments sur le système sympathique
ot-Sympathomimétiques,a-Sympatholytiques
Les a-sympathomimétiques peuventêtre utilisés :— par voie systémique dans certainesconditions où la pression artérielle esttrop faible pour augmenter cette pres-sion (p.308);- localement pour obtenir une décon-gestion nasale ou du tissu conjonctif del'œil (p. 318, p. 320), ou comme adju-vant d'une anesthésie locale, pour pro-duire une diminution localisée de la cir-culation sanguine. Lors de l'applicationlocale, la réduction du flux sanguin peutconduire à une pénurie d'oxygène (A).A la limite, l'hypoxie locale peut provo-quer une nécrose du tissu. Ce sont parti-culièrement les extrémités qui sont ex-posées à ce danger et notamment lesdoigts, les pieds et les oreilles. Pour uneanesthésie locale effectuée aux extré-mités, il ne faudra pas utiliser de vaso-constricteur.
La vasoconstriction par un a-sym-pathomimétique est suivie d'une phased'augmentation de la circulation san-guine (hyperémie réactionnelle, A).Cette reaction peut être observée lors del'administration d'un sympathomimé-tique (naphtazoline, tétryzoline, xylo-métazoline) sous forme de gouttes na-sales. Tout d'abord, par suite de lavasoconstriction, l'irrigation de la mu-queuse nasale est diminuée et par làmême la pression capillaire. Le liquideaccumulé dans l'espace interstitiel, res-ponsable de la congestion de la mu-queuse, peut s'écouler par les veines. Lasécrétion du mucus nasal diminue parsuite de la réduction du fluide dispo-nible. Au cours d'un rhume, la respira-tion par le nez redevient possible.Cependant, après la disparition de l'effetvasoconstricteur, on observe de nouveaudans la phase d'hypérémie un passagedu liquide plasmatique dans l'espace in-terstitiel, le nez est à nouveau« bouché » et le patient se voit obligéde recommencer l'administration degouttes nasales. C'est alors la menaced'un cercle infernal conduisant à la prise
chronique de gouttes dans le nez. La ca-rence persistante en oxygène peut en-traîner une dégradation irréversible de lamuqueuse nasale.
a-Sympatholytiques (B). L'inter-action de la noradrenaline avec les ré-cepteurs ci-adrenergiques peut êtrebloquée par les a-sympatholytiques(antagonistes a-adrénergiques, cn-blo-quants). Cet effet est utile dans le casd'une pression artérielle trop élevée (va-sodilatation —• \ résistance périphé-rique, \ pression artérielle, p. 118). Lespremiers a-sympatholytiques bloquaientl'effet de la noradrenaline non seule-ment au niveau des récepteurs a,, post-synaptiques mais également au niveaudes récepteurs a^-présynaptiques (a.-bloquants non spécifiques, par ex. laphénoxybenzamine ou la phentolamme).
Les récepteurs a;-presynaptiquesservent de détecteur pour la mesure de laconcentration de noradrenaline dans lafente synaptique, et règlent par rétrocon-trôle la libération de noradrenaline. Lastimulation des récepteurs a^-présynap-tiques inhibe la libération ultérieure denoradrenaline. Au contraire, leur blo-cage a pour conséquence une libérationincontrôlée de noradrenaline. Ceci estvisible dans les synapses du muscle car-diaque où sont présents des récepteursadrénergiques P| : tachycardie et arythmie.
cii-Sympatholytiques sélectifs(cii-bloquants, par ex. prazosine ou descomposés à action plus durable, térazo-sine et doxazosine). Ces composés neprovoquent pas d'inhibition de la libéra-tion de noradrenaline.
Les cii-bloquants seront employéschez les hypertendus (p. 306). Commeils rendent impossible une contraction-des vaisseaux dans l'organisme, le sangpeut s'accumuler dans les jambes aumoment du passage à la station debout(mauvaise régulation orthostatique,p. 308).
Dans le cas d'une hyperplasie bé-nigne de la prostate, les cii-bloquants(par ex. térazosine, alfuzosine) peuventêtre utilisés pour diminuer le tonus desmuscles lisses dans la région de la pros-tate et favoriser la miction (p. 250).
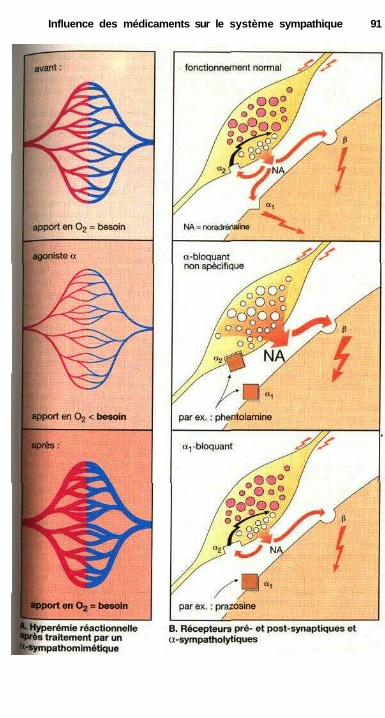
Influence des médicaments sur le système sympathique 91

92 Influence des médicaments sur le système sympathique
P-Sympatholytiques (P-bloquants)
Les P-sympatholytiques sont des antago-nistes de l'adrénaline et de la noradréna-line au niveau des récepteurs (3, ils nepossèdent aucune affinité pour les récep-teurs a.
Effets thérapeutiques.En bloquant les récepteurs ?,, lesP-bloquants mettent le cœur à l'abri deseffets d'une stimulation sympathique surla consommation d'oxygène, (p. 302).Dans ces conditions, une augmentationdu travail cardiaque n'est pratiquementplus possible (cœur en activité modérée).Cette propriété sera utilisée dans le casd'une angine de poitrine, pour empêcherune surcharge cardiaque, qui pourraitprovoquer une crise (prophylaxie del'angine de poitrine, p. 304). Les (3-blo-quants servent aussi à diminuer la fré-quence cardiaque (tachycardie sinusale,p. 134) et diminuent une pression arté-rielle trop élevée. Le mécanisme de leuraction hypertensive est complexe. LesP-bloquants seront utilisés localementpour diminuer la pression interne del'œil {glaucome} ; ils diminuent la sécré-tion de l'humeur.
Effets indésirables.Les P-bloquants sont très souvent utiliséset en général bien supportés, si l'on tientcompte de leurs contre-indications. Ilexiste un risque lors du traitement par lesP-bloquants dans les conditions où l'orga-nisme a besoin de 1' activation permanentedes récepteurs pour le bon fonctionne-ment d'un organe.
Insuffisance cardiaque. En cas defaiblesse du muscle cardiaque, le cœurpeut dépendre d'une stimulation perma-nente du système sympathique pourfournir un débit suffisant. Une augmenta-tion du débit cardiaque sera obtenue lorsd'une activation sympathique, grâce à uneélévation de la fréquence et de la forced'éjection. En présence de P-bloquants, lastimulation sympathique est supprimée, levolume d'éjection et la fréquence décrois-sent : une insuffisance cardiaque latentese révèle, une insuffisance cardiaque déjàmanifeste s'aggrave (A).
D'un autre côté, des études cli-niques ont montré que dans des condi-tions données les P-bloquants peuventégalement agir en cas d'insuffisance car-diaque.
Bradycardie, bloc AV : la dispari-tion de la stimulation sympathique peutdéclencher une diminution trop impor-tante de la fréquence cardiaque ainsi quedes perturbations de la transmission del'excitation entre les oreillettes et les ven-tricules.
Asthme bronchique : une activitéélevée du système sympathique empêchele déclenchement d'un broncho-spasmechez des patients ayant une tendance aurétrécissemment spasmodique des bron-ches (asthme, bronchite du fumeur). Dansces conditions, on aboutit à un essouffle-ment par blocage des récepteurs p; (B).
Hypoglycémie en cas de diabètesucré : lorsque survient une hypogly-cémie chez un patient diabétique soustraitement par l'insuline ou un antidiabé-tique oral, l'adrénaline sera libérée et dé-clenchera par stimulation des récepteursP; dans le foie une augmentation de la li-bération de glucose. Les P-bloquants sup-priment aussi bien la contre régulationque les signes annonciateurs d'une hypo-glycémie dus à la libération d'adrénaline(par ex. les battements de cœur) : dangerd'un coma hypoglycémique.
Altérations circulatoires : sous l'ac-tion du blocage des récepteurs ?;, les ef-fets vasodilatateurs de l'adrénaline médiéspar ces récepteurs disparaissent tandis quel'effet vasoconstricteur lié à une stimula-tion a demeure intact : \ circulation péri-phérique : pieds et mains froids.
Les P-bloquants ont une action« anxiolytique », qui peut reposer surl'atténuation des signes caractéristiquesd'une libération d'adrénaline d'originepsychologique (battements de cœur, trem-blements), signes qui de leur côté peuventrenforcer l'angoisse et le «trac».L'attention n'est pas diminuée par lesP-bloquants et c'est pourquoi ils sont uti-lisés occasionnellement par des orateursou des musiciens lors de grandes repré-sentations (C). Le trac<n'est cependantpas une maladie réclamant un traitementmédicamenteux, i
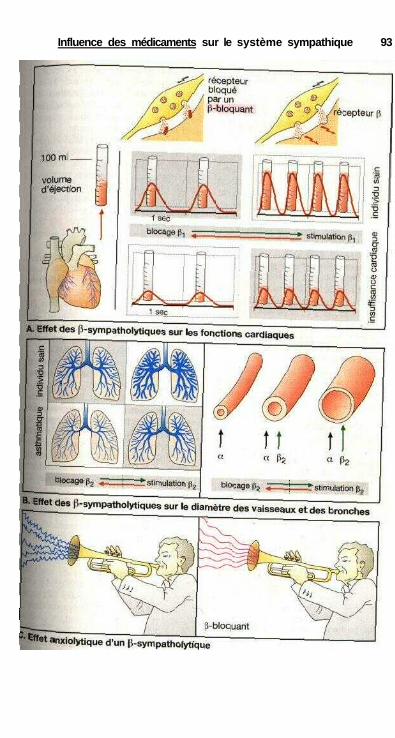
Influence des médicaments sur le système sympathique 93

Influence des médicaments sur le système sympathique94
Différences entre P-bloquants
Les p-sympatholytiques possèdentcomme structure chimique de basecommune la chaîne latérale des p-sym-pathomimétiques (comparez l'isopré-naline aux p-bloquants tels que propra-noiol, pmdolol et aténolol). La structurede base est en général reliée à un sub-stituant aromatique par une liaison-CH^-O-. L'atome de carbone qui portele groupement hydroxyle, constitue uncentre chiral. A part deux exceptions(penbutolol et timolol), tous les p-sym-patholytiques se trouvent sous formeracémique (p. 62).
L'énantiomère lévogyre possèdeune affinité jusqu'à 100 fois plus élevéepour le récepteur P que le composé dex-trogyre, et est donc pratiquement seulresponsable de l'effet de blocage P. Lachaîne latérale et le substituant surl'azote sont importants en ce quiconcerne l'affinité pour le récepteurtandis que le substituant aromatique estprimordial pour définir si la substancemontre encore une activité sympatho-mimétique intrinsèque et est aussi unagoniste / antagoniste partiel. Un ago-nisme (antagonisme) partiel se produitlorsqu'une substance présente une acti-vité intrinsèque, mais si faible, quel'occupation de tous les récepteurs dis-ponibles ne déclenche qu'une partie del'effet obtenu en présence d'un agonistecomplet. En présence d'un agonistepartiel, l'action d'un agoniste complet(par ex. l'isoprénaline) est inhibée carla liaison de l'agoniste complet est em-pêchée. De cette façon, les agonistespartiels agissent aussi comme antago-nistes mais conservent cependant unecertaine capacité de stimuler les récep-teurs. On peut se demander si l'effetagoniste propre des p-bloquants pré-sente un avantage thérapeutique.
Les p-bloquants qui sont desmédicaments cationiques amphiphilespeuvent, en fonction de leur lipophilie.inhiber à concentration plus élevée le
canal sodique et par là même l'excitabi-lité du cœur et la propagation de la sti-mulation : effet de stabilisation demembrane. Aux doses thérapeutiqueshabituelles, la concentration requisepour cet effet n'est pas atteinte.
Il existe des p-sympatholytiquesqui ont une affinité plus élevée pour lesrécepteurs Pi cardiaques que pour lesrécepteurs p, : p-bloquants cardiosé-lectifs (métoprolol, acébutolol, aténololbisoprolol). La cardiosélectivité des p-sympatholy tiques est telle qu'ils peu-vent malgré tout être prescrits par mé-garde à des patients souffrant d'asthmebronchique ou de diabète (p. 92).
La structure chimique des P-blo-quants est également importante pourleurs propriétés pharmacocinétiques.A l'exception des composés hydro-philes (par exemple l'aténolol) les p-bloquants seront complètement ab-sorbés au niveau intestinal. Ils subissentensuite une élimination présysté-mique partielle mais importante (A).
Toutes les possibilités de structurementionnées n'ont qu'une faible impor-tance thérapeutique.
La multiplicité de l'offre donneune impression encore plus curieuse (B).En 1965, avec le propranolol, le premierP-bloquant a été introduit dans l'arsenalthérapeutique ; 30 ans après, environ26 P-bloquants de structure chimiquedifférente sont commercialisés. Ce déve-loppement inquiétant est typique decelui d'un groupe de molécules, quijouent un rôle thérapeutique important etdont la structure active est unique. Pardes modifications de la molécule, onpeut certes produire des substances chi-miques nouvelles (brevetables) maisaucun médicament ayant une action dif-férente. En plus, certaines de ces sub-stances qui ne sont plus protégées par unbrevet, sont vendues par des fabricantsdifférents sous des appellations commer-ciales distinctes. (A lui seul, le propra-nolol a été vendu en 1996 par 13 fabri-cants sous 11 noms différents.)
\
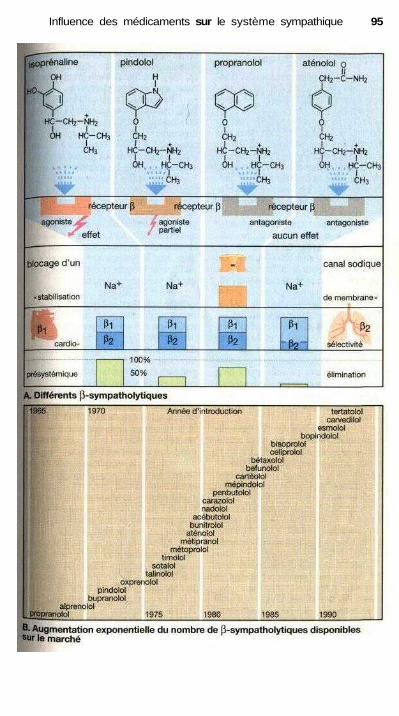
Influence des médicaments sur le système sympathique 95

96 Influence des médicaments sur le système sympathique
Antisympathotoniques
Les antisympathotomques sont des com-posés qui diminuent l'activité du sys-tème nerveux sympathique « le tonussympathique » Ils provoquent une dimi-nution de la pression artérielle (indica-tion hypertension, p 306), bien queleur utilisation en pratique soit très li-mitée du fait d'une mauvaise tolérance
La clonidine est un agoniste c^,qui à cause de sa lipophihe élevée (pré-sence de deux substituants chlore sur lenoyau phénol) traverse la barrière hé-mato-encéphalique La stimulation desrécepteurs oh-post-synaptiques inhibe lecentre vasomoteur dans la medullaoblongata de sorte qu'il accepte oumette en place une pression sanguineplus faible A côte de cela, l'activationdes récepteurs a^-présynaptiques (p 82,p 90) périphériques empêche la libéra-tion de noradrénaline (NA) A côté deson utilisation principale comme antihy-pertenseur elle peut également servirpour atténuer les symptômes de manqueen cas de dépendance aux opioldes
E f f e t s secondaires fatigue, séche-resse de la bouche , l'arrêt brutal d'untraitement par la clonidine déclenche uneffet rebond élévation de la pression ar-térielle à un niveau supérieur au niveauinitial
L'a-méthyl-DOPA (DOPA = di-hydroxyphenylalanme) sera capté active-ment, comme un acide aminé à travers labarrière hémato-encéphalique, sera décar-boxylé dans le cerveau en a-méthyl dopa-mine et finalement hydroxyle en a-methylnoradrénaline La décarboxylation de l'a-méthyl-DOPA occupe une partie de l'acti-vité de la decarboxylase, de sorte que latransformation de DOPA en doparmnesera inhibée et que finalement la quantitéde NA formée sera plus faible L'a-mé-thyl-NA, faux neurotransmetleur, peutêtre stocké, mais possède cependant encomparaison des neurotransmetteurs phy-siologiques une affinité plus élevée pourles récepteurs a^ que pour les récepteursa,, et de ce fait déclenche un effet ana-logue à celui de la clonidine.
E f f e t s secondaires : fatigue, perte
de la régulation orthostatique, symp-tôme parkinsomen extrapyramidal(p 186), réaction cutanée, trouble hépa-tique, anémie hémolytique
Réserpine : c'est un alcaloïde vé-gétal (rauwolfid), qui inhibe la capacitéde stockage des aminés biogenes (NA,dopamme = DA, sérotomne = 5 HT), enbloquant l'ATPase nécessaire à la capture La quantité de NA libérée après stimulation décroît La quantité d'adrénaline libérée par les glandes surrénales estégalement plus faible A doses plus éle-vées, on aboutit a une dégradation irreversible des vésicules de stockage (sym-pathectomie pharmacologique) dont lerenouvellement peut réclamer des jourset même des semaines La réserpine penètre dans le système nerveux central etva là aussi bloquer la capacité de stockage des aminés biogènes
E f f e t f secondaires perturbation dusystème moteur extrapyramidal avecsignes parkinsomens (p 186), sédationrepliement sur soi et dépression (mhibition du stockage des aminés biogenesdans le système nerveux central), congéstion de la muqueuse nasale (rhume réser-pimque), diminution de la libido, impuissance, augmentation de l'appétit
Guanéthidine : elle possède uneaffinité élevée pour le système de transport de la NA dans la membrane axonaleet la membrane des vésicules Elle serastockée à la place de la NA sans pouvoircependant assurer ses fonctions En plus,elle stabilise la membrane axonale defaçon à inhiber la propagation de l'exci-tation électrique à l'extrémité des nertssympathiques Le stockage et la distribu-tion de l'adrénaline des surrénales ne se-ront pas modifiés La guanethidine nepénètre pas dans le SNC
E f f e t s secondaires possibilités demontées tensionnelles à la suite d'unestimulation psychologique du patient,l'adrénaline sera libérée , l'augmenta-tion de pression liée à cette libérationpeut être particulièrement marquée, carchaque interruption de longue durée dutonus sympathique entraîne une hyper-sensibilisation de l'organe cible aux ca-técholamines.
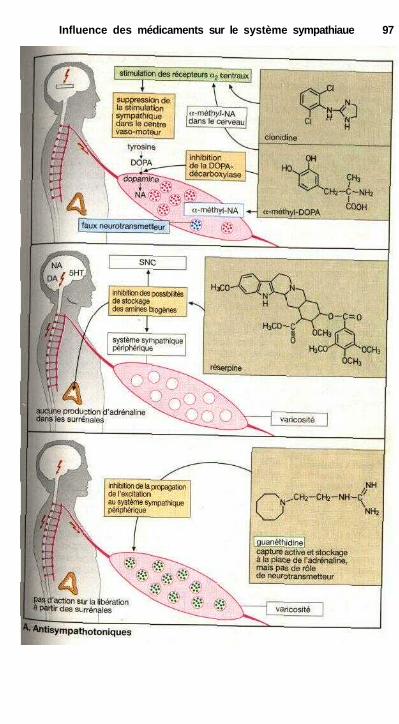
Influence des médicaments sur le système sympathiaue 97

98 Influence des médicaments sur le système parasympathique
Système nerveux parasympathique
Conséquences d'une activation para-sympathique. Le système nerveux pa-rasympathique régule des phénomènesen rapport avec l'absorption (prise denourriture, digestion, absorption) ou lestockage de l'énergie. Ces événementsse déroulent pendant la période derepos de l'organisme, et se contententd'un faible volume respiratoire(bronches rétrécies) et d'une activitécardiaque modérée. Les sécrétions desalive et de l'intestin participent à la di-gestion de la nourriture, le transport ducontenu intestinal est accéléré par suited'une augmentation des mouvementspéristaltiques et d'une diminution detonus des muscles du sphincter. Pourfavoriser la miction, la tension de laparoi de la vessie augmente et le tonusdu sphincter diminue. Une stimulationdes fibres parasympathiques provoqueun rétrécissement de la pupille et unecourbure du cristallin permettant devoir avec plus de précision les objetsproches (accomodation).
Structure du parasympathique.Les corps cellulaires des fibres para-sympathiques preganglionnaires sont
localisés dans le tronc cérébral et larégion sacrée de la moelle. Les fibresémanant du tronc cérébral cheminentpar le nerf crânien III, (nerf oculomo-teur) et le ganglion cilié jusqu'auxyeux, par le nerf VII (nerf facial) et leganglion ptérygopalatin jusqu'auxglandes lacrymales et aux glandes desfosses nasales, par le nerf IX (nerfglosso-pharyngien) et le ganglion sous-maxillaire jusqu'aux glandes salivaireset par le nerf X (nerf vague) jusqu'authorax et aux organes abdominaux,Environ 75 % de toutes les fibres para-sympathiques sont contenues dans lenerf vague. Les neurones du systèmeparasympathique sacre innervent lecôlon, le rectum, la vessie et l'extrémitéinférieure de l'urètre ainsi que les or-ganes génitaux externes.
Neuromédiateur : acétylcholine.L'acétylcholine (ACh) est la substancetransmettrice au niveau des synapsespost-ganglionnaires du parasympa-thique ainsi que des synapses ganglion-naires (du sympathique et du parasym-pathique) ou des plaques motrices(p. 180). Cependant, elle agit dans lessynapses mentionnées ci-dessus sur desrécepteurs distincts :
Localisation
Cellules innervéespar le neuroneparasympathiquede type 2
Corps cellulaire duneurone de type 2dans les ganglionssympathiques etparasympathiques
Plaque motrice,muscles striés
Agoniste
ACh,muscarine
ACh,nicotine
ACh,nicotine
Antagoniste
Atropine
Trimétaphan
d-tubocurarine
Type de récepteur
Récepteur muscari-nique couplé à uneprotéine G
Type ganglionaireRécepteurnicotinique,un canal ioniquestimulé par un ligand
Typemusculaire
L'existence de récepteurs distinctsdans les différentes synapses choliner-
giques permet une action pharmacolo-gique spécifique.
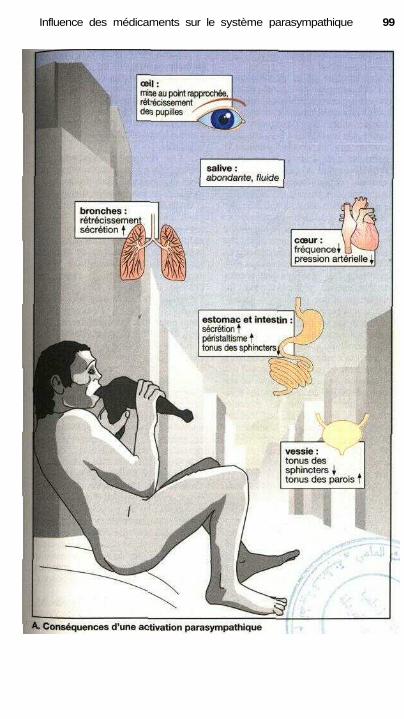
Influence des médicaments sur le système parasympathique 99

100 Influence des médicaments sur le système parasympathique
Synapse cholinergique
L'acétylcholine est le neurotransmet-teur des synapses post-ganglionnairesdes nerfs parasympathiques. Elle eststockée à concentration élevée dans lesvésicules situées dans l'axoplasme et endensité plus importante à la terminaisonnerveuse. Elle est formée à partir de lacholine et d'un acide acétique activé(acétylcoenzyme A) sous l'action del ' enzyme choline-acétyl-transférase.La choline, très polaire, est transportéede façon active dans l'axoplasme. Lesystème de transport spécifique setrouve exclusivement sur la membranedes neurones cholinergiques et les ter-minaisons nerveuses. Le mécanisme dela libération n'est pas connu dans tousses détails. Les vésicules sont ancréesaux filaments du cytosquelette par l'in-termédiaire d'une protéine, la synap-sine, ce qui permet leur accumulation àproximité de la membrane présynap-tique, mais empêche leur fusion aveccette membrane. Lors d'une stimulationdu nerf, la concentration de Ca^ dansl'axoplasme augmente provoquant unestimulation de protéine-kinases et unephosphorylation de la synapsine. Cecipermet une libération des vésiculesproches de la membrane, de leur an-crage et leur fusion avec la membraneprésynaptique. Lors de cette fusion,elles déversent leur contenu dans lafente synaptique. L'acétylcholine dif-fuse rapidement à travers la fente sy-naptique (la molécule d'acétylcholine aune taille un peu supérieure à 0,5 nm, lafente synaptique est large d'environ 30-40 nm). Sur la membrane post-synap-tique qui est aussi la membrane du tissucible, elle se fixe sur des récepteurs.Ces récepteurs peuvent aussi être sti-mulés par un alcaloïde la muscarine : ils'agit de récepteurs muscariniques(récepteurs cholinergiques de typeM). Au contraire, l'action de l'acétyl-choline sur les récepteurs de synapsesganglionnaires (p. 98) et de la plaquemotrice est reproduite par la nicotine :récepteurs nicotiniques de type N.
Après libération dans la fente sy-naptique, l'acétycholine sera hydro-lysée très rapidement et inactivée parune enzyme spécifique l'acétylcholi-nestérase, présente dans la fente, et pardes cholinestérases sériques moinsspécifiques (butyryl-cholinestérase), ensolution dans le sérum ou le liquideinterstitiel.
Les récepteurs muscariniquespeuvent être répartis en plusieurs sous-types en fonction de leur structure mo-léculaire, de leur mode de transductionet de l'affinité respective de divers li-gands. Les récepteurs M), M^ et M3 sontreprésentés ici. Les récepteurs M, setrouvent dans les cellules nerveuses,par exemple les ganglions, où leur acti-vation facilite la transmission d'uneexcitation du neurone de type 1 auneurone de type 2. Les effets del'acétylcholine sur le cœur sont médiéspar des récepteurs M; : l'ouverture d'uncanal potassique conduit à un allonge-ment de la dépolarisation diastolique età une diminution de la fréquence car-diaque. Les récepteurs Mg jouent unrôle dans le tonus des muscles lisses,par exemple de l'intestin et desbronches. Leur stimulation déclencheune activation de la phospholipase C,une dépolarisation de la membrane etune élévation du tonus musculaire. Lesrécepteurs My sont également présentsdans des cellules endocrines, dont lafonction va être stimulée après activa-tion là encore d'une phospholipase C.Dans le tissu cérébral, on met en évi-dence ces trois types de récepteurs mus-cariniques, où ils participent à l'acti-vation de nombreuses fonctions :excitabilité corticale, mémoire, appren-tissage, traitement des signaux doulou-reux et contrôle de l'activité dans letronc cérébral. Ces activités cependantne peuvent pas être attribuées à un sous-type de récepteur unique.
Une activation des récepteurs M,dans 1' endothélium vasculaire peutaboutir à la libération de monoxyded'azote (N0) et provoquer'de façon in-directe une vasodilatation (p. 120).
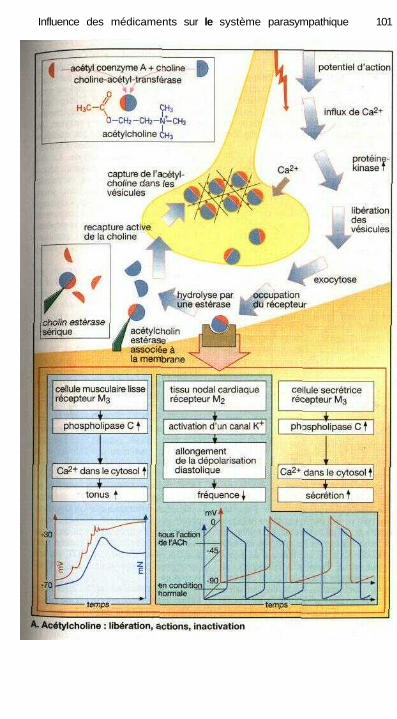
Influence des médicaments sur le système parasympathique 101

102 Influence des médicaments sur le système parasympathique
Parasympathomimétiques
L'acétylcholine (ACh) elle-même neiieutêtre utilisée en thérapeutique en raison leson hydrolyse et de son inactivation iresrapide par l'acétyleholinestérase (AQE),son action peut être imitée de façon di-recte ou indirecte par les parasympatho-mimétiques.
Parasympathomimétiques (i.rects. L'ester de choline carbachol sti-mule le récepteur muscarinique (para-sympathomimétique direct), mais n'estcependant pas dégradé par l'acétyleholi-nestérase. Le carbachol peut donc êtreactif en administration locale sur l'igii(glaucome) ou en administration systé-mique (atonie intestinale ou vésicale).
La pilocarpine, un alcaloïde (dePilocarpus jaborandis) et Yarécoline (deAreca catechu, la noix de Bétel) agissentcomme des parasympathomimétiques di-rects ; ils exercent également par la pré-sence d'une aminé tertiaire une actioncentrale. L'effet central des substancesmuscariniques consiste en une stimula-tion légère et vivifiante, qui est probable-ment l'effet recherché par ceux qui masti-quent la noix de Bétel en Asie du Sud-ggt.Cependant, seule la pilocarpine a une uti^lisation thérapeutique et uniquement enapplication locale, dans le glaucome.
Parasympathomimétiques indi.rects. L'acétyleholinestérase (AChE), en-zyme locale, peut être inhibée de façonsélective. La conséquence de cette inhibi-tion est une élévation de la concentrationd'acétylcholine au niveau des récepteursdans les synapses cholinergiques, le mé-diateur endogène est donc disponible pluslongtemps. Les inhibiteurs de l'enzymesont ainsi des parasympathomimétiquesindirects. L'inhibition de l'enzyme estsensible dans toutes les synapses où l'acé-tylcholine remplit une fonction de neuro-transmetteur. Parmi ces inhibiteurs, ontrouve soit des esters de l'acide carbanii-nique (carbamates, tels \siphysostigmine,la néostigmine) ou les esters de l'acidephosphorique (organophosphates tels lepa.rwx.on = E 600 dérivé de la nitrostis-mine = E 605).
Les substances appartenant à cesdeux groupes de drogue réagissent
comme l'acétylcholine avec l'acétyleholi-nestérase. Elles peuvent être captées parl'enzyme comme des faux substrats,L'ester sous forme d'un complexe avecl'enzyme sera alors attaqué. L'étape limi-tante dans l'hydrolyse de l'acétylcholineest la désacétylation de l'enzyme, unévénement qui ne réclame que quelquesmillisecondes ce qui permet l'activitéélevée de l'acétyleholinestérase. La dé-carbaminoyiation de l'enzyme, néces-saire après hydrolyse d'un carbamaterequiert des heures voire des jours.L'enzyme reste inactive aussi longtempsqu'elle est carbaminoyiée. La coupure durésidu phosphate, c'est-à-dire la déphos-phorylation de l'enzyme est pratique-ment impossible, dans ce cas l'enzyme estbloquée irréversiblement.
Application. La néostigmine, uncarbamate quaternaire, sera utiliséecomme parasympathomimétique indirectdans le cas d'une atonie intestinale ou vé-sicale post-opératoire. A côté de cela, ellesera également employée pour compenserl'insuffisance relative en acétylcholine auniveau de la plaque motrice dans les casde myasthénie grave ou bien pour rac-courcir l'action myorelaxante de sub-stances non dépolarisantes (p. 182) (levéi,de la curarisation avant l'arrêt d'une anes-thésie). La pyridostigmine sera utilisée dela même façon. La physostigmine, un car-bamate tertiaire pénétrant dans le cerveau,peut être utilisée comme antidote lorsd'un empoisonnement par l'atropine oudes substances voisines, car elle atteint
. aussi l'acétyleholinestérase dans le SNC.La néostigmine a une action locale au ni-veau de l'œil pour le traitement des glau-comes. Les carbamates et les organophos-phorés peuvent aussi être utilisés commeinsecticides. Ils se caractérisent certes parune toxicité élevée pour l'homme maisaussi, si on les compare au DDT, par unedécomposition chimique rapide après pul-vérisation.
La tacrine n'est pas un ester et in-terfère uniquement avec le site de liaisonde la choline sur l'enzyme. Elle peut êtreutilisée au cours de la maladied'Alzheimer dans l'espoir d'atténuer lessymptômes de démence.
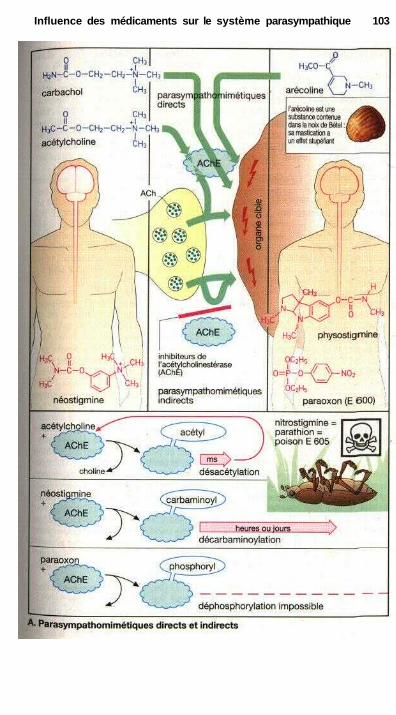
Influence des médicaments sur le système parasympathique 103

104 Influence des médicaments sur le système parasympathique
Parasympatholytiques
La stimulation du parasympathique pro-voque au niveau de la synapse entre unneurone de type 2 et les cellules de l'or-gane cible, une libération d'acétylcholinedont les effets sont représentés sur la fi-gure ci-contre (flèches bleues). Certainesde ces actions parasympathomimétiquwseront utilisées sur le plan thérapeutique(p.102).
Les substances qui agissent commedes antagonistes au niveau des récepteursmuscariniques s'appellent des parasym-pathniytiques (exemple type, l'atropine,un alcaloïde, dont l'action est soulignéeen rouge sur la figure ci-contre).
Leur utilisation thérapeutique estrendue difficile par une mauvaise spécifi-cité d'organe. Pour obtenir un effet précis,il faut :- une application locale- choisir des substances les plus aptes à
traverser la membrane- utiliser des produits spécifiques d'un
sous-type de récepteur.Les parasympatholytiques peuvent êtreutiisés sur le plan thérapeutique :1. Inhibition des sécrétions glandu-laires :Inhibition des sécrétions bronchiques. Laprémédication par l'atropine avant uneanesthésie par inhalation freine une pos-sible hypersécrétion de mucus bron-chique, qui ne pourrait pas être expectoréau cours de l'anesthésie.
Blocage des sécrétions acides del'estomac avec la pirenzépine. La stimu-lation des sécrétions acides de l'estomacpar l'ACh est médiée par un sous-type derécepteurs muscariniques, les récepteursM| (p. 164). La pirenzépine présente uneaffinité supérieure pour les récepteurs M,que pour les autres récepteurs muscari-niques (p. 100). Les cellules pariétales quiproduisent les sécrétions acides possèdentessentiellement des récepteurs M,. Les ré-cepteurs M, ont été mis en évidence en-dehors de la paroi de l'estomac et en par-ticulier dans le cerveau. Cependant lapirenzépine n'y exerce aucune action carelle n'est pas suffisamment lipophile pourtraverser la barrière hémato-encépha-lique. Elle sera utilisée pour le traitement
des ulcères de l'estomac et du duo-dénum (p.164).2. Relaxation de la musculature lisse :Bronchodilatation dans le cas d'une élé-vation de résistance des voies respira-toires (asthme bronchique, bronchitechronique obstructive) en utilisantl'ipratropium, un parasympatholytique.En inhalation cet ammonium quaternairen'affecte pratiquement par les autres or-ganes car son absorption est faible.
Spasmolyse dans le cas de coliquesnéphrétiques ou biliaires avec la N-bu-tyiscopolamine (p. 126). Comme c'est unammonium quaternaire, il ne pénètre pasdans le SNC, et doit être administré parvoie parentérale. La N-butylscopolammea une action spasmolytique particulière-ment marquée, parce qu'elle peut à la foisbloquer les ganglions et relâcher directe-ment les muscles.
Diminution du tonus des muscles del'iris et élargissement de la pupille parl'utilisation locale d'homatropine ou detropicamide (mydriase), de façon à pou-voir examiner le fond de l'œil. Pour effec-tuer un diagnostic, il n'y a besoin qued'une dilatation relativement brève de lapupille. Par comparaison avec l'atropine(dont l'effet peut durer des jours), l'effetdes substances citées plus haut s'estomperapidement.3. Accélération de l'activité du cœur :L'ipratropium sera utilisé pour augmenterla fréquence cardiaque en cas de brady-cardie ou pour élever le seuil d'excitationdans le cas d'un bloc auriculo-ventricu-laire. En tant qu'ammonium quaternaire,cette substance ne pénètre pas dans le cer-veau, ce qui diminue le danger d'une alté-ration du système nerveux central (voirplus loin). Il est également mal absorbé auniveau intestinal (coefficient d'absorption< 30 %) ; pour obtenir un niveau plasma-tique suffisant, il doit être nettement plusdosé que pour l'administration paren-térale.
L'administration d'atropine permetd'éviter un arrêt cardiaque réflexe,comme il peut s'en produire après une sti-mulation du nerf vague par exemple lorsde l'induction d'une anesthésie, d'un la-vage d'estomac ou d'un examen endosco-pique.
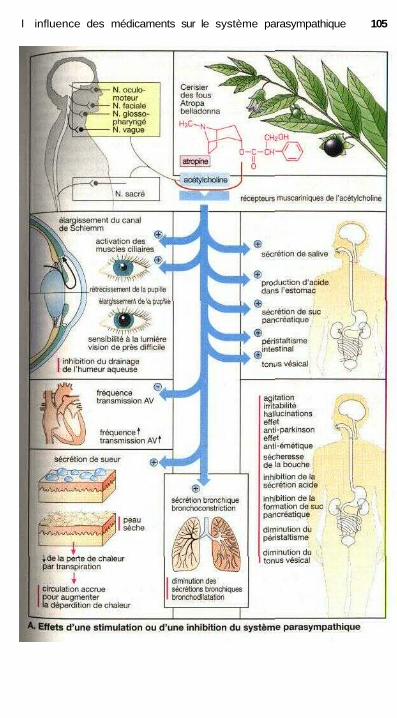
l influence des médicaments sur le système parasympathique 105

106 Influence des médicaments sur le système parasympathique
4. Sédation du système nerveux cen-tral :La scopolamine (éventuellement sousforme d'un emplâtre transcutané) estutilisée dans la prophylaxie d'une kiné-tose (mal des transports, mal de mer,p. 324). La scopolamine (pK,, = 7,2) tra-verse la barrière hémato-encéphaliqueplus vite que l'atropine (pK,, = 9), carune fraction plus importante de la molé-cule reste sous forme non chargée, ca-pable de traverser la membrane.
Sédation dans les états agitésavec la scopolamine qui, au contraire del'atropine, a une action sédative dont onpeut tirer parti avantageusement enl'administrant comme prémédicationd'une anesthésie.
Atténuation des symptômes de lamaladie de Parkinson, qui est liée àune prépondérance relative de l'acétyl-choline dans le corps strié, en utilisantpar exemple la benzatropine (p. 186).Les anticholmergiques utilisés commeagents antiparkinsoniens traversent ai-sément la barrière hémato-encépha-lique. Pour une même action centrale,les effets périphériques sont moins mar-qués que dans le cas de l'atropine.Contre-indications à l'usagedes parasympatholytiquesGlaucome : en effet, le relâchement desmuscles des sphincters de la pupillebloque l'écoulement de l'humeuraqueuse et augmente la pression intra-oculaire.
Problèmes de miction dans le casd'un adénome prostatique, car le relâ-chement des muscles de la vessie, réglépar le parasympathique, aggrave lesdifficultés.
Empoisonnement par l'atro-pine. Les parasympatholytiques se ca-ractérisent par une fenêtre thérapeu-tique importante. Les rares casd'empoisonnement par l'atropine, sus-ceptibles de mettre la vie en danger,sont reconnaissables par les effets péri-phériques ou centraux suivants :
E f f e t s périphériques : tachycar-die, sécheresse de la bouche ; éléva-tion de la température du corps (hyper-
thermie) due à une inhibition de latranspiration. L'excitation se transmetégalement aux glandes sudoripares cho-linergiques, bien qu'elles soient inner-vées par le système sympathiqueL'inhibition de la sécrétion de la sueurenlève à l'organisme la possibilitéd'éliminer la chaleur produite au coursdes réactions du métabolisme, en vapo-risant la sueur (chaleur de vaporisationp. 200). En compensation, se produitune dilatation des vaisseaux de la peaudestinée à favoriser le dégagement dechaleur par une augmentation du débitsanguin cutané, rougissement de lapeau. La conséquence d'un blocage dupéristaltisme intestinal est une consti-pation.
E f f e t s centraux : agitation motricequi peut s'amplifier jusqu'à la folie fu-rieuse, altérations psychiques, halluci-nations et état de confusion (enAllemagne, le nom de la plante dontprovient l'atropine est le cerisier desfous).
Les vieillards sont particulière-ment sensibles aux effets centraux d'unempoisonnement. Rappelons-nous legrand nombre de substances possédasdes effets secondaires atropiniques :antidépresseurs tncycliques, neurolep-tiques, antihistaminiques, anti-aryth-miques, anti-parkinsoniens.
Le traitement d'un empoisonne-ment grave par l'atropine com-porte, à côté des mesures symptoma-tiques générales (lavage d'estomac,diminution de la température pardes bains froids) l'administrationd'un parasympathomimétique indirect(physostigmine, p. 102) qui passedans le SNC contrairement à la néo-stigmine.
Les empoisonnements par l'atro-pine peuvent survenir par exemple chezdes enfants ayant avalé les baies de labelladone (cerisier des fous) ou bienlors d'une absorption excessive d'anti-dépresseurs tricycliques (tentative desuicide).
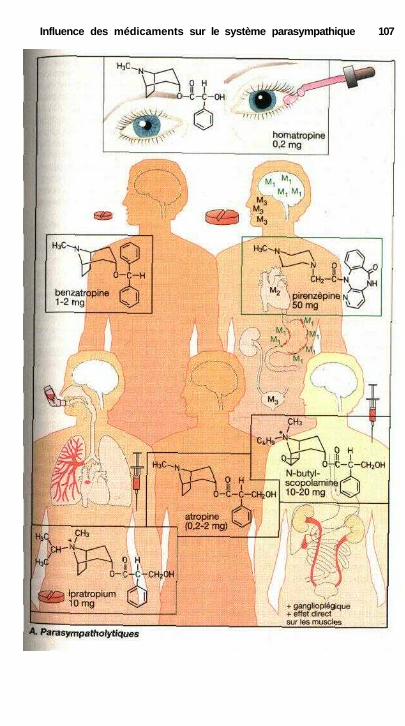
Influence des médicaments sur le système parasympathique 107

108 Nicotine
Transmission ganglionnaire
Un nerf végétatif efférent, qu'il soitsympathique ou parasympathique, secompose en principe de deux neuronesdisposés l'un à la suite de l'autre. Lepoint de contact (synapse) entre le neu-rone 1 et le neurone 2 est situé dans unganglion, c'est pourquoi on parlerapour les neurones 1 et 2 de neurone pré-ou post-ganglionnaire. L'excitationélectrique (potentiel d'action) du pre-mier neurone entraîne la libérationd'acétylcholme (ACh) dans le gan-glion. L'acétylcholine stimule des ré-cepteurs présents, sur la membrane duneurone 2, dans la région synaptique.La stimulation de ces récepteurs ouvreles canaux ioniques non spécifiquesprésents dans le récepteur (p. 64), detelle sorte que le potentiel de membranedécroît. Si une quantité suffisante deces récepteurs est stimulée en mêmetemps, on atteint un seuil de potentielauquel se déclenche une dépolarisationrapide, qui provoque ensuite un poten-tiel d'action se propageant le long duneurone 2. En temps normal, tous lespotentiels d'action qui parviennent auneurone préganglionnaire ne génèrentpas un potentiel d'action qui se propagede nouveau dans le neurone 2. La sy-napse ganglionnaire a une fonction defiltre (A).
Au niveau des récepteurs de lamembrane neuronale situés dans la ré-gion de la synapse ganglionnaire l'effetde l'acétylcholine peut être égalementdéclenché par la nicotine : récepteursnicotiniques.
Actions de la nicotine au niveauganglionnaire. Si la nicotine est intro-duite dans l'organisme en faible quan-tité, elle stimule les récepteurs gan-glionnaires. On obtient une dépola-risation partielle mais pas la générationde potentiels d'action. A ce moment,cependant, il suffit d'une libérationd'acétylcholine plus faible que dansune circonstance normale pour déclen-cher la propagation d'un potentiel d'ac-tion. La nicotine à faible concentrationstimule la transmission ganglionnaire,
elle change la capacité de filtration duganglion, la fréquence des potentielsd'action du deuxième neurone se rap-proche de celle observée dans le neu-rone 1 (B). A concentration plus élevéela nicotine agit en bloquant le ganglion.La stimulation simultanée d'une quan-tité plus élevée de récepteurs nicoti-niques entraîne une dépolarisationmembranaire si prononcée qu'un poten-tiel d'action ne peut plus se produire,même quand se produit une libérationintensive et coordonnée d'acétylcholine(C).
La nicotine imite en effet l'actionde l'ACh au niveau des récepteurs maisavec elle il n'est pas possible d'obtenirles changements fréquents de concen-tration de l'agoniste dans la fentesynaptique qui sont nécessaires à lastimulation ganglionnaire. La concen-tration de nicotine dans la fente synap-tique ne peut augmenter aussi rapide-ment que celle d'acétylcholine aprèslibération par les terminaisons ner-veuses et la nicotine n'est pas éliminéeaussi rapidement de la fente synaptiqueque l'acétylcholine.
Les récepteurs ganglionnaires del'ACh peuvent être bloqués par le trimé-taphan (ganglioplégique) qui n'a au-cune activité intrinsèque et se comportecomme un véritable antagoniste.
L'' hexaméthonium est un ganglio-plégique ayant un autre mode d'action :il bloque le canal ionique non spéci-fique du récepteur.
Certains neurones de type 1 abou-tissent, sans avoir été relayés, à l'extré-mité de la voie nerveuse jusqu'aux cel-lules des surrénales. En terme dedéveloppement, ces cellules ont lamême origine que les corps cellulairesde neurones sympathiques post-synap-tiques. La stimulation d'un neurone detype 1 entraîne aussi dans les glandessurrénales une libération d'acétylcho-line qui induira dans les cellules une sé-crétion d'adrénaline dans le sang (D)De faibles doses de nicotine qui indui-sent seulement une dépolarisation par-tielle, provoquent maigre tout une libé-ration d'adrénaline (p. 110, p. 112).
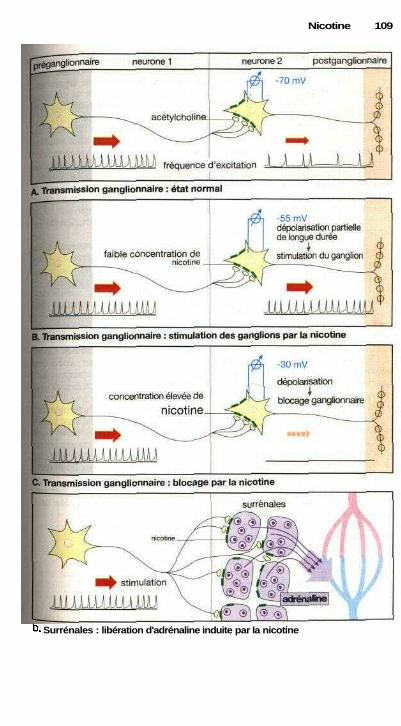
Nicotine 109
D- Surrénales : libération d'adrénaline induite par la nicotine

110 Nicotine
Actions de la nicotine sur les fonc-tions de l'organisme
L'alcaloïde du tabac, la nicotine, estcapable à faible concentration de dé-clencher via une stimulation des récep-teurs nicotiniques de l'acétylcholineune dépolarisation partielle au niveaudes ganglions : stimulation ganglion-naire (p. 108). La nicotine exerce uneaction de même type dans de nombreuxterritoires nerveux. Ces différentes ac-tions sont examinées de plus près ci-dessous selon la structure concernée.
Ganglions végétatifs. La stimula-tion des ganglions touche aussi bien lapartie sympathique que la partie para-sympathique du système nerveux végé-tatif. L'activation du parasympathiqueest visible au niveau de l'estomac parune augmentation des sécrétions (inter-diction de fumer en cas d'ulcère), et parune élévation de l'activité de l'intestin(« effet laxatif » de la première ciga-rette matinale ; défécation ; diarrhéechez les « débutants »).
La tendance à une diminution dela fréquence cardiaque, médiée par leparasympathique, sera contrebalancéepar une stimulation simultanée du sym-pathique et des surrénales.
La stimulation des nerfs sympa-thiques entraîne par suite de la sécrétionde noradrénaline une vaso-constriction,la résistance périphérique augmente.
Glandes surrénales. La libérationd'adrénaline a en premier lieu un effetsur la circulation : élévations de la/re-quence cardiaque et de la résistancepériphérique. D'un autre côté, on noteégalement une action sur le métabo-lisme : par la dégradation du glycogèneet la libération d'acides gras sont mis enplace des substrats favorables à la pro-duction d'énergie. La sensation de faimest abolie. L'état métabolique répondpar une activation de l'organisme à un« stress silencieux ».
Barorécepteurs. La dépolarisa-tion partielle des barorécepteurs leurpermet déjà de réagir à une augmenta-tion relativement faible de la pressionsanguine par une réduction de l'activitésympathique.
Post-hypophyse. La libération devasopressine (ADH) a un effet antidiu-rétique (p. 162) ; l'effet vasoconstric-teur est seulement sensible pour desconcentrations d'hormone très élevées.
Glomus carotidien. La sensibilitéde la réponse à une augmentation de laconcentration de CO; augmente et apour conséquence une élévation de lafréquence respiratoire.
Récepteurs à la pression, à latempérature ou à la douleur. La sen-sibilité aux stimuli correspondants estaccrue.
Area postrema. La sensibilisa-tion des chémorécepteurs entraîne uneexcitation des centres du vomissement.
La nicotine peut aussi, à faibleconcentration, augmenter l'excitabilitéau niveau des plaques motrices. Cetteaction peut se manifester chez lesgrands fumeurs par des crampes, parexemple des muscles du mollet, et uneraideur musculaire.
L'action centrale de la nicotine nepeut pas être attribuée à une zone parti-culière du cerveau. La nicotine aug-mente la vigilance et la concentration.Cet effet peut être décrit comme une ca-pacité accrue d'appréhender les événe-ments extérieurs et de réagir.
En raison de la multiplicité de seseffets, la nicotine ne peut pas être uti-lisée à des fins thérapeutiques.
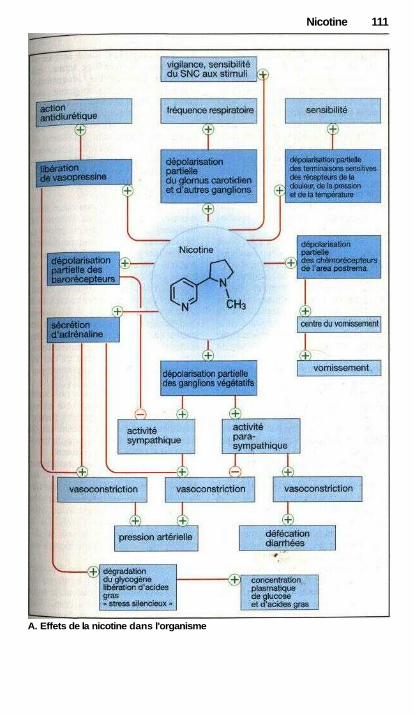
Nicotine 111
A. Effets de la nicotine dans l'organisme

112 Nicotine
Conséquences du tabagisme
Les feuilles séchées et fermentées deNicotiana tabacum, une plante de la fa-mille des solanacées, sont désignéessous le nom de tabac. Le tabac est es-sentiellement fumé, plus rarement priséou chiqué. Au moment où le tabac seconsume, se forment en quantité détec-table, environ 4 000 substances, si bienque l'absorption par le fumeur dépendnon seulement de la qualité du tabac etde la présence d'un filtre mais aussi dela vitesse avec laquelle il se consume(température du foyer) et de la profon-deur de l'inhalation.
Le tabac contient de 0,2 à 5 % denicotine. Dans la fumée du tabac sontégalement dispersées des particules degoudron. La nicotine est absorbée trèsrapidement dans les bronches et lespoumons (environ 8 secondes après lapremière inhalation on peut mettre enévidence la présence de nicotine dans lecerveau). La concentration plasmatiquede nicotine après une cigarette atteintun niveau d'environ 25-50 ng/ml pourlequel peuvent se produire les effets dé-crits p. ll0. La concentration de nico-tine dans le plasma décroît dès la fin dela cigarette par suite d'une distributiontrès rapide, l'élimination finale s'ef-fectue avec une demi-vie d'environ2 heures. La nicotine est dégradée paroxydation.
Il est vraisemblable que l'augmen-tation du risque cardio-vasculaire ob-servée chez les fumeurs est une consé-quence de l'action chronique de lanicotine : maladies coronaires (entreautre infarctus), altérations centrales(apoplexie) ou périphériques (membresinférieurs) de la circulation sanguine.Le rôle de la nicotine comme un desfacteurs favorables au développementd'une athérosclérose est encore discuté.Elle augmente par le biais d'une libéra-tion d'adrénaline la concentration deglucose et d'acides gras libres, sans queces substrats énergétiques soient immé-
diatement nécessaires à une activité del'organisme. A plus long terme, elleaugmente l'agrégabilité plaquettaireabaisse l'activité fibrinolytique san',guine et favorise la coagulation.
Ce n'est pas seulement la nicotinemais aussi l'ensemble des autres sub-'stances contenues dans la fumée dutabac qui sont responsables des consé-quences de la tabagie. Parmi ces sub-stances, quelques-unes possèdent defaçon démontrable des propriétés can-cérigènes.
Les particules de poussière inha-lées avec la fumée du tabac doivent êtreéliminées du tractus respiratoire enmême temps que le mucus recouvrantl'épithélium cilié. Cependant l'activitédes cils vibratiles est inhibée par lafumée : le transport mucociliaire est at-teint. Ceci favorise une infection bacté-rienne et constitue une des causes de labronchite chronique, qui se développechez les fumeurs réguliers (toux du fu-meur). La lésion chronique de la mu-queuse bronchique peut être une causeimportante du risque accru qu'ont lesfumeurs de déclarer un carcinome pul-;monaire.
Des études statistiques ont établila relation impressionnante qui existeentre le nombre des cigarettes fuméesquotidiennement et l'augmentation durisque de mourir d'un infarctus du myo-carde ou d'un cancer du poumon.
D'un autre côté, les statistiquesmontrent aussi que les risques d'in-farctus ou d'un autre accident cardio-vasculaire, tombent à un niveau prochede ceux des non-fumeurs dans un délaide cinq à dix ans après l'arrêt. De lamême façon, le danger de voir se dé-clencher un carcinome bronchique s'es-tompe.
L'arrêt brutal chez les fumeursn'est pas associé à des symptômes desevrage importants. En général, le sujetse plaint d'une nervosité accrue, d'unmanque de concentration et d'une prisede poids.
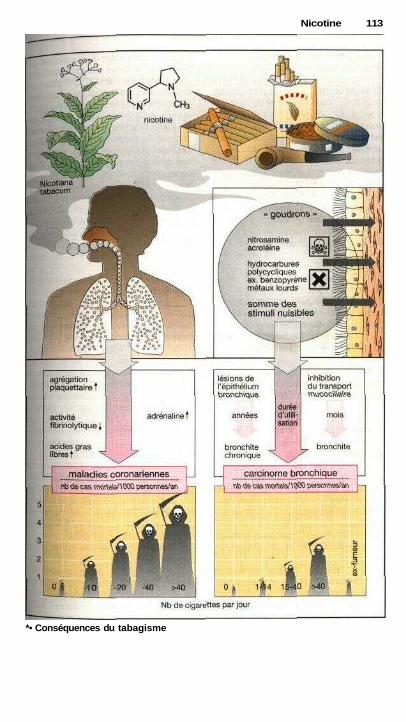
Nicotine 113
*• Conséquences du tabagisme

114 Aminés biogènes
Aminés biogènes - Actions et rôlespharmacologiques
Dopamine (A). La dopamine précur-seur de la noradrénaline et de l'adréna-line (p. 82), est présente dans les neu-rones sympathiques et les glandessurrénales. Dans le système nerveuxcentral, la dopamine joue un rôle deneurotransmetteur : elle module dans lestriatum l'activité motnce extrapyrami-dale (p. 186), gouverne dans l'areapostrema l'envie de vomir (p. 324), in-hibe dans l'anté-hypophyse la libéra-tion de prolactine (p. 240).
Il existe plusieurs sous-types derécepteurs de la dopamine, couplés àune protéine G. D'un point de vue thé-rapeutique on distingue les récep-teurs D, (sous-types D| et D,) et les ré-cepteurs D; (sous-types D;, D3 et D4).Les effets sur le SNC passent par la sti-mulation des récepteurs D,. Après uneperfusion de dopamine, la stimulationD[ provoque une dilatation des artèresrénales et mésentérique (utile dans lesétats de choc). A concentration plusélevée on observe des effets cardiaquesdus à une stimulation des récepteurs P|puis une vasoconstriction par stimula-tion a.
Ne pas confondre la dopamine etla dobutamine qui stimule les récep-teurs a et p mais pas les récepteurs do-paminergiques (p. 62).
Analogues de la dopamine.L'administration de L-DOPA, un pré-curseur de la dopamine, augmente lasynthèse endogène de celle-ci (p. 186,maladie de Parkinson). La bromocrip-tine stimule les récepteurs D^ (indica-tion : maladie de Parkinson, blocagede la prolactine en cas d'aménorrhée ;acromégalie, p. 240). Les effets secon-daires classiques de ces substancessont des nausées et des vomissements.Les neuroleptiques (p. 234) et la méto-clopramide (p. 324) agissent commedes antagonistes dopaminergiques. Laréserpine, un antihypertenseur, et l'a-méthyl- DOPA (p. 96) bloquent égale-ment ces récepteurs. Ces molécules in-
hibitrices entraînent fréquemment desaltérations motrices extrapyramidales.
Histamine (B). L'histamine eststockée dans les mastocytes circulantsou tissulaires et joue un rôle dans les ré-actions inflammatoires et allergiques(p. 320). Elle provoque bronchocons-triction, augmentation du péristaltisrneintestinal, vasodilatation et augmenta-tion de la perméabilité capillaire. Dansla muqueuse gastrique, elle peut être li-bérée à partir de cellules proches descellules entérochromaffines et stimulerla sécrétion acide. Dans le SNC, l'hista-mine joue également un rôle de neuro-transmetteur. Il existe deux types derécepteurs importants sur le plan théra-peutique, les récepteurs H| et H; impli-qués dans les actions vasculaires del'histamine et couplés à une protéine G.n existe aussi des récepteurs H,.
Antagonistes. Les antihistami-niques H, bloquent aussi d'autresrécepteurs (récepteurs muscariniques,récepteurs dopaminergiques) et sontutilisés comme agents anti-allergiques(par ex. bamipme, chlorphénoxamme,démostil, phéniramine, dimétindène) ;comme antiémétique (méclozme,dimenhydrate, p. 324) ; comme somni-fère sans ordonnance (par ex. diphen-hydramine, p. 220). La prométhazineconstitue l'intermédiaire vers les neuro-leptiques du type phénothiazine(p. 234). Principaux effets secondaires :fatigue (diminution de l'attention auvolant !), effets de type atropinique(constipation, sécheresse de la bouche).Les antihistaminiques H^ (cimétidine,famotidine, ranitidine) inhibent la sé-crétion acide de l'estomac (traitementde l'ulcère peptique, p. 166).
Inhibiteurs de la libération d'his-tamine. Le cromoglycate et le nédo-cromil stabilisent les mastocytes et blo-quent la libération d'histamine (p. 320).Ils seront appliqués localement.
Quelques anti-H, peuvent aussibloquer la libération d'histamine parles mastocytes : l'oxatomide et le kéto-tifène seront utilisés par voie systé-mique.
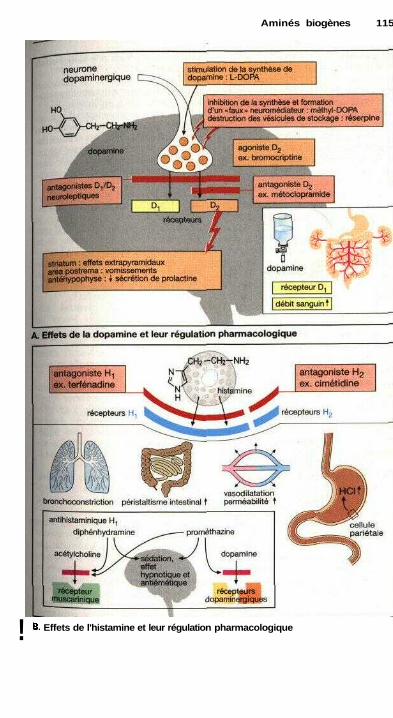
Aminés biogènes 115
! B. Effets de l'histamine et leur régulation pharmacologique

116 Aminés biogènes
Sérotonine
Sérotonine (5-hydroxy-tryptamine, 5HT).Origine. La 5HT est synthétisée à partirdu L-tryptophane dans les cellules enté-rochromaffines de l'épithélium intes-tinal. La sérotomne est égalementformée et joue un rôle dans les cellulesnerveuses du plexus mésentérique et dusystème nerveux central. Les plaquettessanguines ne sont pas capables de syn-thétiser la sérotonine, mais elles peuventla capter et la stocker.
Récepteurs de la sérotomne. Onpeut distinguer plusieurs sous-types derécepteurs, selon leurs propriétés biochi-miques et pharmacologiques. Les plusimportants sur le plan pharmaco-théra-peutique sont les récepteurs 5HTi et5HT; ainsi que les sous-types 5HÏ3 et5HÏ4 dans certains cas. La plupart de cestypes de récepteurs sont couplés à uneprotéine G. Le sous-type 5ïfT^ contientun canal ionique non sélectif (p. 64,canal ionique activé par un ligand).
E f f e t s de la sérotonine. Systèmecardiovasculaire. Les effets de la séro-tonine sur le système cardiovasculairesont complexes car elle peut déclencherdes effets différents voire opposés enagissant via des récepteurs distincts endes sites différents. Elle exerce parexemple un effet vasoconstricteur directvia les récepteurs 5HT, sur les cellulesmusculaires lisses. Elle peut égalementindirectement et de plusieurs façons di-later les vaisseaux et diminuer la pres-sion artérielle. Elle peut par l'intermé-diaire des récepteurs 5îfî^ bloquer lesneurones sympathiques périphériquesou ceux du tronc cérébral et faire dimi-nuer le tonus sympathique. Dans l'endo-thélium vasculaire et via les récepteurs5HTi, elle stimule la libération de mé-diateurs vasodilatateurs (EDRP, p. 120 ;prostacycline, p. 148). La sérotonine li-bérée par les plaquettes participe à laformation du thrombus, à l'hémostase età l'apparition d'une hypertension gravi-dique.
La kétansérine est un antihyperten-seur qui agit comme antagoniste des ré-cepteurs 5HT;. On peut cependant se de-mander si son action hypertensive est
due à ce blocage, car elle bloque aussiles récepteurs a.
Le sumatriptan est un traitementde la migraine, qui agit comme agonistedes récepteurs 5HTio (p. 316).
Tractus gastro-intestinal. La sé-rotonine provenant des neurones duplexus mésentérique ou des cellules en-térochromaffines agit sur la motilité in-testinale et les sécrétions de fluide dansl'intestin par l'intermédiaire des récep-teurs 5HÎ4.
Le cisapride est un produit qui sti-mule la motilité de l'estomac, de l'in-testin grêle et du gros intestin. On lenomme aussi agent procinétique. Il serautilisé dans les cas d'altérations de lamotilité gastro-intestinale (reflux gastro-œsophagien par ex.). Son mécanismed'action n'est pas complètement établimais passe vraisemblablement par unestimulation des récepteurs 5ÏTT,.
Système nerveux central. Lesneurones sérotoninergiques jouent unrôle dans plusieurs fonctions du systèmenerveux central, comme on peut lemettre en évidence en analysant l'actionde plusieurs substances interférant avecla sérotonine.
La fluoxétine inhibe la recaptureneuronale de la sérotonine libérée et agitcomme antidépresseur. Elle a un effetexcitant assez fort et participe, dans legroupe des antidépresseurs, aux traite-ments de deuxième intention. Un de seseffets annexes est également une dimi-nution de l'appétit.
La buspirone est une moléculeanxiolytique ; la stimulation des récep-teurs 5HTn centraux semble jouer unrôle important dans son action.
L' ondansétron présente un effetmarqué contre les nausées et vomisse-ments accompagnant un traitement parles cytostatiques. C'est un antagonistedu récepteur WTy Le tropisetron et legranisetron ont une action équivalente.
Les agents psychédéliques (LSD)et psychomnnétiques (par exemple mes-caline, psilocybine) peuvent provoquerun changement du niveau de conscience,des hallucinations et des manifestationsd'angoisse, probablement sous l'in-fluence des récepteurs 5HT.
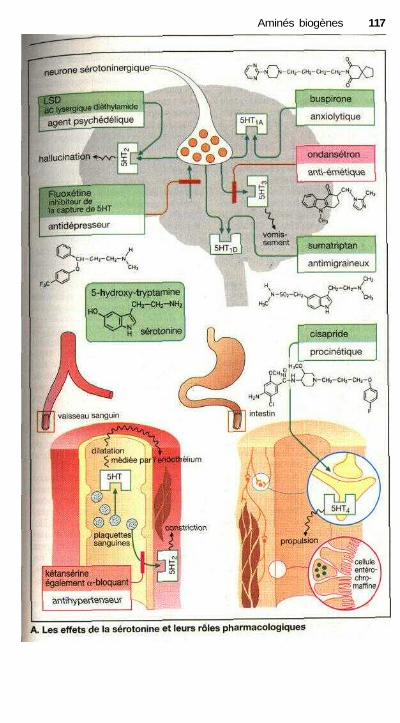
Aminés biogènes 117

118 Vasodilatateurs
Vasodilatateurs : vue d'ensemble
La taille des vaisseaux régule la distribu-tion du sang dans la circulation. Le dia-mètre du lit vasculaire veineux condi-tionne l'apport sanguin au cœur,c'est-à-dire le volume d'éjection et ledébit cardiaque. La taille des artèresconditionne la résistance périphérique.Résistance périphérique et débit car-diaque sont deux paramètres cruciauxpour la pression artérielle (p. 308).
Les Vasodilatateurs les plus impor-tants sur le plan thérapeutique sont pré-sentés en A ; l'ordre correspond à peuprès à la fréquence d'emploi. Certains deces produits exercent une activité diffé-rente dans les territoires veineux ou arté-riels de la circulation (largeur des co-lonnes).
Utilisations possibles. Vaso-dilatateurs des territoires artériels :diminution de la pression en cas d'hyper-tension (p. 306), réduction du travail car-diaque dans l'angine de poitrine (p. 304),diminution de la résistance à l'éjectiondans l'insuffisance cardiaque (p. 132).Vasodilatateurs des territoires veineux :diminution de l'apport de sang au cœurdans l'angine de poitrine (p. 304) oul'insuffisance cardiaque (p. 132).L'utilisation thérapeutique réelle seradonnée pour chacun des groupes de sub-stances.
Mise en œuvre d'une contre-régu-lation lors d'une chute de pression arté-rielle provoquée par les Vasodilatateurs(B). L'activation du système sympathiqueproduit dans l'organisme une augmenta-tion de la pression artérielle par l'intermé-diaire d'une augmentation de la fréquencecardiaque (tachycardie réflexe) ou dudébit cardiaque. Les patients remarquent«les battements du cœur». L'activationdu système rénine-angiotensine-aldosté-rone (RAA) aboutit à une augmentationdu volume sanguin et par là également àcelle du débit cardiaque.
Les phénomènes de contre-régula-tion peuvent être inhibés pharmacologi-quement (p-bloquants, inhibiteurs del'enzyme de conversion, diurétiques).
Mécanismes d'action. Le tonus desmuscles lisses vasculaires peut être di-
minué de différentes manières. Dans lecas de signaux stimulants comme l'angio-tensine II ou la noradrénaline, on utiliserades inhibiteurs de l'enzyme de conversionou des antagonistes a. Les analogues de laprostacycline comme l'iloprost, ou de laprostaglandine E] comme l'alprostadilreproduisent l'action de médiateurs vaso-'dilatateurs. Les antagonistes calciques,qui bloquent l'influx calcique dépolai-bsant, et les activateurs des canaux potas-siques qui stimulent l'efflux potassiquehyperpolarisant agissent au niveau desprotéines-canal. Les nitrates organiqueslibérant du monxyde d'azote influencentle métabolisme cellulaire.
Différents Vasodilatateurs.Seront évoqués par la suite, les nitrates(p. 120), les antagonistes calciques(p. 122), les antagonistes a, (p. 90) et lenitroprussiate de sodium (p. 120).
La dihydralazine et le minoxidil(plus exactement un métabolite associéà un sulfate) dilatent les artérioles etseront utilisés pour le traitement del'hypertension. Etant donné les possibi-lités de contre-régulation de l'orga-nisme ils ne conviennent pas à une mo-nothérapie. Le mécanisme d'action dela dihydralazine est mal connu, le mi-noxidil stimule l'ouverture de canauxpotassiques. Les principaux effets se-condaires sont pour l'hydralazine l'ap-parition d'un lupus érythémateux etpour le minoxidil le développement dela pilosité. En application locale, il peutaider les chauves.
Après administration intraveineusede diazoxide on obtient essentiellementune dilatation des artérioles ; ce produitpeut être utilisé lors de poussées d'hyper-tension. Par voie orale, on observe égale-ment une inhibition de la sécrétion d'insu-line, de sorte que le diazoxide peutégalement être utilisé dans le cas de tu-meurs du pancréas sécrétant de l'insuline.Ces deux effets sont médiés par une acti-vation de canaux potassiques.
Parmi les Vasodilatateurs, oncompte également une méthylxanthine, lathéophylline (p. 320), un inhibiteur dephosphodiestérase, l'amrinone (p. 132), laprostacycline (p. 148) et les dérivés del'acide nicotinique (p. 154).
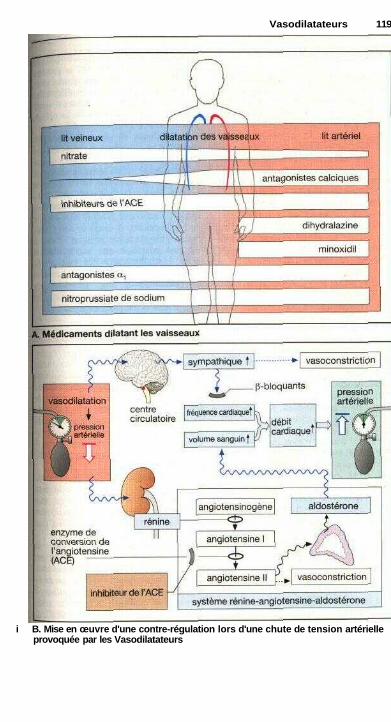
Vasodilatateurs 119
i B. Mise en œuvre d'une contre-régulation lors d'une chute de tension artérielleprovoquée par les Vasodilatateurs

120 Vasodilatateurs
Nitrates organiques
Différents esters de l'acide nitrique(HNO.)) avec des polyalcools agissent enrelaxant les muscles lisses, ce sont parexemple le trinitrate de glycérol ou le di-nitrate d'isosorbide. Leur e f f e t est plusmarqué dans le lit vasculaire veineuxque dans les territoires artériels.
On utilise sur le plan thérapeutiqueles conséquences de ces effets vascu-laires au niveau du cœur. La diminutionde l'apport de sang veineux et de la ré-sistance artérielle soulage le cœur (dimi-nution de la pré et de la post-charge,p. 304). De ce fait, le bilan en oxygènes'améliore. Le rétrécissement spasmo-dique des principales artères coronaires(spasme coronaire) est bloqué.
Indication. Principalement l'an-gine de poitrine (p. 302), plus rarementune forme sévère d'insuffisance car-diaque chronique ou aiguë. L'adminis-tration régulière de doses élevées abou-tissant à des niveaux sanguins constantsdiminue l'efficacité du traitement parsuite d'une accoutumance de l'orga-nisme : augmentation de la tolérance. La« tolérance au nitrate » peut être évitéesi on ménage chaque jour une périodesans nitrate par exemple la nuit.
Effets indésirables. Au début dutraitement se manifestent souvent desmaux de têtes dus à la dilatation desvaisseaux dans la région du crâne. Ceteffet s'estompe également par suited'une accoutumance malgré la mise enplace d'un intervalle sans nitrate. Pourdes doses plus élevées surviennent deschutes de tension, tachycardie réflexe etcollapsus.
Mécanisme d'action. La diminu-tion du tonus des cellules musculaireslisses vasculaires dépend d'une activa-tion de la guanylate cyclase et d'uneaugmentation de la concentration deGMP cyclique. Cette activation est due àla libération de monoxyde d'azote. N0peut être produit comme un médiateurphysiologique par les cellules endothé-liales et influencer les cellules muscu-laires lisses voisines (endothelium de-rived relaxing factor, EDRF). Lesnitrates emprunteraient ainsi une voie
déjà établie ce qui explique leur activiyélevée. La libération de N0 se produitdans les muscles lisses vasculaires avecutilisation de groupements sulfhydriles(SH) ; la « tolérance aux nitrates » seraitdue à un appauvrissement de la celluleen donneurs de groupements SH.
Trinitrate de glycérol (nitrogiv.cérine). Il se caractérise par une capacitéélevée à traverser les membranes et unefaible stabilité ; c'est le médicament dechoix pour le traitement de l'angine depoitrine. Pour cela, il est placé sur la mu-queuse buccale (comprimé sécablespray) ; l'action se produit en l'espacede 1 à 2 minutes. A cause de son élimi-nation présystémique presque totale, ilest mal adapté à une administrationorale. L'administration par voie trans-dermique (sous forme de timbre) permetde contourner le foie. Le dinitrated'isosorbide traverse facilement lesmembranes et est plus stable que la ni-troglycérine. Il sera converti en partie en5-mononitrate d'isosorbide dont l'actionest plus faible mais aussi plus longue. Ledinitrate d'isosorbide peut être égale-ment administre par voie sublinguale,mais sa forme principale d'administra-tion est la forme orale dont le but est uneaction de plus longue durée. Comptetenu de sa polarité élevée et de sa faiblevitesse d'absorption le mononitrated'isosorbide ne permet pas une admi-nistration sublinguale. Par voie orale, ilsera bien absorbé et ne subira pas d'éli-mination présystémique.
Molsidomine. Elle est inactive parelle-même. Après prise orale, elle seratransformée dans l'organisme en unesusbtance active. Dans ces conditions,on a moins à craindre l'apparition d'une« tolérance aux nitrates ».
Nitroprussiate de sodium. Ilcontient un groupement N0 mais n'estpas un ester. Il relaxe de la même ma-nière les lits vasculaires veineux ou arté-riels. Il peut être utilisé sous surveillanceétroite pour maintenir la pression arté-rielle à une valeur constante et contrôlée.Le thiosulfate de sodium peut servir à in-activer les groupements cyanures libéréspar le nitroprussiate (p. 300).
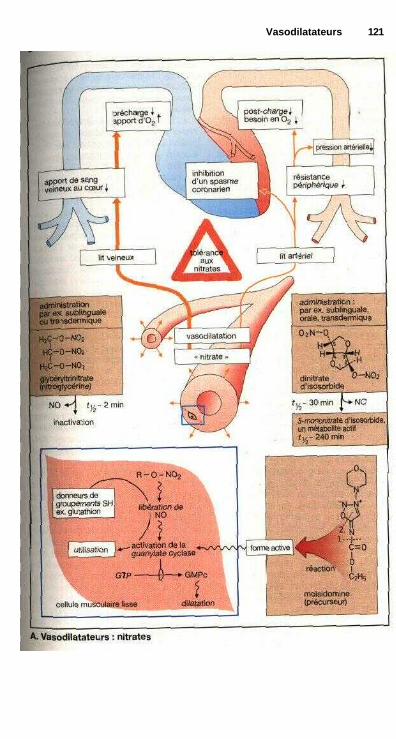
Vasodilatateurs 121

122 Vasodilatateurs
Antagonistes calciques
Lors d'une stimulation électrique de lamembrane des cellules du muscle car-diaque ou bien des cellules de musclelisse se produisent différents flux ioniqueset entre autres un influx de calcium. Sontconsidérées comme des antagonistes cal-ciques les substances qui inhibent l'influxde calcium et seulement faiblement lesautres flux ioniques comme par exemplel'influx de Na+ ou l'efflux de K\ On lesappelle aussi bloqueurs des canaux cal-ciques ou inhibiteurs de l'influx calcique.Les antagonistes calciques utilisés sur leplan thérapeutique peuvent être divisés endeux groupes selon leur action sur le cœuret les vaisseaux.
I. Les dérivés des dihydropyri-dines. Les dihydropyridines, par exemplela nifédipine, sont des substances hydro-phobes, non chargées. Elles produisent enparticulier une relaxation des muscleslisses vasculaires du lit artériel. Auxconcentrations thérapeutiques, ne se ma-nifeste pratiquement aucune action car-diaque (dans des expériences pharmaco-logiques sur des préparations de musclecardiaque isolé se déclenche une actioncardiaque avérée). Ces molécules se sontimposées dans le domaine thérapeutiqueen tant qu''antagoniste's calciques vaso-sélectifs. La conséquence d'un relâche-ment des résistances vasculaires est unediminution de la pression artérielle. Auniveau cardiaque, la post-charge diminue(p. 302) avec par conséquent une réduc-tion du besoin en oxygène. Les spasmesdes artères coronaires sont bloqués.
Les indications de la nifédipinesont V angine de poitrine (p. 304) et l'hy-pertension (p. 306). En ce qui concernel'angine de poitrine, elle convient nonseulement pour la prophylaxie mais aussipour le traitement des crises. Les effetssecondaires sont : battements de cœur(tachycardie réflexe liée à la chute de lapression artérielle), maux de tête, oedèmedes membres inférieurs.
Les substances énumérées ci-des-sous ont en principe les mêmes effets :
La nitrendipine, Yisradipine et la/e-lodipine servent également au traitementde l'hypertension. La nicardipine et la ni-
soldipine seront utilisées pour le traite-ment de l'angine de poitrine. La nimodi-pine est administrée en cas d'hémorragiesous-arachnoïde pour éviter les vasos-pasmes.
II. Vérapamil et autres antago-nistes calciques cationiques et amphi-philes. Le vérapamil contient un atomed'azote chargé positivement dans lagamme des pH physiologiques etconstitue ainsi une molécule cationiqueamphiphile. Il agit chez les malades nonseulement en bloquant les muscles tissesvasculaires mais aussi le muscle car-diaque. Dans le cœur, un influx de cal-cium est important pour la dépolarisationdu nœud sinusal (formation de l'excita-tion électrique), dans le nœud auriculo-ventriculaire (propagation de l'excitationdes oreillettes aux ventricules) ainsi quepour le myocarde (couplage électroméca-nique). Le vérapamil agit donc comme unchronotrope négatif, un inotrope et undromotrope négatifs.
Indications. Le vérapamil est utilisécomme anti-arythmique dans les tachya-rythmies de type supra-ventriculaire. Encas de fibrillation ou de troubles durythme des oreillettes ((lutter auriculaire),il peut diminuer grâce à son action sur laconduction auriculo-ventriculaire la fré-quence au niveau du ventricule. Le véra-pamil est aussi utilisé pour la prophylaxiedes crises d'angine de poitrine (p. 304)ainsi que comme antihypertenseur(p.306).
Effets secondaires. A cause de soneffet sur le nœud sinusal, la diminution dela pression artérielle ne sera pas associée àune tachycardie réflexe ; la fréquence nechange pas, ou on observe même une bra-dycardie. Un bloc auriculo-ventriculaireet une insuffisance cardiaque peuvent semanifester. Les patients se plaignent sou-vent de constipation car le vérapamil in-hibe aussi les muscles lisses de l'intestin.
Gallopamil (méthoxy-vérapamil). Ilest très proche du vérapamil à la fois parsa structure et par son effet biologique.
Diltiazem. C'est un dérivé desbenzodiazépines, cationique et amphi-phile, caractérisé par un spectre d'ac-tion très proche de celui du vérapamil.
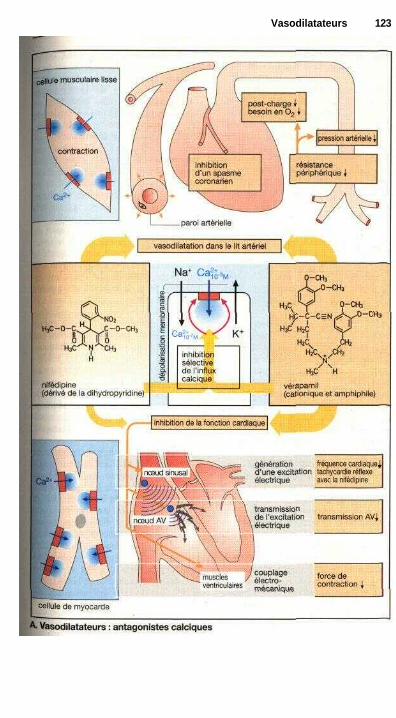
Vasodilatateurs 123

124 Inhibiteurs du système rénine-angiotensine-aldostérone
Inhibiteurs de l'enzymede conversion
L'enzyme de conversion de l'angioten-sine (ACE) appartient au système decontrôle de la pression artérielle, le sys-tème rénine-angiotensine-aldostérone.La rénine est sécrétée par des cellulesde l'appareil juxtaglomérulaire du né-phron, qui jouent un rôle importantdans le contrôle des fonctions du né-phron. La sécrétion de rénine est sti-mulée par une diminution de la pressionde perfusion, une diminution de laconcentration de NaCl dans l'orga-nisme et une stimulation sympa-thique p. La rénine est une glycopro-téine qui clive l'angiotensinogènecirculant dans le sang pour libérer undécapeptide, l'angiotensine I. L'enzymede conversion transforme ce décapep-tide en angiotensine II, biologiquementactive.
L'enzyme de conversion est unepeptidase non spécifique, capable decliver un dipeptide à l'extrémité C-ter-minale de différents peptides. Elle pro-voque par exemple l'inactivation de labradykinine. Cette enzyme est égale-ment présente dans le plasma mais c'estla forme située sur la face luminale descellules endothéliales qui contribue à laformation à'angiotensine I I . Cet octo-peptide contribue à l'élévation de lapression artérielle : 1. vasoconstrictiondans la partie artérielle, mais aussiveineuse du réseau sanguin ; 2. f de lasécrétion d'aldostérone et donc de laréabsorption d'eau et de sodium —> fdu volume sanguin ; 3. î du tonussympathique central, stimulation péri-phérique de la sécrétion et de l'actionde la noradrénaline.
Inhibiteurs de l'ACE. Ces inhi-biteurs (captopril, énalapril) sont defaux substrats qui occupent le site actifde l'enzyme. L'énalapril dont l'affinitépour l'enzyme est plus importante estplus actif et agit plus longtemps que lecaptopril.
Indications : hypertension, insuf-fisance cardiaque. La diminution de lapression artérielle est due essentielle-ment à l'inhibition de la formationd'angiotensine II, mais peut être enpartie due au blocage de la dégradationdes kinines (action vasodilatatrice)Dans le cas d'une insuffisance car-diaque, la résistance à l'éjection car-diaque diminue par suite d'une baissedes résistances périphériques ; diminu-tion de la sécrétion d'aldostérone et dutonus des veines capacitives —» dimi-nution de l'apport veineux ; la stase vei-neuse en amont du cœur disparaît.
Effets indésirables. La plupart dutemps, les inhibiteurs de l'ACE se révè-lent comme des médicaments actifs etbien tolérés. On note fréquemment unetoux sèche, probablement due à une di-minution de la dégradation des kininesdans la muqueuse bronchique. Dans cer-tains cas où le système rénine-angioten-sine-aldostérone est déjà activé (perted'eau et de sels après un traitement pardes diurétiques, insuffisance cardiaque,stérose de l'artère rénale), les inhibi-teurs de l'ACE peuvent au début du trai-tement provoquer une chute de pressiontrop importante. Autres inhibiteurs del'ACE mis sur le marché ou en dévelop-pement : lisinopril, périndopril, rami-pril, fosinopril, benazépril, cilazapnl,trandolapril.
Antagonistes des récepteurs del'angiotensine II, II existe deux sous-types de récepteurs de l'angioten-sine II : les récepteurs AT| qui médientles effets connus de l'angiotensine etles récepteurs AT;, dont le rôle resteobscur. Le losartan est un antagonistedes récepteurs AT, utilisé pour le trai-tement de l'hypertension. Son actionprincipale et ses effets secondairessont semblables à ceux des inhibiteursde l'ACE, à l'exception de la toux (pasde blocage de la dégradation des ki-nines).
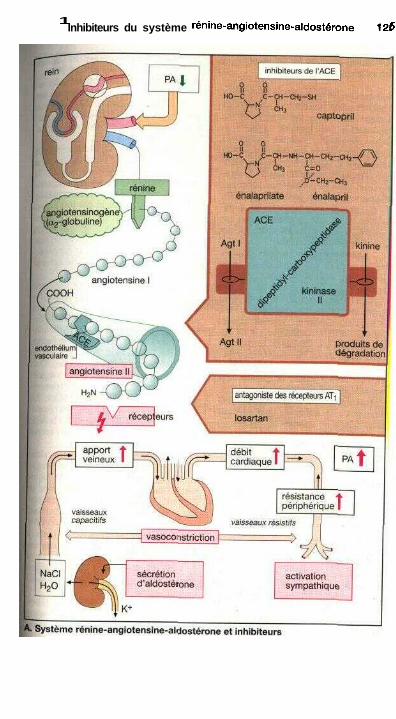
1Inhibiteurs du système rénine-angiotensine-aldostérone 126

126 Médicaments actifs sur les muscles lisses
Substances agissant sur les organesmusculaires lisses
Substances bronchodilatatrices. Unecontraction des bronches augmente la ré-sistance des voies respiratoires, commepar exemple dans l'asthme ou les bron-chites spastiques. Quelques substancesdont les propriétés sont décrites plus endétail dans d'autres chapitres sont utili-sées comme bronchodilatateurs '. la théo-phylline (une méthylxanthine, adminis-trée par voie orale ou parentérale, p. 320),les ^-sympathomimétiques (p. 84, en in-halation ou par voie parentérale) ainsi queYipratropium, un parasympatholytique(p. 104 et 107 ; en inhalation).
Spasmolytiques. Dans les crampesdouloureuses du canal cholédoque ou del'urètre, on utilisera la N-butylscopola-mine (p. 104). Compte tenu de son ab-sorption faible (présence d'un ammoniumquaternaire, proportion absorbée < 10 %),on doit l'administrer par voie parentérale.Comme l'effet thérapeutique est en gé-néral faible, on administre souvent enmême temps un analgésique puissant, parexemple un opiolde tel la péthidine. Il fautnoter que dans beaucoup de spasmes de lamusculature intestinale les nitrates orga-niques (par exemple en cas de colique hé-patique) ou la nifédipine (par exempledans l'achalasie : spasmes de l'œsophage)sont également actifs.
Substances bloquant les contrac-tions utérines (tocolyse). Les flysympa-thomimétiques, comme par exemple le fé-notérol conviennent en cas de menaced'accouchement prématuré ou bien en casde complications dangereuses en coursd'accouchement, qui rendraient néces-saire une césarienne, de façon à inter-rompre les contractions (administrationparentérale ou parfois orale). Le principaleffet secondaire est une tachycardie (ré-flexe, en raison de la dilatation médiée parles récepteurs (3;, mais également via unestimulation des récepteurs P| cardiaques).
Substances déclenchant l'accou-chement L'ocyfocme, hormone sécrétéepar la post-hypophyse (p. 240), sera utiliséeen premier lieu, par voie parentérale, (ouégalement nasale ou buccale) pendant ouaprès la naissance, pour déclencher ou ren-
forcer les contractions utérines. En utilisantcertaines prostaglandines (p. 194, PGF, •dinoprost, PGE; : dmoprostone, sulpros-tone) on peut provoquer à tout moment descontractions rythmiques de l'utérus et unedilatation du col. Elles servent essentielle-ment à l'interruption de grossesse (applica-tion locale ou parentérale).
Alcaloïdes de l'ergot de seigle. Cesont des substances synthétisées parSecale comutum (ergot du seigle), laforme végétative d'un des champignonsparasites des céréales. L'alimentationavec une fanne contenant des épis conta-minés a provoqué autrefois des empoison-nements de masse (ergotisme) avec destroubles circulatoires et des pertes de sen-sibilité des pieds et des mains (gangrène)ainsi que des troubles du système nerveuxcentral (hallucinations).
Les alcaloïdes de l'ergot de seiglecontiennent des acides lysergiques (la for-mule en A montre un amide). Ils agissentsur la musculature de l'utérus et des vais-seaux. L' ergométrine agit plus particuliè-rement sur l'utérus. Elle déclenche facile-ment une contraction prolongée de lamusculature utérine (tétanie utérine). Ceciréduit de façon dangereuse le flux sanguinparvenant au placenta et donc l'approvi-sionnement en oxygène de l'enfant. Ledérivé semi-synthétique méthylergomé-trine ne sera donc utilisé qu'après la déli-vrance lorsque les contractions de l'utérussont insuffisantes.
L'ergotamme ainsi que les alca-loïdes ergotoxines (ergocristme, ergo-cryptine, ergoconnne) agissent de façonprépondérante sur les vaisseaux. Selon lediamètre des vaisseaux, on pourra ob-server une contraction ou une dilatation.Le mécanisme d'action est mal connu.L'effet agoniste partiel sur les récepteursa peut être important. L'ergotamine estutilisée pour le traitement des migraines(p. 316). Son dérivé la dihydroergotaminesera en plus administré pour les malaisesorthostatiques (p. 308).
D'autres dérivés de l'acide lyier-gique sont la méthysergide un antagonistesérotoninergique, la bromocnptine unagoniste dopaminergique (p. 114) et lecomposé hallucinogène acide lysergiquediéthylamide (LSD, p. 238).
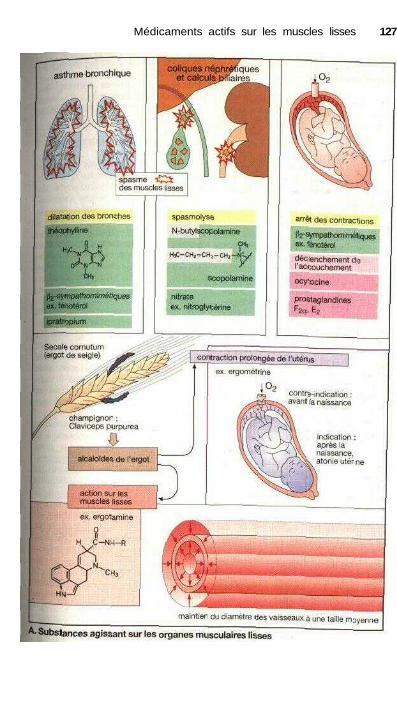
Médicaments actifs sur les muscles lisses 127

128 Médicaments actifs sur le cœur
Vue d'ensemble sur les possibilitésde moduler la fonction cardiaque (A)
1. Le travail cardiaque est régulé parl'activité des systèmes sympathique etparasympathique (p. 84, p. 105). Il estdonc possible d'exercer une influencesur les fonctions du cœur à l'aide demolécules actives sur le système végé-tatif. C'est ainsi que certains anxio-lytiques du type benzodiazépines(p. 224), par exemple le diazépam, se-ront utilisés en cas d'infarctus du myo-carde pour empêcher une activation dusympathique due à l'angoisse et doncune augmentation du travail cardiaque.Sous l'influence des antisympathoto-niques (p. 96) utilisés pour diminuerune tension artérielle élevée, le travaildu cœur décroît. Les ganglioplégiques(p. 108) étaient autrefois utilisés en casde crise hypertensive. Les parasympa-tholytiques (p. 104) ou les (3-blo-quants (p. 92) inhibent la transmissionde la stimulation végétative aux cellulesdu muscle cardiaque en bloquant les ré-cepteurs correspondants.
2. Un cœur isolé et de ce fait sé-paré de son innervation végétative,continue à battre pendant des heures sion lui apporte des solutions nutritivesvia l'aorte et les artères pulmonaires(préparation de Langendorff). Sur unetelle préparation, seules les moléculesagissant directement sur les cellulescardiaques peuvent exercer une in-fluence sur la force des contractions ouleur fréquence. Les parasympathomi-métiques et les sympathomimétiquesagissent au niveau des récepteurs pourles neurotransmetteurs des nerfs végé-tatifs. De même, les sites d'actions desglycosides cardiaques (Na-K ATPase,p. 130), des antagonistes calciques (lescanaux calciques, p. 122) ainsi queceux des substances à action anesthé-sique locale, bloquant les canaux so-diques (p. 134, p. 202) sont situés sur lamembrane plasmique. La cible des sub-stances bloquant la phosphodiesté-rase est intracellulaire (par ex. l'amri-none,p.132).
3. Il faut également mentionner lapossibilité d'agir sur la fonction car-
diaque, dans le cas d'une angine de poi-trine (p. 302) ou d'une insuffisancecardiaque (p. 132) en utilisant dessubstances vasodilatatrices qui vontdiminuer l'apport de sang veineux et/oula résistance périphérique.
Le cycle cardiaque : contractionet relaxation (B)Le signal de la contraction est un po-tentiel d'action émis par les nœuds si-nusaux (PA). La dépolarisation duplasmalemme déclenche une augmen-tation brutale de la concentration decalcium cytosolique, qui provoque àson tour le raccourcissement des fila-ments contractiles (couplage électro-mécanique). La valeur de la concentra-tion de calcium atteinte conditionnel'importance du raccourcissement,c'est-à-dire la force de la contractionLes sources de calcium sont : a) le cal-cium extracellulaire qui pénètre dans lacellule par l'ouverture de canaux cal-ciques ; h) le calcium stocké dans lescavités du réticulum endoplasmique ,c) le calcium lié sur la face interne dela membrane plasmique. La membraneplasmique, par de nombreuses invagi-nations, pénètre profondément dans lescellules du muscle cardiaque (tubulestransverses).
Le signal de la relaxation est leretour du potentiel de membrane à lavaleur de repos. Au cours de cettephase de repolarisation, la concentra-tion de calcium tombe au-dessous duseuil d'activation des myofilaments(3 x 10-'M) : les sites de liaison de lamembrane plasmique peuvent à nou-veau fixer le calcium, le réticulum ré-accumule le calcium à l'intérieur deses cavités ; les Ca-ATPases présentesdans la membrane plasmique transpor-tent, en utilisant de l'énergie, le cal-cium entré dans la cellule pendant lasystole vers l'espace extracellulaire.En plus, intervient un transporteur(carrier capable d'utiliser l'énergiepotentielle liée au gradient transmem-branaire de Na'1' : il transporte en effethors de la cellule un ion Ca^ enéchange d'un ion Na* entrant (échangeNa^/Ca^).
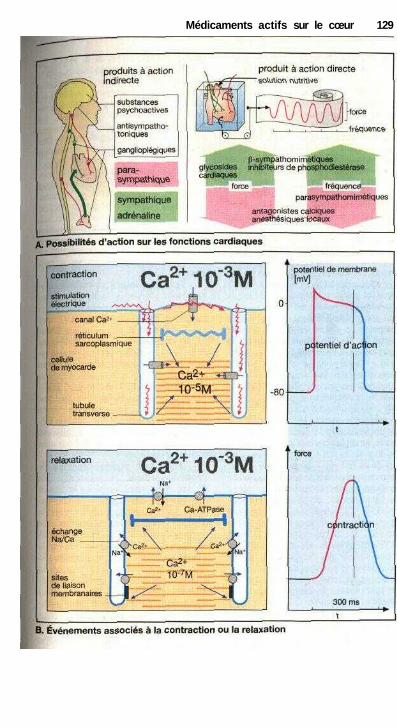
Médicaments actifs sur le cœur 129

130 Médicaments actifs sur le cœur
Glycosides cardiaques
On peut extraire de certaines plantes (A)des molécules contenant une fraction glu-cidique liée à un noyau stéroïde (formulep. 133) qui augmentent la contraction dumuscle cardiaque (B) : glycosides cardio-toniques, cardio^téroïdes ou digitaliques.
La fenêtre thérapeutique de ces mo-lécules est très étroite : le dépassement dela dose augmentant la contraction car-diaque peut aboutir à un empoisonne-ment : arythmie et contracture (B).
Mécanisme d'action. Les glyco-sides cardiaques (GC) se lient, sur la faceexterne, aux ATPases Na-K dépendantesdes cellules du muscle cardiaque et blo-quent leur activité. Ces enzymes expul-sent de la cellule les ions Na+ qui y ont pé-nétré et font entrer de nouveau les ions K*qui en étaient sortis ; elles maintiennentainsi les gradients ioniques (Na4^ et K^, lepotentiel de repos négatif de la membraneet l'excitabilité électrique de la cellule. Enprésence de glycosides cardiaques, unepartie des Na-K ATPases est inhibée, lesenzymes libres peuvent assurer un trans-port « normal » du Na-^ et du K* par uneaugmentation de leur activité. Le stimulusactivateur est une élévation de quelquesmM de la concentration intracellulaire deNa+ (concentration normale environ7 mM). On observe simultanément uneaugmentation de la quantité de Ca^ li-bérée durant la systole (Ça2* de couplage)et donc de la force de contraction. Ceciest dû au fait que l'augmentation du Na+
intracellulaire entraîne une diminution dugradient transmembranaire de Na+ qui estla force motrice de l'échange Na-VCa^(p. 128). La conséquence est donc uneaugmentation du contenu en calcium de lacellule. Si la proportion des Na-KATPases bloquées est trop importante,l'homéostasie des échanges Na^ estdéréglée et le potentiel de membrane di-minue -» apparition d''arythmies. Le Ça2*en excès bloque la relaxation pendant ladiastole : contracture.
Les actions des glycosides car-diaques dans le système nerveux centralsont également liées à une occupation dela Na-K ATPase (C). La stimulation dunerf vague entraîne une diminution de la
fréquence cardiaque et de la vitesse deconduction auriculoventriculaire (AV).Chez un patient souffrant d'une insuffi-sance cardiaque, l'amélioration de la si-tuation circulatoire contribue aussi à la ré-duction de la fréquence cardiaque. Lastimulation de l'area postrema provoquedes nausées et des vomissements. Destroubles de la vision des couleurs peuventse manifester.
Les indications des glycosides car-diaques sont : 1. l ' insuffisance cardiaquechronique, t. la fibrillation et le flutterauriculaire. La conséquence de l'actioninhibitrice sur la conduction AV est unediminution de la fréquence ventriculairece qui améliore l'efficacité de la contrac-tion cardiaque (D). Dans certains cas, onvoit également réapparaître un rythmesinusal.
Les symptômes d'un empoisonne-ment sont : 1. une arythmie potentielle-ment dangereuse, par exemple une brady-cardie sinusale, un bloc AV, desextrasystoles ventriculaires ou une fibril-lation ventriculaire. 2. Des altérationscentrales : vision «jaune» caractéris-tique puis par exemple fatigue, confusionet hallucinations. 3. Nausées, vomisse-ments et diarrhées. 4. Au niveau rénal :perte d'eau et de sel, qu'il ne faut pasconfondre avec l'élimination du fluidedes œdèmes provoquée par une dose thé-rapeutique. Ces liquides s'accumulentlors d'une insuffisance cardiaque par suited'un stase veineuse.
Traitement pharmacologique del'empoisonnement. Administration deK* qui, entre autres, empêche la liaisondes glycosides cardiaques ; cette adminis-tration peut cependant aussi altérer laconduction AV. Administration d'un anti-arythmique comme la phénytoïne ou la li-docaine (p. 136). Traitement oral par lacolestyramine (p. 152) pour empêcher lafixation et la résorption des digitoxines setrouvant dans l'intestin (circulation enté-rohépatique). Injection des fragmentsd'anticorps, F^, qui lient et donc inacti-vent la digoxine et la digitoxine. Ces frag-ments ont une pénétration beaucoup plusrapide dans les tissus, une élimination ré-nale et une antigénicité plus faible que lesanticorps entiers.
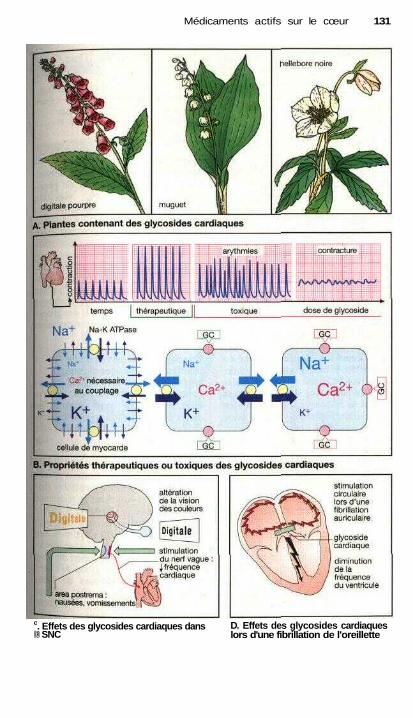
Médicaments actifs sur le cœur 131
c. Effets des glycosides cardiaques dansl® SNC
D. Effets des glycosides cardiaqueslors d'une fibrillation de l'oreillette

132 Médicaments actifs sur le cœur
La pharmacocinétique des glycosidescardiaques (A) est fonction de leur po-larité, c'est-à-dire du nombre de grou-pements hydrophiles: la g-strophantinea une capacité à traverser les mem-branes quasi nulle, celle de la digoxineest bonne et celle de la digitoxine en-core meilleure. La g-strophantine nepénètre pas dans les cellules que ce soitcelles de l'épithélium intestinal, du tu-bule rénal ou du foie. Elle convientdonc parfaitement pour l'induction parvoie intraveineuse d'un traitement parles glycosides cardiaques. L'absorptionde la digoxine dépend de sa forme gale-nique et des conditions d'absorptiondans l'intestin. La forme galénique estaujourd'hui si bien adaptée que les dé-rivés méthyldigoxine ou acétyldigoxinen'offrent plus aucun intérêt. Au niveaudu rein, la reabsorption complète n'estpas possible, environ 30 % de la quan-tité présente dans l'organisme (encoreappelée dose efficace) sont éliminés parjour. Dans le cas d'une altération de lafonction rénale survient un risque d'ac-cumulation. La digitoxine est absorbéede façon presque parfaite au niveau del'intestin et du rein. Dans le foie, ellesubit une transformation : hydrolyse dusucre, hydroxylation sur le Cl 2 (don-nant la digoxine), conjugaison parexemple à un acide glucuronique. Lesproduits de conjugaison éliminés par labile subiront un cycle entéro-hépatique(p. 38), ceux passés dans le sang serontéliminés par le rein. Dans le cas d'uneinsuffisance rénale, on n'aura pas d'ac-cumulation plus importante. En cas desurdosage, l'effet décroît après arrêt del'administration du produit mais pluslentement que pour la digoxine.
Autres molécules inotropes positivesL'amrinone, un inhibiteur de phos-phodiestérase (p. 66, élévationd'AMPc) ne peut être administré quepar voie parentérale et, à cause d'unemauvaise tolérance, que pour une duréemaximale de 14 jours. Il est réservé auxcas les plus graves d'insuffisance car-diaque. Il en est de même pour la nilri-none. L'effet inotrope positif des p-sympathomimétiques n'est qu'à peineutilisé : ils provoquent des arythmieset la sensibilité du système des récep-teurs P décroît lors d'une stimulationprolongée.
Principes de traitement d'uneinsuffisance cardiaque chroniqueLa faiblesse du muscle cardiaque en-traîne une diminution du volumed'éjection et une stase veineuse enamont du cœur, ce qui conduit à la for-mation d'un œdème. L'administrationd'un glycoside cardiaque qui a pourbut d'augmenter la force du cœur estune thérapie presque causale. L'usagedes diurétiques (thiazidiques) (p. 160)constitue une deuxième possibilité thé-rapeutique. En diminuant la résistancepériphérique et la résistance à l'éjec-tion, elle permet également une aug-mentation du volume d'éjection. Cetteaction et l'effet sur l'élimination des li-quides (diminution de l'apport de sangveineux) inhibent l'apparition de stasesveineuses. Les inhibiteurs de l'en-zyme de conversion (p. 124) agissentde façon semblable, en effet ils inhibentla formation de l'angiotensine II à pro-priété vasoconstrictrice et réduisent lasécrétion d'aldostérone qui favorise larétention des liquides.
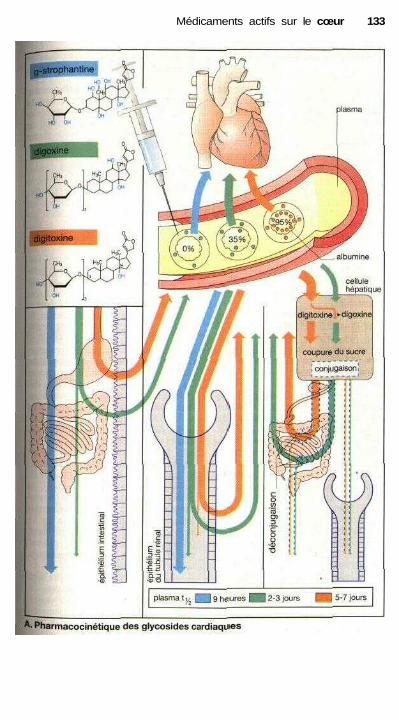
Médicaments actifs sur le cœur 133

134 Médicaments actifs sur le cœur
Traitement des arythmies cardiaques
L'impulsion électrique nécessaire à lacontraction, sous forme d'un potentield'action qui se propage (p. 136), estémise par les cellules « pacemaker » dunœud sinusal et diffuse dans le tissu car-diaque à travers l'oreillette, le nœudatrioventriculaire (AV), les différentsfaisceaux du système de transmission del'excitation jusqu'aux ventricules (A).Les irrégularités des battements du cœurpeuvent altérer de façon dangereuse sonrôle de pompe.
I. Substances ayant une influencespécifique sur les nœuds sinusaux ouatrioventriculaires. Dans certainesformes d'arythmies, on peut utiliser dessubstances qui peuvent agir spécifique-ment sur la fonction des nœuds, sinusal etatrioventriculaire, en les stimulant (flècherouge) ou au contraire en les inhibant(flèche verte).
Bradycardie sinusale. Une tropfaible fréquence d'impulsion du nœud si-nusal (< 60/minute) peut être accéléréepar un parasympatholytique. L'ipratro-pium, un ammonium quaternaire, possèdepar rapport à l'atropine l'avantage de nepas atteindre le système nerveux central(p. 107). Les sympathomimétiques agis-sent également comme des substanceschronotropes positives ; leur inconvénientest d'augmenter de façon générale l'exci-tabilité du myocarde de sorte que d'autrescellules myocardiques de l'oreillette oudes ventricules peuvent envoyer des im-pulsions supplémentaires (tendance à uneextrasystolie). Dans le cas d'une pause oud'un arrêt cardiaque Y adrénaline estutilisée pour susciter une nouvellecontraction.
Tachycardie sinusale (fréquenceau repos > 100/minute). Les ^-bloquantsinhibent l'influence sympathique et abais-sent la fréquence cardiaque.
Fibrillation ou Butter auriculaireont pour conséquence une fréquence decontraction des ventricules trop élevée quipeut être diminuée par le vérapamil(p. 122) ou les glycosides cardiaques(p. 130). Ils inhibent la conduction desimpulsions au niveau du nœud atrioven-
triculaire, de telle sorte qu'une quantitéplus faible de ces impulsions parvienne auventricule.
II. Influences non spécifiques surla genèse des excitations et sur laconduction. Dans les cas à'extrasystolessupraventriculaires et ventriculaires, detachycardie, de fibrillation ou de flutterauriculaires aussi bien que ventri-culaires, il existe des impulsions prove-nant d'autres sites que le nœud sinusal.Dans ces formes d'arythmies on utiliseracomme agents thérapeutiques ou prophy-lactiques des anesthésiques locaux oudes substances bloquant les canaux so-diques (B). Les anesthésiques locaux blo-quent l'excitation électrique des nerfssensitifs (p. 202). Un effet inhibiteur surle cœur n'est pas souhaitable (cardiodé-pression), cet effet est cependant utiledans certaines formes d'arythmies (voirplus haut). Les anesthésiques locaux sontfacilement dégradés (voir les flèches) etpeu utilisables par voie orale (procaïne. li-docaïne). A dose contrôlée, la lidocainepeut être utilisée comme antiarythmiqueen injection intraveineuse. La procaina-mide et la méxilétine, ont un métabolismeplus faible, et sont considérées commedes anesthésiques locaux et comme desexemples d'antiarythmiques à action parvoie orale. En même temps que l'effetsouhaité se manifestent des effets indési-rables. Au niveau du cœur ces antiaryth-miques non seulement inhibent l'excitabi-lité des cellules (effet bathmotropenégatif, stabilisation de membrane) maisils diminuent également la fréquence si-nusale (effet chronotrope négatif), laconduction auriculoventriculaire (dromo-tropie négative) et la force de contraction( e f f e t inotrope négatif). L'action sur lesphénomènes électriques peut également,selon un paradoxe qui n'est qu'apparent,donner lieu à des arythmies cardiaques,actions arythmogènes.
^'inhibition des fonctions des nerfsest à l'origine des effets secondaires ob-servés dans le système nerveux centralcomme par exemple vertiges, assoupisse-ment, confusion, troubles sensoriels, alté-rations motrices (tremblements, instabi-lité de la démarche, crampes).
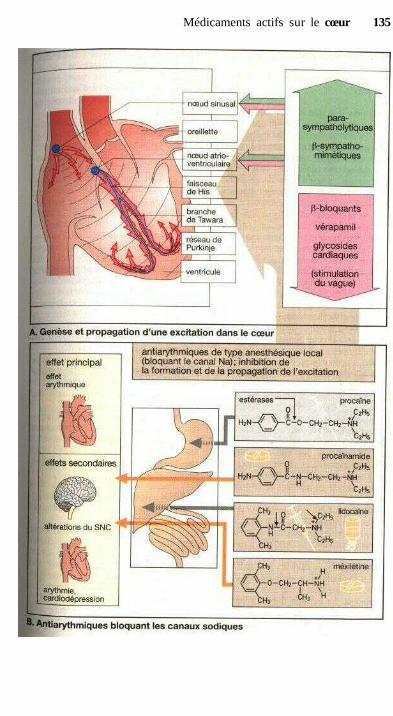
Médicaments actifs sur le cœur 135

136 Médicaments actifs sur le cœur
Propriétés électrophysiologiquesdes antiarythmiques appartenantà la famille des bloqueurs de canauxsodiques
Potentiels d'action et flux ioniques. Al'aide d'une microélectrode intracellu-laire on peut mesurer la tension électrique(le potentiel) de part et d'autre de la mem-brane d'une cellule cardiaque. Lors d'unestimulation électrique, on observe unchangement caractéristique de ce poten-tiel de membrane : le potentiel d'action.L'origine de ce potentiel est une succes-sion coordonnée de flux ioniques.Pendant la dépolarisation rapide(phase 0), on observe principalement uninflux de courte durée d'ions Na* à traversla membrane. Ensuite, la dépolarisationest maintenue par un influx temporaired'ion Ca24^ (ainsi que de Na4') (phase 2,plateau du potentiel d'action PA). Unefflux de K^ est responsable du retourdu potentiel de membrane (phase 3, repo-larisation) à la valeur de repos (phase 4).La rapidité de cette phase de dépolarisa-tion, dépend de la vitesse à laquelle le po-tentiel d'action se propage dans les cel-lules adjacentes du myocarde.
Les flux ioniques transmembra-naires s'effectuent à travers des poresprotéiques : canaux Na, Ça ou K. En (A)est résumée la façon dont l'état fonc-tionnel du canal Na varie au cours des dif-férentes phases d'un potentiel d'action.
Propriétés des antiarythmiques.Les antiarythmiques de type bloqueursdes canaux Na empêchent l'ouverture ducanal sadique au moment de la stimula-tion électrique (effet de stabilisation demembrane). Ceci peut avoir pour consé-quence (A, panneau du bas) : a) La vi-tesse de dépolansaUon diminue et par là-même l'extension de l'excitation dans lemyocarde. La propagation d'une excita-tion « parasite » devient plus difficile.b) La dépolarisation est totalement blo-quée. La formation d'une stimulation pa-thologique par exemple dans la zone bor-dant un infarctus est impossible, c) Lapériode avant la génération d'une nou-velle dépolarisation, la période réfrac-taire s'allonge. Une durée accrue du
potentiel d'action (voir ci-dessouscontribue à un allongement de ]période réfractaire avec pour conséquence une impossibilité d'une stimu-lation prématurée et donc du danger defibrillation.
Mécanisme d'action. Les anti-arythmiques, bloqueurs des canaux Nasont comme la plupart des anesthésiqu»locaux des molécules amphiphiles catio-niques (p. 206, à l'exception de la phény.toïne, p. 188). Les mécanismes molécu.laires possibles de cette action inhibitricesur la fonction du canal sodique sont ex-pliqués de façon détaillée p. 202. La faiblespécificité de structure se reflète en unefaible spécificité d'action : ce n'est passeulement la fonction du canal sadiquequi peut être altérée, mais aussi celles descanaux calciques et potassiques. Parconséquent, les antiarythmiques catio-niques amphiphiles perturbent non seule-ment la dépolarisation mais aussi la phasede repolansation. Selon la substance, ladurée du potentiel d'action pourra être al-longée (classe IA), raccourcie (classe IB)ou demeurer constante (classe IC).
Les antiarythmiques de ce typesont IA : quimdme, procainamide, aJma-line, disopyramide, propafénone ; IB : li-docaine, mexilétine, tocaimde ainsi quephénytome ; IC : flécamide.
On rangera dans la classe 111 l'amio-darone ainsi que le sotalol, un p-bloquantqui déclenchent de façon spécifique un al-longement de la durée du potentiel d'ac-tion avec une faible action sur la vitessede dépolarisation.
Notons que les (3-bloquants fontpartie de la classe II et que les antago-nistes calciques, vérapamil et diinazemconstituent la classe IV.
Utilisation thérapeutique. Comptetenu de Vétroitesse de la fenêtre théra-peutique, ces antiarythmiques ne serontutilisés que lorsque les altérations durythme sont tellement marquées qu'ellesaltèrent la fonction de pompe du cœur, oubien lorsque surviennent d'autres compli-cations. L'association de plusieurs anti-arythmiques est rare. Certains produitscomme par exemple l'amiodarone sontréservés à des cas particuliers.
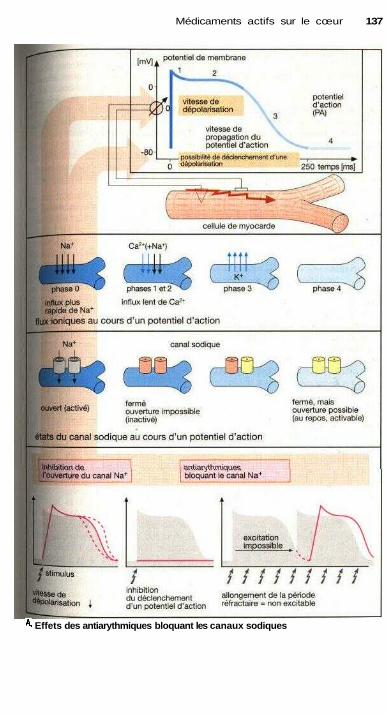
Médicaments actifs sur le cœur 137
A- Effets des antiarythmiques bloquant les canaux sodiques

138 Anti-anémiques
Traitement de l'anémie
L'anémie correspond à une diminution ducontenu du sang en globules rouges ou en-core de l'hémoglobine contenue dans cesglobules. La capacité de transport del'oxygène du sang est diminuée.
Erythropoïèse (A). Les érythrocytesse forment à partir des cellules souches parde multiples divisions. Ensuite viennentla synthèse d'hémoglobine et finalementl'expulsion du noyau cellulaire. L'érythro-poïèse est stimulée par une hormone,Yérythropoïétine, une glycoprotéine sécré-tée par les reins lorsque la pression par-tielle en C>2 du tissu diminue.
Lorsque la production d'érythro-poïétine est suffisante, une altération del'érythropoïèse peut avoir en principedeux origines.
1. L'inhibition de la multiplica-tion cellulaire liée à une synthèse d'ADNinsuffisante. Ceci peut se produire en casde carence en vitamine B,^ ou en acide-folique (anémie macrocytaire hyper-chrome). 2. La synthèse d'hémoglobineest perturbée. Ceci se produit en cas decarence enfer car l'hémoglobine contientdu fer (anémie microcytaire hypochrome).Vitamine B], (B)La vitamine B,; (cyanocobalamine) estsynthétisée par des bactéries. La vitamineB[; présente dans le gros intestin ne peutcependant pas être absorbée (voir ci-des-sous). Le foie, la viande, le poisson, lesproduits laitiers et les œufs sont riches envitamine B|;. Le besoin journalier mi-nimal est d'environ .1 y,g. Le transport dela vitamine B|; de l'intestin vers le sangnécessite un facteur appelé facteur in-trinsèque produit par les cellules parié-tales de l'estomac. Sous forme d'uncomplexe avec cette glycoprotéine, lavitamine a,, est en effet transportée parendocytose hors de l'iléum. Liée à uneprotéine de transport, la transcobalamine,la vitamine B 12 aboutit ensuite dans un or-gane de réserve, le foie, ou dans les cel-lules de l'organisme.
Origine d'une carence en vita-mine B];. Il s'agit dans la plupart des casd'une anomalie de l'absorption liée à uneatrophie gastrique avec une carence en
facteur intrinsèque. A côté d'une anémiemacrocytaire se manifestent des lésionsdes muqueuses et des altérations neurolo-giques dues à une dégénérescence de lagaine de myéline (anémie pernicieuse)La meilleure thérapeutique consiste enun apport parentéral de cyanocobala.mine ou d'hydroxycobalamine (vit. B,,remplacement du groupe CN par un OIftOn observe très rarement comme effet se-condaire une réaction d'hypersensibilité.Acide folique (B)Les légumes verts et le foie sont riches enacide folique. Le besoin minimal estd'environ 50 ^g/jour. L'acide folique sousforme polyglutamate, apporté par la nour-riture est transformé en monoglutamateavant l'absorption. L'acide folique est dé-truit par la chaleur. L'origine d'une ca-rence peut être : un apport insuffisant, destroubles de l'absorption associés à des ma-ladies intestinales, des besoins accrus pen-dant la grossesse. Les anti-épileptiques(phénytoïne, primidone, phénobarbital) etles contraceptifs oraux peuvent diminuerl'absorption de l'acide folique vraisembla-blement en inhibant la formation de laforme monoglutamate. Les inhibiteurs dela dihydrofolate réductase (ex. métho-trexate, p. 294) ralentissent la synthèse dela forme active, le tétrahydrofolate. Lessymptômes de la carence sont une anémiemacrocytaire et des lésions des mu-queuses. Le traitement consiste en l'ad-ministration orale d'acide folique.
L'administration d'acide foliquepeut masquer une carence en vitamineBI;. Cette vitamine catalyse la transfor-mation du méthyl-tétrahydrofolate en té-trahydrofolate nécessaire à la synthèsed'ADN (B). Une inhibition de cette réac-tion par suite d'une carence en vitamineB^ peut être compensée par une augmen-tation de la dose d'acide folique admi-nistrée. L'anémie liée à la carence envitamine B|; persiste, les troubles neuro-logiques se poursuivent inchangés et leurorigine est maintenant difficile à diagnos-tiquer à cause de l'absence de change-ment de la formule sanguine. L'admi-nistration inconsidérée de préparationsmultivitaminiques contenant de l'acidefolique peut également être nuisible.
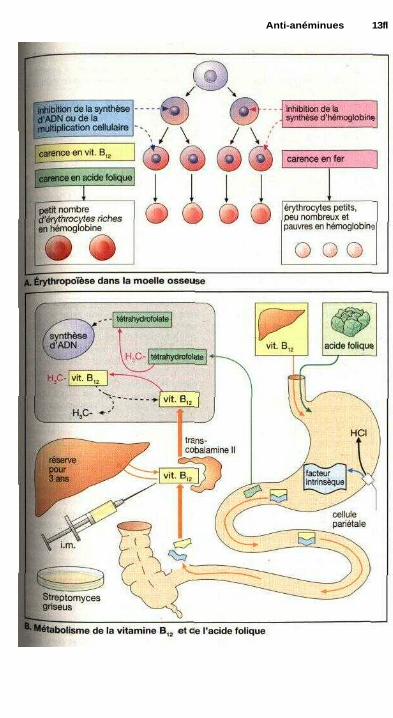
Anti-anéminues 13fl

140 Anti-anémiques
Métabolisme du fer
Le fer contenu dans la nourriture peutêtre absorbé de façon variable. Sousforme ferrique, Fe^, il n'est pratique-ment pas absorbé à partir du contenuneutre de l'intestin grêle ; à ce niveau,la forme Fe24- est beaucoup plus facile-ment absorbée. Le passage du fercontenu dans un hème (présent dansl'hémoglobine, la myoglobine), s'ef-fectue également assez bien. Dans lescellules de l'épithélium intestinal le ferest oxydé et, soit stocké sous forme deferritine (voir plus bas) ou bien fixé àune protéine de transport, la transfer-rine, une pi-glycoprotéine. Il ne par-vient dans l'organisme pas plus de ferqu'il n'est nécessaire pour compenserles pertes (par desquamation des cel-lules de la peau et des muqueuses, oules pertes de sang). (On parle de blo-cage muqueux.) Cette quantité corres-pond pour les hommes à environ1 mg/jour et pour les femmes à2 mg/jour (pertes de sang mens-truelles) ; elle correspond à peu près à10 % du fer présent dans la nourriture.Les complexes fer-transferrine sontprincipalement captés par les érythro-cytes au cours d'un processus d'endo-cytose et le fer (Fe14-) utilisé pour la syn-thèse de l'hémoglobine. 70 % ducontenu en fer de l'organisme, en-viron 5 g, se trouvent dans les érythro-cytes. Après digestion des érythrocytespar les macrophages du système réti-culo-endothélial le fer de l'hémoglo-bine est libéré. Sous forme de ferritine(protéine apoferritine + Fe^) le fer fer-rique peut être stocké, ou bien il peutêtre introduit de nouveau dans l'éry-thropoïèse par la transferrine.
Origine d'une carence en fer :elle provient souvent d'une perte desang chronique (ex. ulcère gas-trique / duodénal, tumeurs) - 1 litre desang contient 500 mg de fer. Endépit d'une augmentation importantedu pourcentage d'asorption (jusqu'à50 %), ce phénomème ne peut contre-balancer la perte et le contenu de l'orga-
nisme en fer diminue. Le manque de fpentraîne une altération de la synthèsed'hémoglobine : anémie par carenceen fer (p. 138).
Le traitement de choix (aprèsidentification et suppression de l'ori.gine du saignement) est l'administra-tion orale de composés ferreux parex. de sulfate de fer (dose journalière100 mg de fer, correspondant à environ300 mg de FeSÛ4, répartie en plusieursprises). Le remplissage du stock de ferpeut réclamer des mois. L'adminis-tration orale a cependant l'avantage derendre impossible une surcharge en ferde l'organisme, dans le cas où les mu-queuses sont intactes, à cause de la rela-tion entre l'absorption et les besoins(blocage muqueux).
E f f e t s secondaires. Les troublesgastro-intestinaux les plus fréquents(constipation, diarrhée, douleur abdo-minale) nécessitent une prise au mo-ment des repas, bien que l'absorptionsoit plus élevée dans un estomac vide.
Interactions médicamenteuses.Les antiacides inhibent l'absorption dufer. L'association avec l'acide ascor-bique (vitamine C) pour protéger Fe2^de l'oxydation en Fe3'1' est en théorie in-génieuse, mais pas indispensable enpratique.
L'administration parentéralesous forme de composés ferriquesn'est envisagée que lorsque le traite-ment oral n'est pas possible. Elle pré-sente un danger de surdosage avecdépôt de fer dans les tissus (hémosidé-rose). La capacité de liaison de la trans-ferrine est limitée et le fer libre sousforme Fe34- est toxique. C'est pourquoion utilisera des formes complexées dufer, les atomes de fer seront transférésà la transferrine soit directement, soitaprès phagocytose par les macrophagesde telle sorte que le fer parvienne jus-qu'aux reserves de ferritine. Les e f f e t ssecondaires possibles sont : en injec-tion i.m. : douleur au site d'injection etcoloration de la peau ; en injection i.v. :fièvre, chute de pression et choc ana-phylactique.
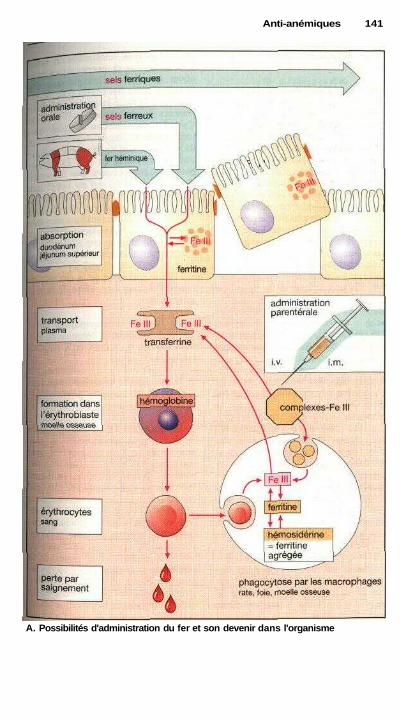
Anti-anémiques 141
A. Possibilités d'administration du fer et son devenir dans l'organisme

142 Anti-thrombotiques
Prophylaxie et traitementdes thromboses
Après la lésion d'un vaisseau, le sys-tème de coagulation est activé afin deconstituer à l'aide des thrombocyteset des molécules de fibrine un « bou-chon » qui bouche la plaie et permet leretour à une circulation normale. Laformation sans nécessité d'un caillotdans un vaisseau, c'est-à-dire unethrombose, peut constituer un dangermortel : si le thrombus se forme à partirdes lésions athéromateuses d'une artèrecoronaire, un infarctus peut se produire ;formé dans une veine profonde de lajambe, il peut se détacher, être emportéjusqu'aux artères pulmonaires etdonner naissance à une embolie pulmo-naire en les bloquant.
La prophylaxie de la thromboseest réalisée en utilisant des médica-ments qui diminuent la coagulation dusang : coumariniques et héparines(A). A côté de cela, on cherchera à in-hiber avec l'acide acétylsalicylique{'agrégation des plaquettes, qui parti-cipe à la formation du thrombus, en par-ticulier dans les artères (p. 148). Letraitement des thromboses utilise desmolécules qui détruisent la « trame defibrine » : les fibrinolytiques (p. 146).
(A) montre une vue d'ensembledu système de coagulation et descibles de l'action des coumariniques etde l'héparine. La cascade de la coagula-tion peut être déclenchée par deux voiesdistinctes (B). 1. A l'intérieur des vais-seaux aux emplacements qui ne sontpas recouverts d'endothélium, partransformation du facteur XII en fac-teur activé XIIa (système intrinsèque).2. Sous l'influence d'une lipoprotéineprovenant des tissus (thrombokinasetissulaire) par transformation du fac-teur VII en facteur VIIa (système ex-trinsèque). Les deux voies se rejoignentau niveau du facteur X pour une mêmeportion terminale.
Les facteurs de coagulation sontdes molécules protéiques. « Activa-tion » signifie pour la plupart coupure
de fragments et transformation (saufpour la fibrine) en enzymes protéoly.tiques (protéases). Quelques-uns desfacteurs activés ont besoin pour mani.tester leur activité protéolytique de laprésence de phospholipides (PL) et deÇa2 +. Les ions calcium réalisent vrai-semblablement la jonction entre H[)facteur et la surface phospholipidiquecomme représenté en (C). Les phos-pholipides sont présents dans le facteurplaquettaire 3 (FP,) qui est libéré lorsde l'agrégation plaquettaire, et dans lathrombokinase tissulaire (B). L'acti-vation successive de plusieurs enzymesaboutit à ce que les réactions initiales sedéveloppent comme une avalanche etconduisent finalement à une formationmassive de fibrine.
Le déroulement de la cascade decoagulation peut être inhibé de la ma-nière suivante :
1. les dérivés coumariniques dimi-nuent la concentration sanguine en fac-teurs inactifs II, VII, IX et X dont ilsinhibent la synthèse ;
2. le complexe héparine-antithrom-bine III abolit l'activité protéolytiquedes facteurs activés ;
3. les chélateurs du calcium empê-chent l'activité enzymatique des fac-teurs dépendants du calcium ; ce sontdes substances contenant des groupe-ments COO qui lient les ions calcium(C) : citrate et EDTA (acide éthylènediamine tétra-acétique) forment avec lecalcium des complexes solubles, l'oxa-late précipite avec le calcium sousforme de complexe insoluble. La com-plexation du calcium n'est pas utili-sable sur le plan thérapeutique, car laconcentration de calcium doit être dimi-nuée de façon tellement importantequ'elle n'est plus compatible avec lasurvie (hypocalcémie tétanique). Cescomposés ne seront donc utilisés (patex. sous forme de sels sodiques) quepour bloquer la coagulation du sang endehors de l'organisme.
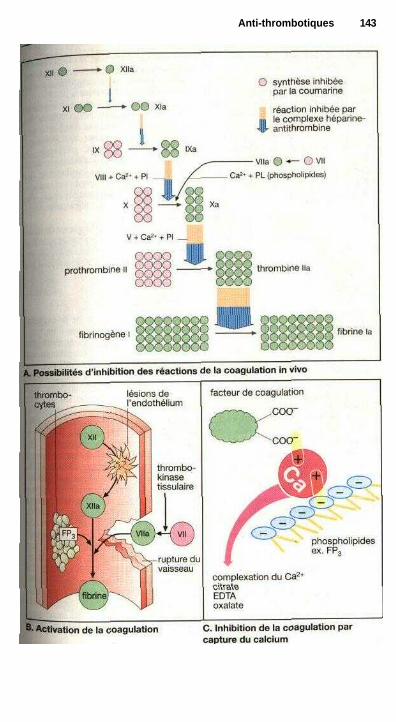
Anti-thrombotiques 143

144 Anti-thrombotiques
Dérivés coumariniques (A)
La vitamine K active dans le foie la fixa-tion d'un groupement carboxyle sur lesrésidus glutamiques des précurseurs desfacteurs II, VII, IX et X ; les groupementsCOOH sont indispensables à la fixationaux phospholipides par l'intermédiaire ducalcium (p. 142). Il existe différents dé-rivés de la vitamine K d'origine distincte :vitamine K| (phytoménadione) dans lesplantes, vitamine K; issue des bactéries del'intestin, vitamine K.) (ménadione) pro-duite par synthèse chimique. Toutes sonthydrophobes et nécessitent les acides bi-liaires pour être absorbées.
Anticoagulants oraux. Les 4-hy-droxy-coumarines de structure prochede celle de la vitamine K participentcomme « fausse vitamine K » dans ces ré-actions et bloquent la synthèse des fac-teurs dépendants de la vitamine K.
Les coumariniques sont bien ab-sorbés après administration orale. Leurdurée d'action est très variable. La syn-thèse des facteurs de la coagulation dé-pend du rapport des concentrations entrela vitamine K et les coumariniques exis-tant dans les cellules du foie. La dose né-cessaire pour obtenir une inhibition suffi-sante de la coagulation doit être ajustéepour chaque patient individuellement(contrôle par le temps de Quick).Finalement, le malade ne doit pluschanger de façon importante son alimen-tation en légumes verts (changement de laconcentration de vitamine K), ne doit pasprendre des médicaments supplémen-taires modifiant l'absorption ou l'élimina-tion du coumarinique (changement deconcentration du coumarinique) ou bienne doit pas provoquer une inhibition de lafonction des plaquettes par la prise d'aspi-rine.
L'effet indésirable le plus impor-tante est un saignement. Dans ce cas,l'effet des coumariniques peut être anta-gonisé par l'administration de vitamineK| ; la capacité de coagulation du sangn'est cependant restaurée qu'après desheures ou des jours lorsque la reprise de lasynthèse hépatique a permis d'aboutir àune élévation suffisante de la concentra-tion plasmatique des facteurs de coagula-
tion. En cas d'urgence, il est nécessaird'apporter les facteurs manquants (par ex6
par une transfusion de sang frais, ou d'unconcentré de prothrombine).
Parmi les autres effets secondairesnotables on peut citer : des nécroses hé-morragiques de la peau au début du traite-ment ainsi qu'une chute des cheveux-administrés pendant la grossesse, ilspeuvent provoquer des altérations de laformation des os et des cartilages et deslésions cérébrales (à cause des saigne-ments) ; on peut également observer, plusrarement, un risque de saignement rétro-placentaire.
Compte tenu de l'étroitesse de la fe-nêtre thérapeutique et du problème délicatdes interactions médicamenteuses, les an-ticoagulants oraux ne sont aujourd'huique rarement employés.
Héparine (B)
L'activation d'un facteur de la coagula-tion correspond à l'hydrolyse d'un frag-ment protéique par un facteur situé plushaut dans la cascade de la coagulation, quipermet de libérer le site actif de l'enzymeCe phénomène peut être de nouveau inac-tivé par un moyen physiologique sousl'action de l'antithrombine III (AT III,une des glycoprotéines circulant dans leplasma). L'héparine inhibe la coagulationen augmentant d'un facteur 1 000 la vi-tesse d'action de l'antithrombine III.L'héparine est stockée en même tempsque l'histamine dans les vésicules desmastocytes. Son rôle physiologique estmal connu. L'héparine utilisée dans le do-maine thérapeutique est extraite d'intestinde porc ou de poumon de veau. Les molé-cules d'héparine sont des chaînes desucres aminés portant des groupementsCOO- et SO,-, contenant 10 à 20 desunités représentées en (B) (poids molécu-laire moyen environ 20 000). L'activitéinhibitrice de la coagulation est différenteselon la longueur de la chaîne. Pour stan-dardiser, l'activité d'une préparationd'héparine est donnée en unité internatio-nale (UI) par comparaison avec une pré-paration de référence.
Les nombreuses charges négativessont importantes à plusieurs points de
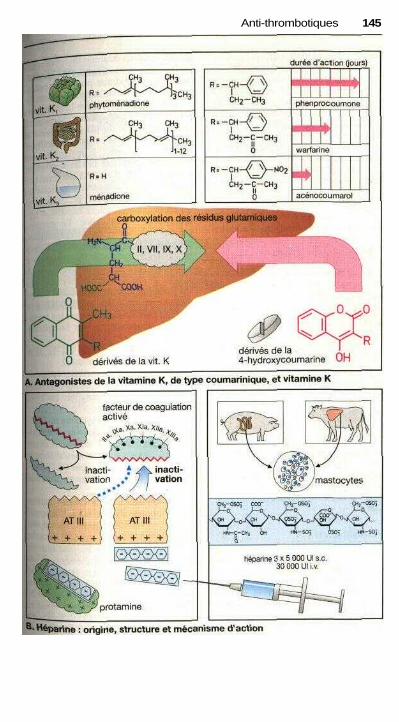
Anti-thrombotiques 145

146 Anti-thrombotiques
vue : 1. elles empêchent le passage à tra-vers les membranes : l'héparine est inac-tive après administration orale ou cutanéeet doit être injectée ; 2. leur interactionavec des charges positives des résidus ly-sine est utile à la formation du complexeavec AT III ; 3. elles permettent la liaisonde l'héparine à son inhibiteur (antidote),la protamine (une protéine polycatio-nique du sperme du saumon).
Lors de saignements dus à l'hépa-rine, l'injection de protamine suffit àbloquer immédiatement l'action del'héparine.
Un autre effet secondaire à citerest l'apparition de réactions allergiquesavec, entre autres, une tendance àl'agrégation des plaquettes et à lathrombopénie (cliniquement : throm-boses et saignements).
Pour la prophylaxie des throm-boses, il suffit d'une faible dose de5 000 UI, 2 à 3 fois par jour en injectionsous-cutanée. L'héparine de petit poidsmoléculaire (poids moléculaire moyenenviron 5 000) a une durée d'actionplus longue et ne doit être administréequ'une fois par jour (certoparine, dalte-parine, enoxaparine, reviparine, tinza-parine).
Le risque de saignement est sifaible pour de petites doses d'héparinequ'il est déjà possible de faire la pre-mière injection deux heures avant uneopération. Pour empêcher la croissancedes caillots au niveau d'une thrombosedéjà constituée, il est nécessaire d'uti-liser des doses journalières plus impor-tantes d'héparine en injection intravei-neuse.
Fibrinolyse (A)La fibrine provient du fibrinogène parcoupure de deux oligopeptides sousl'action protéolytique de la thrombine(facteur lia). Les molécules de fibrinepolymérisent pour former un réseau.Celui-ci peut être découpé en fragmentssolubles par une enzyme endogène laplasmine. La plasmine dérive d'un pré-curseur inactif, le plasminogène parcoupure d'un fragment protéique. Pourdissoudre les thrombi (par exemple
dans le cas d'un infarctus du myocardeon injectera de l'activateur du plasnnnogène. La condition pour une throm'bolyse réussie est de donner l'activateuaussitôt que possible après la formationdu thrombus. L'urokinase est un ach.valeur endogène du plasminogène nn,peut être extrait de cultures de cellulesrénales humaines. L'urokinase estmieux supportée que la streptokinaseCelle-ci n'a pas d'activité enzymatiquepropre ; c'est à la suite de l'associationavec une molécule de plasminogènequ'apparaît un complexe capable d'hy-drolyser le plasminogène. La strepto-kinase, dérivée de streptocoquesdemeure malgré tout une protéine bac-'térienne. Ceci explique l'apparition defréquentes réactions anaphylactiques. Ala suite d'infections antérieures par desstreptocoques, des anticorps anti-strep-tokinase peuvent être présents dans leplasma. Leur interaction avec les molé-cules de streptokinase leur fait perdretoute activité.
Un autre activateur est le tPA(tissue plasminogen activator).
Le fait que les fibrinolytiques,comme on pouvait s'y attendre, favori-sent les saignements constitue un effetsecondaire indésirable.
L'inactivation du système de fibri-noiyse peut être réalisée avec des inhi-biteurs de pla.srniiie comme l'acideS.-aminocaproïque, p-aminoinéthyl-brn-zoïque, Y acide tranexamique ou Yapro-.tinine qui inhibe également d'autresprotéases.
Diminution de la concentrationde fibrinogène dans le sang. L'ancrodest un composant du venin d'un serpentmalais vivant dans les trous. Il détachedu fibrinogène par voie enzymatique unseul fragment, donnant naissance à uncomposé inutilisable. La coagulabilitédu sang diminue en même temps que laconcentration de fibrinogène. Comme lefibrinogène (poids moléculaire environ340000) contribue à la viscosité dusang, on aboutit également à une amélio-ration de la fluidité du sang. On espèrepouvoir utiliser ces deux effets dans cer-taines anomalies de l'hémostase.
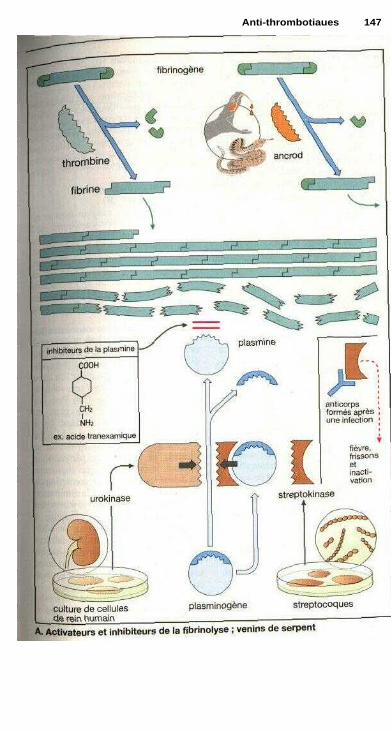
Anti-thrombotiaues 147

148 Anti-thrombotiques
Inhibition de l'agrégationplaquettaire (A)
Dans le lit artériel, les thrombi se com-posent principalement d'agrégats pla-quettaires ; en effet, les plaquettes san-guines se déposent facilement auniveau des lésions d'athérosclérose desvaisseaux du cerveau ou de la circula-tion cardiaque ; il se produit un in-farctus cardiaque ou cérébral. L'acideacétylsalicylique (AAs, p. 196) inhibel'agrégation des plaquettes. Le groupe-ment acétyl, labile, se fixe de façon co-valente à la cyclooxygénase plaquet-taire et inhibe de façon irréversiblecette enzyme et la synthèse de throm-boxane A; (p. 194). L,e thromboxaneA, libéré provoque dçux événements,favorables à l'hémostase dans le casd'une lésion vasculaire mais néfastes encas d'athérosclérose, {'agrégation desplaquettes et le rétrécissement des vais-seaux.
La synthèse de prostacycline, unantagoniste endogène du thromboxaneest inhibée en même t^mps. La prosta-cycline est formée dans les cellulesendothéliales sous l'action de la cy-clooxygénase (p. 194). Les cellules en-dothéliales peuvent cependant rem-placer la cyclooxygén^se inactivée viaune nouvelle synthèse de l'enzyme,tandis que celle-ci n'çst pas possibledans les plaquettes dépourvues denoyau. L'effet d'une ac-étylation uniquepersiste ainsi durant la durée de vie desplaquettes (environ unç semaine). Uneinhibition préférentielle de la synthèsede thromboxane peut igtre obtenue parl'administration d'AAg en dose plusfaible (30.300 mg/j).
Indications : prévention d'un in-farctus du myocarde, p^r ex dans le casd'une angine de poitr-ine instable, oubien après un infarctus (préventiond'une récidive), prévention d'un acci-dent cérébral en cas d'altérations dudébit sanguin cérébral. Effets indési-rables : ils dérivent de la propriété prin-cipale de l'aspirine : risque d'hémor-ragie (hémorragies c-érébrales !), etégalement de l'inhibition de la synthèse
de prostaglandines (p. 196) : c'est-'dire lésions des muqueuses de l »tomac et de l'intestin.
La triclopidine et l'abciximabsont utilisés pour des indications particulières ; ils interfèrent avec un site dpfixation du fibnnogène sur les plaquettes (glycoprotéme IIb/IIIa), nyl'intermédiaire duquel les plaquette<peuvent être reliées les unes aux autres
Inhibition de l'agrégationdes érythrocytes (B)La vitesse du flux sanguin dans la ré-gion des veinules post-capillaires est àson niveau le plus faible, ce qui entraînedans cette zone une tendance particu-lière des érythrocytes à l'agrégation.On aboutit alors à une stagnation duflux (stase) et donc un apport insuffi-sant d'oxygène. Une faible vitesse duflux sanguin favorise aussi la formationd'un thrombus.
En diminuant la concentrationdes érythrocytes on peut augmenterla vitesse de circulation du sang. Unedilution significative du sang (hémodi-lution) peut être obtenue par une prisede sang et un échange par une solutionremplaçant le plasma (p. 150). Pour quel'approvisionnement en oxygène dutissu demeure correct en dépit de ladilution du sang, il faut que le débitsanguin augmente : les vaisseaux sedilatent, la résistance périphériquediminue, le débit cardiaque augmente.En même temps que l'augmentation dela vitesse du flux sanguin, la diminutionde la concentration des érythrocytescontribue à diminuer la tendance àl'agrégation. Les indications de l'hé-modilution sont : troubles circulatoiressévères des jambes, accident vasculairecérébral, prévention de la thrombose.Ce traitement réclame un cœur en bonétat et pouvant subir une surcharge.
Il est parfois souhaitable de modi-fier la déformabilité des érythrocytes etpar là même la fluidité du sang à l'aidede la pentoxyfilline. Ce produit a uneutilité avérée dans le cas des oblitéra-tions artérielles de la jambe (claudica-tion intermittente).
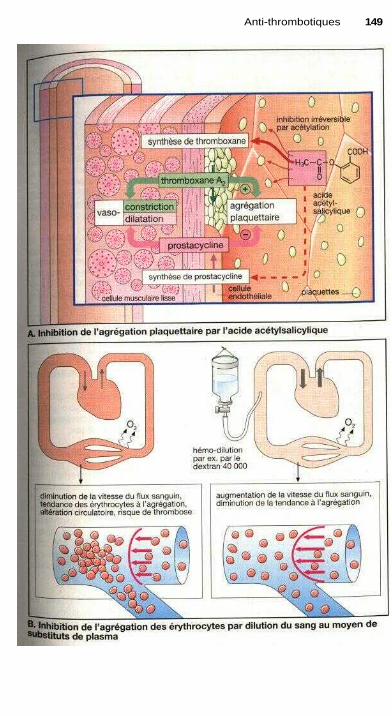
Anti-thrombotiques 149

150 Substituts du plasma
Substituts du plasma
Une perte de sang importante peut en-traîner une défaillance circulatoire ouchoc, susceptible de mettre en danger lavie du patient. Le danger provient moinsde la perte des érythrocytes, c'est-à-diredu transport d'oxygène que de la dimi-nution du volume sanguin.
Pour remédier au risque de chochypovolémique, il est nécessaire d'aug-menter le volume circulant. Dans le casoù la perte de sang n'est pas trop impor-tante, la perfusion d'une solution d'unsubstitut du plasma est suffisante. Leplasma sanguin se compose en principed'eau, de sels et des protéines plasma-tiques. Un substitut du plasma n'a pascependant besoin de contenir des pro-téines. Les macromolécules (colloïdes)conviennent comme substitut car ellesont des propriétés voisines de celles desprotéines plasmatiques : 1. elles d i f f u -sent mal hors du lit vasculaire et sontdifficilement filtrées au niveau gloméru-laire, 2. elles lient les sels dissous etl'eau en raison de leurs propriétés decolloïdes osmotiques. Elles sont donccapables d'assurer pendant de nom-breuses heures un remplissage durabledu système circulatoire. D'un autre côté,les colloïdes doivent pouvoir malgrétout être complètement éliminés parl'organisme.
Comparés au sang total ou auplasma, les substituts du plasma ont plu-sieurs avantages : ils sont d'une utilisa-tion plus aisée et moins onéreuse, plusfaciles à stocker, et ne contiennent aucunvirus susceptible de provoquer une in-fection comme le HIV ou le virus del'hépatite B.
Trois colloïdes sont utilisés aujour-d'hui comme succédanés du plasma :deux polysaccharides, le dextran oul'amidon modifié, et un polypeptide, lagélatine.
Le dextran est un polymère d'ori-gine bactérienne composé de moléculesde glucose enchaînées en 1 -» 6 (au lieude 1 -* 4 comme normalement). Lessubstituts de plasma disponibles sur lemarché contiennent des solutions de
masse moléculaire comprises en»60000 (dextran 60) et 40000(dextran 40 de petit poids moléculaireD.). La longueur de chaîne de chaquemolécule varie cependant de façon importante. Les plus petites des moléculede dextran peuvent encore être filtrée»au niveau glomérulaire et sont lentementéliminées par le rein ; les molécules plu,grosses sont phagocytées par les cellulesdu système réticulo-endothélial et dégra-dées. En dehors de la compensation despertes de sang, les solutions de dextranservent également à des opérations d'hé-modilutions dans certains troubles circu-latoires (p.148).
Il faut également signaler que ledextran de petit poids moléculaire, aucontraire du dextran 60, peut diminuerdirectement la tendance des érythrocytesà l'agrégation par une modification deleurs propriétés de surface. Pendant uneadministration de longue durée, etcompte tenu de l'élimination plus rapidedes petites molécules par le rein, les mo-lécules de grande taille s'accumulentde telle sorte que se constitue dans lesang au cours du temps une populationde molécules de masse moléculairemoyenne toujours plus élevée.
Les effets secondaires les plus im-portants dérivent du caractère antigé-nique des dextrans et correspondent àune réaction anaphylactique.
L'hydroxyéthylamidon est dérivéde l'amidon. Compte tenu de la présencedes groupes hydroxyéthyl, il sera hydro-lyse plus lentement et persiste plus long-temps dans le sang que ne le faitl'amidon. L'hydroxyéthylamidon pos-sède les mêmes propriétés pharmacolo-giques et les mêmes utilisations théra-peutiques que les dextrans. Un effetsecondaire particulier est la survenue dedémangeaisons qui peuvent persisterlongtemps.
Gélatine : elle se compose dechaînes peptidiques hydrophiles, déri-vées du collagène. Elle est utiliséecomme succédané plasmatique mais ce-pendant pas pour une hémodilution dansle cas de troubles circulatoires.
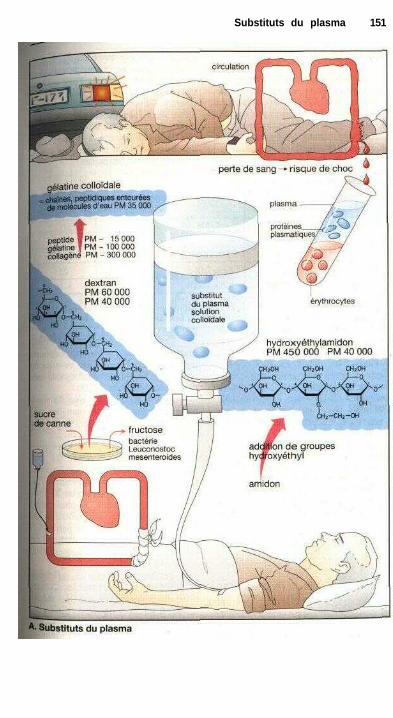
Substituts du plasma 151
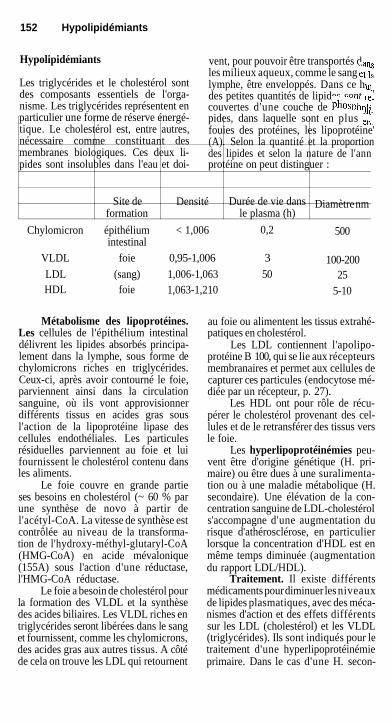
152 Hypolipidémiants
Hypolipidémiants
Les triglycérides et le cholestérol sontdes composants essentiels de l'orga-nisme. Les triglycérides représentent enparticulier une forme de réserve énergé-tique. Le cholestérol est, entre autres,nécessaire comme constituant desmembranes biologiques. Ces deux li-pides sont insolubles dans l'eau et doi-
vent, pour pouvoir être transportés dles milieux aqueux, comme le sang ei?8
lymphe, être enveloppés. Dans ce hdes petites quantités de lipides sont ut>
couvertes d'une couche de phosphnl6'pides, dans laquelle sont en plus g1'fouies des protéines, les lipoprotéine'(A). Selon la quantité et la proportiondes lipides et selon la nature de l'annprotéine on peut distinguer :
Chylomicron
VLDLLDLHDL
Site deformationépithéliumintestinal
foie(sang)foie
Densité
< 1,006
0,95-1,0061,006-1,0631,063-1,210
Durée de vie dansle plasma (h)
0,2
350
Diamètre nm
500
100-20025
5-10
Métabolisme des lipoprotéines.Les cellules de l'épithélium intestinaldélivrent les lipides absorbés principa-lement dans la lymphe, sous forme dechylomicrons riches en triglycérides.Ceux-ci, après avoir contourné le foie,parviennent ainsi dans la circulationsanguine, où ils vont approvisionnerdifférents tissus en acides gras sousl'action de la lipoprotéine lipase descellules endothéliales. Les particulesrésiduelles parviennent au foie et luifournissent le cholestérol contenu dansles aliments.
Le foie couvre en grande partieses besoins en cholestérol (~ 60 % parune synthèse de novo à partir del'acétyl-CoA. La vitesse de synthèse estcontrôlée au niveau de la transforma-tion de l'hydroxy-méthyl-glutaryl-CoA(HMG-CoA) en acide mévalonique(155A) sous l'action d'une réductase,l'HMG-CoA réductase.
Le foie a besoin de cholestérol pourla formation des VLDL et la synthèsedes acides biliaires. Les VLDL riches entriglycérides seront libérées dans le sanget fournissent, comme les chylomicrons,des acides gras aux autres tissus. A côtéde cela on trouve les LDL qui retournent
au foie ou alimentent les tissus extrahé-patiques en cholestérol.
Les LDL contiennent l'apolipo-protéine B 100, qui se lie aux récepteursmembranaires et permet aux cellules decapturer ces particules (endocytose mé-diée par un récepteur, p. 27).
Les HDL ont pour rôle de récu-pérer le cholestérol provenant des cel-lules et de le retransférer des tissus versle foie.
Les hyperlipoprotéinémies peu-vent être d'origine génétique (H. pri-maire) ou être dues à une suralimenta-tion ou à une maladie métabolique (H.secondaire). Une élévation de la con-centration sanguine de LDL-cholestérols'accompagne d'une augmentation durisque d'athérosclérose, en particulierlorsque la concentration d'HDL est enmême temps diminuée (augmentationdu rapport LDL/HDL).
Traitement. Il existe différentsmédicaments pour diminuer les niveauxde lipides plasmatiques, avec des méca-nismes d'action et des effets différentssur les LDL (cholestérol) et les VLDL(triglycérides). Ils sont indiqués pour letraitement d'une hyperlipoprotéinémieprimaire. Dans le cas d'une H. secon-
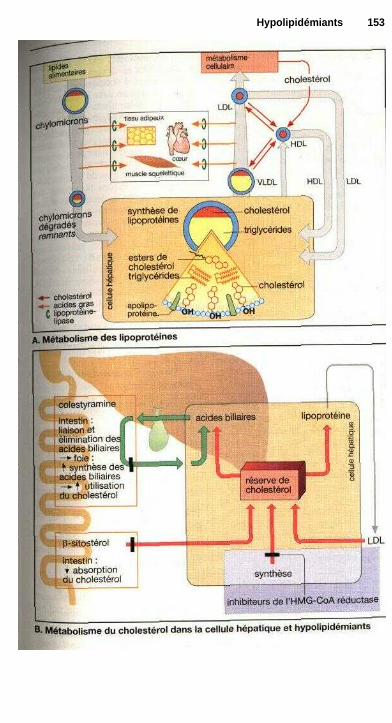
Hypolipidémiants 153

154 Hypolipidémiants
daire on doit d'abord chercher à dimi-nuer les lipides plasmatiques par un ré-gime ou par le traitement de la maladiesous-jacente.
Molécules actives (B). Lacolestyramine et le colestipol sont desrésines échangeuses d'anions, non ab-sorbables. Elles lient les acides biliairesdans l'intestin inhibant ainsi leur réab-sorption et stimulent donc indirecte-ment leur formation. La cellule hépa-tique pourvoit à ses besoins accrusgrâce à une augmentation de l'expres-sion de l'HMG-CoA réductase et desrécepteurs des LDL (rétrocontrôle né-gatif),
Aux doses nécessaires (3 x 10-15g/j) les résines provoquent destroubles intestinaux. Elles empêchentl'absorption de graisses et des vita-mines liposolubles (A, D, E, K). Dansl'intestin, elles absorbent certains médi-caments comme les glycosides car-diaques, les antivitamines K et les diu-rétiques et diminuent ainsi leurbiodisponibilité. La consistance sa-bleuse des résines échangeuses d'ionsest ressentie par l'utilisateur commetrès désagréable.
Le ft-sitostérol est un stéroïde vé-gétal qui n'est pas absorbé par voieorale et peut à dose suffisante empêcherl'absorption entérale du cholestérol.
Les statines, lovastatine (L), sim-vastatine (S), pravastatine (P) etfluvas-tatine (F) inhibent l'HMG-CoA réduc-tase. Chacune de ces quatre moléculescomporte une chaîne latérale semblableau substrat physiologique de l'HMG-CoA réductase (A). L et S existent sousforme lactone, qui est absorbée rapide-ment après prise orale, est extraite enproportion importante lors du premierpassage hépatique et y est convertie enmétabolite actif. P et F existent déjàsous forme active et seront captés defaçon active par l'intermédiaire d'untransporteur d'anion spécifique des cel-lules hépatiques (servant à la captationdes acides biliaires dans le sang et uti-lisé également pour l'absorption sélec-tive de l'a-amanitine un poison de
l'amanite phalloïde) (A). L'extractinhépatique élevée, désignée en génér'isous le terme d'élimination présvsrmique, est utilisée dans le cas des st6'tines pour limiter l'action de ces or 'duits sur le foie. Malgré l'inhibition ril'HMG-CoA réductase, le contenu encholestérol des hépatocytes ne diminuppas car, lors d'une baisse de la concentration de cholestérol, se produitcompensation (en plus de la réductaseune augmentation des récepteurs desLDL. Comme les molécules de réduc-tase nouvellement formées sont égale-ment bloquées en présence de statinesl'hépatocyte couvre l'ensemble de sesbesoins en cholestérol en puisant celuides LDL plasmatiques (B). La concen-tration des LDL circulantes décroîtainsi que la durée de séjour des LDLdans le plasma, ce qui diminue le risqued'oxydation en une LDL oxydée favori-sant l'athérosclérose.
En associant une Staline avec unerésine échangeuse d'ions, on peut dimi-nuer de façon encore plus importante, laconcentration de LDL.
Un effet secondaire rare mais dan-gereux des statines est une lésion desmuscles squelettiques. Le risque estaccru en cas d'association aux fibrates(voir ci-dessous).
L'acide nicotinique et ses dérivés(pyridylcarbinol, xanthinol-nicotinate,
. acipimox) activent la lipoprotéine li-pase endothéliale et diminuent de cettefaçon principalement le niveau de tri-glycérides. Parmi les effets secondaireson observe au début du traitement unedilatation vasculaire médiée par lesprostaglandines (flush -> chute de pres-sion sanguine), qui peut être bloquéepar l'administration de faibles dosesd'acide acétylsalicylique.
Le clofibrate et ses dérivés (béza-fibrate, étofibrate, gemfibroz.il) dimi-nuent les concentrations plasmatiquesde lipides par un mécanisme mal défini.Ils peuvent entre autres provoquer desaltérations hépatiques et des lésions desmuscles squelettiques (myalgies, myo-pathies, rhabdomyolyse).
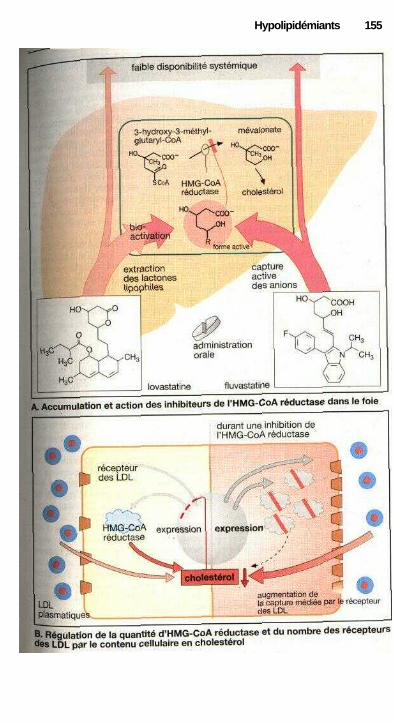
Hypolipidémiants 155

156 Diurétiques
Diurétiques : vue d'ensemble
Les diurétiques (saliurétiques) déclen-chent une augmentation de l'émissiond'urine (diurèse). Au sens strict, il s'agitde substances qui agissent directement surles reins. Principalement en raison de l'in-hibition de la réabsorption d'eau et deNaCl, ils augmentent l'émission d'urine.
Les principales indications des diu-rétiques sont :
Résorption des œdèmes (A) : lesœdèmes correspondent à un gonflementdes tissus dû à une augmentation de leurcontenu en liquide, essentiellement loca-lisé dans l'espace extracellulaire (volumeinterstitiel). Après administration d'undiurétique, le volume du plasma décroîtpar suite de l'augmentation de l'élimina-tion rénale d'eau et de sel, le sang est« condensé ». La conséquence est uneaugmentation de la concentration des pro-téines du sang et par là-même de la pres-sion osmotique. La présence dans le litcapillaire de cette force qui attire lesliquides, augmente le passage du liquidedes tissus dans la circulation. C'est ainsique le fluide tissulaire décroît et que dis-paraît l'œdème. La diminution du volumeplasmatique et du volume interstitielconduit à une réduction du volume extra-cellulaire (VEC). Selon les symptômes dela maladie, on utilisera : les thiazidiques,les diurétiques de l'anse, les antagonistesde l'aldostérone, les osmodiurétiques.
Baisse de la tension artérielle : lesdiurétiques constituent les médicamentsde choix pour diminuer une pression arté-rielle élevée (p. 306). Déjà à doses faibles,ils diminuent les résistances périphé-riques (sans diminution notable de l'es-pace extracellulaire) et diminuent ainsi lapression sanguine.
Traitement d'une insuffisance car-diaque : la baisse des résistances périphé-riques provoquée par les diurétiques faci-lite l'éjection du sang par le cœur(diminution de la post-charge, p. 132,p. 302), le débit cardiaque et la capacitéde travail de l'organisme augmentent.L'élimination accrue des liquides entraîneune réduction des volumes extracellu-laires et par là-même une diminution del'apport sanguin au cœur (réduction de la
pré-charge, p.302). Les symptômes de ]astase veineuse en amont du cœur, telgonflement du foie et un œdème des ch11
villes disparaissent. On utilise principale'ment les thiazidiques (parfois en associa'tion avec des diurétiques épargnant lepotassium) ou des diurétiques de l'anse
Prévention d'une insuffisante ré.nale : en cas de défaillance circulatoire(choc), par exemple à la suite d'un saigne-ment massif, existe le danger d'une inter-ruption de le production d'urine par lesreins (anurie). A l'aide de diurétique, oncherchera à rétablir le flux urinaire. Onutilisera dans ce cas les osmodiurétiquesou les diurétiques de l'anse.
Les effets secondaires (A) d'uneadministration massive d'un diurétiquesont: 1. la diminution du volume sanauinpeut entraîner une chute de pression et uncollapsus ; 2. l'élévation de la concentra-tion des érythrocytes et des plaquettesaugmente la viscosité du sang et accroît ledanger d'une coagulation intravasculaireet d'une thrombose (A).
Lorsque sous l'action d'un diuré-tique on aboutit à une fuite d'eau et de selou à une diminution de l'espace extracel-lulaire, l'organisme peut déclencher enréaction (B) une activation du systèmerénine-angiotensine-aldostérone (p. 124) :à cause de la diminution du volume san-guin on aboutit à une réduction de la cir-culation rénale. Ceci entraîne une sécré-tion par le rein de la renine, une hormone,dont l'activité enzymatique stimule la for-mation dans le sang d'angiotensine I.L'angiotensine 1 est transformée en an-giotensine II sous l'action de l'enzyme deconversion angiotensine converting en-zyme, ACE. L'angiotensine II à son tourdéclenche, entre autres, la libération d'al-dostérone. Ce minéralocorticoïde aug-mente la reabsorption d'eau et de sodiumpar le rein et s'oppose donc à l'action desdiurétiques. Les inhibiteurs de l'enzymede conversion (p. 124) empêchent cettecontre-régulation et renforcent l'activitédes diurétiques.
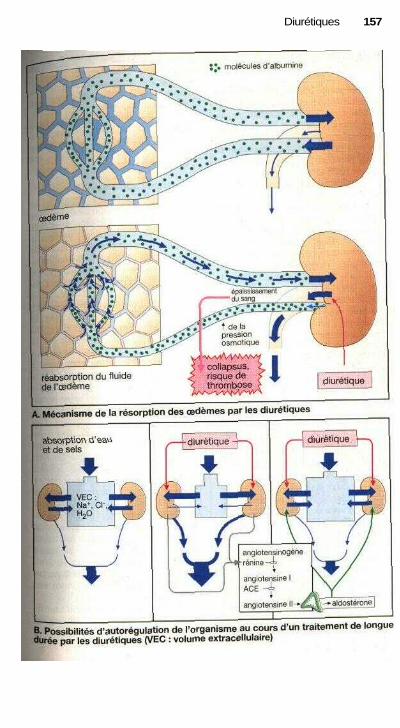
Diurétiques 157

158 Diurétiques
Réabsorption du sodium au niveaudes reins (A)
La plus petite unité fonctionnelle durein est le néphron. Dans le réseau glo-mérulaire, le liquide plasmatique estfiltre dans la capsule de Bowman (CB)d'un néphron (p. 40) pour donner nais-sance à l'urine primitive. Au niveau dutubule proximal (Tp), environ 70 % duvolume filtre sont réabsorbés, avec unereabsorption simultanée d'eau et deNaCl. Dans la partie épaisse de labranche ascendante de l'anse de Henle,seul le NaCl est reabsorbé, l'eau ne peutpas suivre. Ce phénomène est la condi-tion de l'existence d'une circulation àcontre-courant qui permet l'accumula-tion d'une concentration importante deNaCl dans la medulla rénale. Dans letubule distal (Td), l'eau et le NaCl se-ront de nouveau réabsorbés de façon si-multanée. A la fin du néphron, cette ré-absorption s'effectue sous le contrôlede l'aldostérone, avec échange de Na*contre K4' et H^ Dans le tubule collec-teur (Te), la vasopressine (ADH) aug-mente la perméabilité à l'eau de laparoi. L'eau, attirée par la concentra-tion élevée de NaCl dans la medulla,diffuse dans cette direction et demeuredonc dans l'organisme. C'est ainsiqu'une urine concentrée parvient fina-lement dans le bassinet.
Le transport de Na+ à travers lescellules du tubule s'effectue pratique-ment de façon identique dans tous lessegments du néphron. La concentrationintracellulaire de Na+ est nettement plusfaible que celle de l'urine primitive. Cegradient de concentration est donc laforce motrice pour l'entrée de Na+ dansle cytosol de la cellule tubulaire. Unsystème de transport inclus dans lamembrane (carrier) assure l'entrée duNa^ L'énergie libérée par cet influxpeut être utilisée pour exporter enmême temps une autre molécule contreson gradient. Le sodium sera expulsédans l'espace extracellulaire sous l'ac-tion d'une Na-K ATPase consommantde l'énergie (hydrolyse de l'ATP). Lesmolécules d'enzymes sont situées uni-
quement sur la partie de la membranpdirigée vers l'interstitium (basolaté-rale), et non sur la partie luminale, dptelle sorte que le Na4' ne puisse plues'échapper vers l'urine.
Les diurétiques inhibent tous la ré-absorption de Na4'. Les principales pos-sibilités d'action sont au niveau de l'in-flux et du transport de Na4^ versl'extérieur.
Diurétiques osmotiques (B)
Molécules : mannitol, sorbitol. Sited'action : principalement le tubuleproximal. Mode d'action : puisque leNaCl et l'eau sont reabsorbés ensembledans le tubule proximal, la concentra-tion de sodium dans la lumière tubu-laire ne change pas en dépit de l'ab-sorption massive de Na+ et d'eau. Lescellules de l'organisme ne possèdentpas de système de transport pour lemannitol (structure p. 169) et le sorbitolet ces molécules ne traversent pas lamembrane cellulaire. Ces substancesdoivent donc être introduites dans lacirculation par perfusion. Après filtra-tion glomérulaire, elles ne sont égale-ment pas réabsorbées de l'urine primi-tive à cause de leur faible capacité depassage à travers les membranes. Cesalcools, proches de sucres, fixent lesmolécules d'eau et les retiennent dansla lumière tubulaire. Lorsque les ionsNa4' sont transportés dans les cellulestubulaires, l'eau ne peut plus suivre enquantités normales. La concentration deNa'1' dans l'urine diminue. Ceci diminuela réabsorption du Na'1'. Une des raisonsest que la différence de concentrationavec l'intérieur des cellules tubulairesdécroît et que diminue en même tempsla force motrice pour l'influx de Na*.Le résultat d'une diurèse osmotique estl'émission d'un volume importantd'urine diluée.
Indications : prévention d'un col-lapsus rénal, glaucome, diminutiond'un œdème cérébral.
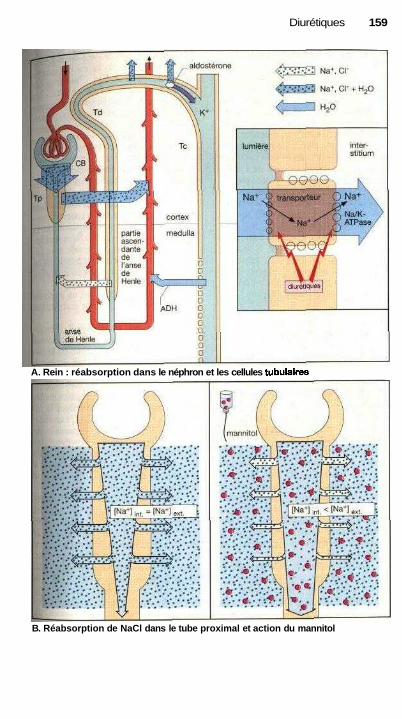
Diurétiques 159
A. Rein : réabsorption dans le néphron et les cellules tabulaires
B. Réabsorption de NaCl dans le tube proximal et action du mannitol

160 Diurétiques
Diurétiques de type sulfonainide
Ces substances contiennent un groupe-ment sulfonamide -SO^NIL et peuventêtre utilisées par voie orale. Dans le rein,les diurétiques sont filtrés au niveau duglomérule et, en plus, sécrétés par les cel-lules tubulaires. Leur concentration dansl'urine est supérieure à celle du sang. Ilsagissent sur les cellules tubulaires du côtéluminal, c'est-à-dire du côté de l'urine.Les plus actifs sont les diurétiques del'anse, les plus fréquemment utilisés sontles thiazides. Les inhibiteurs de l'anhy-drase carbonique ne sont aujourd'hui plusutilisés comme diurétiques.
L'acétawlamide est un inhibiteurde l'anhydrase carbonique. Il agit es-sentiellement dans le tubule proximal.Mécanisme d'action : l'enzyme anhy-drase carbonique (AC) accélère l'établis-sement de l'équilibre de la reaction :H^HCO^^ lïf:0,^ ILO+CO;.
Elle favorise dans les cellules tubu-laires l'apparition d'ions H*, qui sont en-suite éliminés dans l'urine en échange del'entrée d'un Na^ Ces H4' se combinentalors dans l'urine à des anions HC03- et leCO; peut pénétrer à travers la membranede la cellule tubulaire. La cellule contientalors de nouveau W et HCO^. En casd'inhibition de l'enzyme, cette réaction alieu trop lentement et la quantité de Na4',de HCÛ3 et d'eau réabsorbée de cetteurine primitive, dont l'écoulement est ra-pide, sera plus faible La perte de ïïCOy'conduit à une acidose. L'activité diuré-tique des inhibiteurs de l'AC disparaîtau cours d'une administration à longterme. L'anhydrase carbonique sert égale-ment pour la production d'eau dans lachambre de l'œil. Aujourd'hui ne sub-sistent comme indications pour lessusbtances de ce type que le glaucome etl'épilepsie.
Le dorwtamide est utilisé en appli-cation locale lors d'un glaucome pour di-minuer la pression intérieure de l'œil.
Diurétiques de l'anse. Ce sont lefurosémide, le pirétanide et d'autres.Après administration orale, apparaît enmoins d'une heure une diurèse impor-tante, qui cependant ne dure qu'environ4 heures. L'effet est rapide, violent et
court : « diurèse forcée ». Le site d'actionest la partie épaisse de la branche ascen-dante de l'anse de Henle. C'est là qu'ilsinhibent un cotransport Na\ K*, Cl-. Laconséquence est une élimination accruede ces électrolytes et d'eau. De mêmel'élimination rénale de Ça2* et Mg^ aug^mente. Les e f f e t s secondaires propressont : diminution (réversible) de l'audi-tion, potentialisation de l'activité de mé-dicaments néphrotoxiques. Indications :œdème pulmonaire (dans le cas d'une in-suffisance cardiaque gauche, ils présen-tent en outre l'avantage de provoquer unedilatation immédiate des vaisseaux vei-neux capacitifs et une diminution de lapost-charge) ; cas où les thiazides sont in-efficaces, ex. insuffisance rénale avec ré-duction de la clearance de la créatinine audessous de 30 ml/min ; prévention de l'in-suffisance rénale aiguë.
Bien que ce ne soit pas un sulfona-mide, on peut ranger l'acide étacryniquedans ce groupe.
Diurétiques thiazidiques (benzo-thiazides). Ce sont par ex. l'hydrochloro-fhiazide, le trichlorométhiadde, ou le bu-twde. La chlortalidone est un analoguethiazidique d'action longue. Ces sub-stances agissent sur la partie moyenne dutubule distal mais leur site d'action molé-culaire n'est pas connu. Ils inhibent la re-absorption d'eau et de NaCl. L'excrétionrénale de Ça24- diminue tandis que celle deMg2 + augmente. Indications : hyperten-sion, insuffisance cardiaque, résorptiondes œdèmes. Ils sont souvent combinés àun diurétique épargnant le K^ triamtérèneou amiloride (p. 162).
Les effets indésirables des diuré-tiques de type sulfonamide peuvent êtrea) une hypokaliémie qui est la consé-quence d'une perte accrue de K1' dans lapartie terminale du tubule distal où a lieuune augmentation de l'échange Na*contre K^ ; b) une hyperglycémie ; c) unehyperuricémie : augmentation de laconcentration d'acide urique dans le sangavec un risque de crise de goutte chez lessujets prédisposés. Les diurétiques detype sulfonamide entrent en compétitionavec l'acide urique pour le système desécrétion des acides.
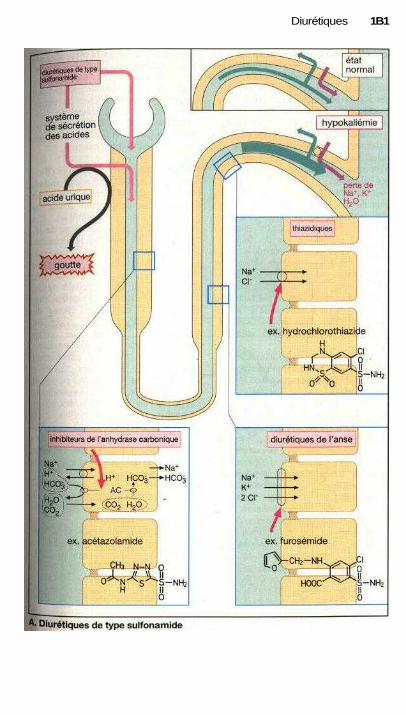
Diurétiques 1B1

162 Diurétiques
Diurétiques antikaliurétiques (A)Ces substances agissent à la partie termi-nale du tubule distal ou à la partie prox-imale du tube collecteur, où Na* est ré-absorbé et échangé contre K^ ou ïî+.L'activité diurétique est relativementfaible. Au contraire des diurétiques detype sulfonamide (p. 160) ceux-ci n'en-traînent pas de perte de t^ ; bien plus, ilexiste un danger d'hyperkaliémie. Cesmolécules peuvent être administréespar voie orale.
a) Triamtérène et amiloride : enplus de la filtration glomérulaire, ilssont également sécrétés dans le tubuleproximal, ils agissent du côté luminal(urinaire) sur les cellules des tubules.Tous les deux inhibent l'entrée de Na+
et donc son échange contre ï^ ou K^Ils sont essentiellement utilisés en asso-ciation avec les diurétiques thiazidiques(ex. l'hydrochlorothiazide) car leurs ef-fets opposés sur l'élimination de K^ secompensent tandis que leurs actions surl'excrétion d'eau et de NaCl s'addition-nent.
b) Antagonistes de l 'aldostérone :l'aldostérone, stéroïde minéralocorti-coïde, stimule la réabsorption de Na* enéchange de K^ (Cl- et l'eau suivent).Son action sur la synthèse protéique apour conséquence une augmentation dela capacité de transport des cellules tu-bulaires. La spironolactone ainsi queson métabolite principal la canrénonesont des antagonistes des récepteurs del'aldostérone et bloquent son action.L'effet diurétique de la spironolactonene se manifeste pleinement qu'aprèsplusieurs jours d'administration. Onpeut penser à deux explications de cephénomène : a) la transformation de laspironolactone en son métabolite lacanrénone, dont l'élimination est pluslente et qui donc s'accumule (p. 48) ;b) le fait qu'une inhibition de la syn-thèse protéique ne devient sensible quelorsque les protéines déjà présentes sontdevenues non fonctionnelles et doiventêtre remplacées par néosynthèse.
Un effet secondaire particulipl'interférence avec l'action des l̂1roïdes androgènes ; on peut observl'apparition de gynécomasties (gros-^sements de la poitrine chez l'homnnIndications : principalement les e
constances associées à une augmem^'tion de la libération d'aldostérone n3'ex. cirrhose du foie avec ascites '
Vasopressine (ADH) et dérivés (B)L'ADH, un peptide de 9 acides aminésest libérée par la post-hypophyse etaugmente la réabsorption rénale del'eau (hormone antidiurétique). CMeffet est médié par le sous-type de ré-cepteur V;. Elle augmente la perméabi-lité à l'eau de l'épithélium du tube col-lecteur (mais pas aux sels), de sorte quel'eau attirée par la pression osmotiqueélevée qui règne dans la medulla rénalediffuse hors de l'urine. La nicotine aas-mente (p. 110) tandis que l'éthanol di-minue la libération d'ADH. À concen-trations plus élevées que cellesnécessaires pour son action antidiuré-tique, l'ADH stimule la musculaturelisse et, entre autres, les vaisseaux(« vasopressine »). Cet effet passe parles récepteurs Vi. La pression artérielleaugmente ; une constriction des artèrescoronaires peut déclencher une crised'angine de poitrine.
La lypressine (8-L-lysine-vaso-pressine) agit comme l'ADH. D'autresdérivés de l'ADH ne montrent plus quel'une des deux actions.
La desmopressine sert au traite-ment du diabète insipide (carence enADH) ; elle est utilisée en injection ouen pulvérisation nasale.
Felypressine ou ornipreswe sontutilisées comme vasoconstricteurs enplus des anesthésiques locaux (p. 204).
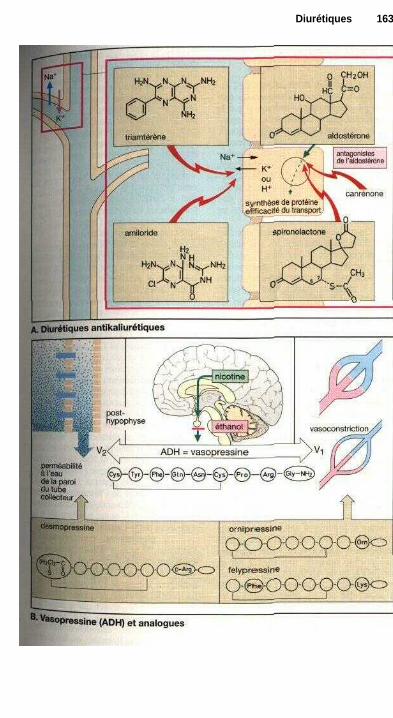
Diurétiques 163

164 Produits contre les ulcères gastriques
Traitement des ulcèresde l'estomac et du duodénum
Dans la région d'un ulcère (ulcus) de l'es-tomac ou du duodénum, la muqueuse (mu-cosa) est attaquée si profondément par lesuc digestif que la couche de tissu sous-jacente (sub-mucosa) se trouve exposée.Cette « autodigestion » se déclenchelorsque l'équilibre entre l'acide chlorhy-drique corrosif et le mucus neutralisant,qui recouvre la muqueuse d'un film pro-tecteur, est déplacé en faveur de l'acide.Une lésion de la muqueuse peut êtreaccentuée par la bactérie Helicobacterpylori qui réside dans le mucus.
On utilise des médicaments dont lesbuts thérapeutiques sont les suivants :1. soulagement de la douleur ; 2. accélé-ration de la cicatrisation ; 3. diminutiondes récidives. Les moyens thérapeutiquessont : I. atténuation des agents corrosifsen diminuant la concentration d'ions H*(A), II. renforcement de l'action protec-trice grâce à des produits protégeant lamuqueuse, III. élimination d'Helicobacterpylori (p. 166).
I. Produits diminuant la concentrationd'ions H*
I. a. Produits neutralisants. Lesgroupements liant les ions H^ commeC(V', HCC>3- ou OH- font partie avecleurs contre-ions de la famille des anti-acides. Les réactions de neutralisation quise produisent dans l'estomac après l'ad-ministration de CaCO, ou de NaHCOjsont figurées dans la partie gauche de lafigure (A). Pour les anti-acides qui ne sontpas absorbés, le contre-ion ne reste en so-lution qu'en milieu acide, en amont de laréaction de neutralisation. Après libéra-tion par le pancréas d'une sécrétion neu-tralisante, ces ions précipitent en grandepartie en se combinant de nouveau à desgroupements basiques, sous forme par exde CaC03 ou de A1P04, et sont éliminésavec les fèces. L'organisme est de ce faità peine gêné par ces contre-ions ou lesgroupements basiques. En cas d'insuffi-sance rénale, la faible absorption suffitmalgré tout pour provoquer une augmen-tation de la concentration sanguine ducontre-ion (ex. empoisonnement par le
magnésium avec des perturbations cardiaques sous forme de ralentissement!Des effets secondaires sont associés à laprécipitation dans l'intestin : diminutionde l'absorption d'autres produits pharma-ceutiques par adsorption à la surface de<précipités ; perte de phosphate lors de laprise de quantités importantes d'Al(OH)
Les ions Na4^ restent en solution enprésence des sécrétions pancréatiquesriches en HCOf et peuvent être réab-sorbés comme le HC03-. Compte tenu decette absorption de sodium, l'administra-tion de NaHCO, doit être prohibée dansles maladies où l'administration de NaCldoit aussi être réduite : hypertension, in-suffisance cardiaque, œdème.
Comme le bol alimentaire possèdeune action tampon, les anti-acides serontpris entre les repas (par ex. 1 et 3 h aprèsles repas et pendant la nuit). Les diiti-acides non absorbés seront préférés.Comme le Mg(OH)^ a un effet laxalif (àcause de son action osmotique et/ou del'effet de Mg2 + sur la sécrétion de cholé-cystokinine, p. 168) et que l'AI(OH), estconstipant (à cause de l'effet astringent del'Al^, p. 176), ces deux anti-acides serontle plus souvent utilisés en association.
I. b. Inhibiteurs de la productiond'acide. Le neurotransmetteur acétylcho-line, l'hormone gastrine et l'histamine li-bérée au niveau de la muqueuse stimulentles cellules pariétales de la muqueuseintestinale, et augmentent la sécrétiond'HCl. L'histamine provient des cellulesentérochromaffines ; sa libération est dé-clenchée par le nerf vague (via des récep-teurs M|) et par la gastrine. Les effets del'acétylcholine et de l'histamine peuventêtre bloqués par des antagonistes adminis-trés par voie orale et qui parviennent à lamuqueuse par la circulation sanguine.
La pirenzépine, contrairement àl'atropine, bloque préférentiellement lesrécepteurs de l'ACh de type M| et pro-voque donc moins d'effets secondairesanalogues à ceux de l'atropine (p. 104).La cellule pariétale présente cependantdes récepteurs M3, si bien que le site d'ac-tion de la pirenzépine doit se situerailleurs (cellules entérochromaffines ;transmission ganglionnaire, où les récep-teurs M| ont une action modulatrice).
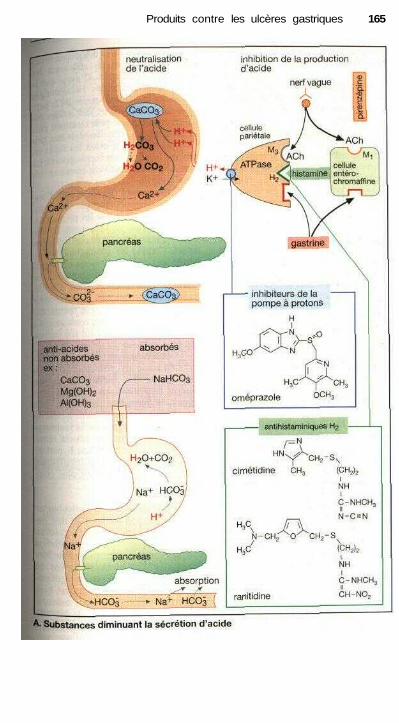
Produits contre les ulcères gastriques 165

166 Produits contre les ulcères gastriques
Les récepteurs de l'histamine descellules pariétales sont de type H,(p. 114) et peuvent être bloqués par lesaiitiliistaiiliniqiics H, (p. 165). A causedu rôle central de l'histamine dans lastimulation des cellules pariétales, lesantihistammiques inhibent égalementl'effet des autres stimuli, par ex. celuide la gastrine dans le cas de tumeurs dupancréas sécrétant de la gastrine (syn-drome de Zollinger-Ellison). Le pre-mier des antihistamimques H;, la cimé-tidine, ne provoque déjà que des effetssecondaires assez rares, entre autres desaltérations du SNC (par ex. confusion),ou des troubles endocriniens chezl'homme (gynécomastie, diminution dela libido, impuissance). La cimétidinepeut aussi bloquer dans le foie la dégra-dation d'autres substances. Les sub-stances apparues plus tard, la ranitidineet la famotidme sont actives à dosesplus faibles. L'inhibition des enzymesmicrosomiales du foie diminue defaçon sensible pour des « charges ensubstances » plus faibles, de sorte queces produits ne perturbent pas les traite-ments par d'autres médicaments.
L'oméprazole (p. 165) peut en-traîner une inhibition maximale de lasécrétion d'acide. Après administrationorale dans des dragées résistantes ausuc gastrique, il parvient via la circula-tion sanguine jusqu'aux cellules parié-tales. Il se forme alors en milieu acideun métabolite actif qui inhibe, grâce à laformation d'une liaison covalente, lapompe qui transporte dans le suc gas-trique H+ en échange de K^ (HWATPase). Le lansoprazole et le panto-prawle agissent de la même manière.
II. Agents protecteurs. Le sucralfate(A) contient de nombreux groupementshydroxyde d'aluminium. Il ne s'agit ce-pendant pas d'un anti-acide, car il ne di-minue pas de façon globale la concen-tration d'acide dans le suc gastrique.Après administration orale, les molé-cules de sucralfate s'imbibent de sucgastrique acide : formation d'un em-plâtre. Celui-ci adhère à l'emplacement
où le revêtement de la muqueuse intes-tinale est altéré et où affleurent ]e<,couches plus profondes. C'est là que lesucralfate séquestre les H^ Protégéedes acides et en même temps de la pep-sine, de la trypsine et des acides bi-liaires, la muqueuse peut cicatriser plusrapidement. Le sucralfate doit être prisl'estomac vide (1 h avant les repas oupendant la nuit). Il est bien supporté, lesions Al^ libérés peuvent entraîner uneconstipation.
Misoprostol (B) : il s'agit d'uneprostaglandine obtenue par hémisyn-thèse et plus stable que les prostaglan-dines naturelles, de telle sorte qu'ellepeut être utilisée par voie orale et resteractive. Comme les prostaglandmes li-bérées localement au niveau de la mu-queuse, elle stimule la formation demucus et bloque la sécrétion d'acide.Les effets systémiques associés (fré-quemment diarrhée, chez la femme en-ceinte déclenchement des contractions),réduisent considérablement son utilisa-tion thérapeutique.
Carbénoxolone (B) : c'est un dé-rivé de l'acide glycyrrhizique, contenudans le suc des racines de réglisse(Succus liquiritiae). La carbénoxolonestimule la formation de mucus. Elleexerce une action comparable à celle del'aldostérone et augmente la réabsorp-tion d'eau et de NaCl au niveau rénal.Ceci peut donc aggraver une hyperten-sion, un œdème ou une insuffisancecardiaque. Ce produit n'est plus com-mercialisé en France.
III. Élimination d'Helicobacterpylori (C). Ce germe joue un rôle pa-thogène important dans les gastriteschroniques et les ulcères. Une solutionclassique est la combinaison d'antibio-tiques avec l'oméprazole. Au cas oùl'amoxicilline (p. 268) ou la clanthro-mycine (p. 274) ne seraient pas biensupportées on peut utiliser le métrom-dazole (p. 272). Les sels de bismuthcolloïdaux sont certes également actifsmais présentent le risque d'une sur-charge en métaux lourds.
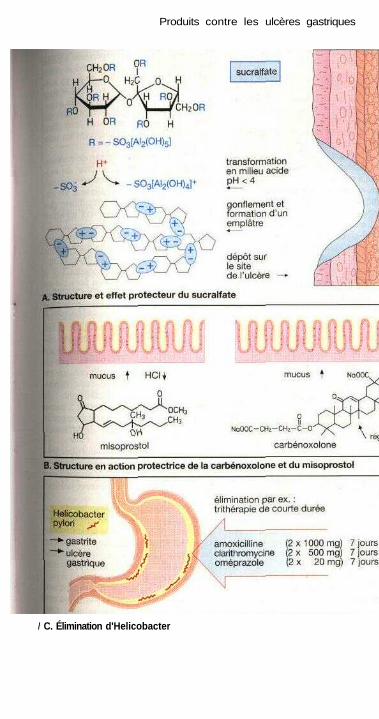
Produits contre les ulcères gastriques 167
I C. Élimination d'Helicobacter

168 Laxatifs
Laxatifs
Les laxatifs accélèrent et facilitent l'éva-cuation des selles en augmentant parsuite d'une action locale le pénstal-tisme intestinal et/ou en ramollissant lecontenu de l'intestin
1. Laxatifs de lest. La distensionde l'intestin par le contenu intestinalstimule les mouvements vers l'avant dela musculature intestinale (le pénstal-tisme) La stimulation des récepteurs à latension, situes dans la paroi de l'intestin,entraîne par voie réflexe une contractiondes muscles (en rouge sur la figure A) si-tués à l'arrière et une relaxation de ceux(en bleu) situés a l'avant du contenu del'intestin, ce qui pousse ce dernier endirection de l'anus
Mucilages et fibres (B). Ce sontdes substances insolubles et non absor-bées, qui s'imbibent de fluide dans l'in-testin et gonflent Dans la nourriture ha-bituelle, ce sont les fibres végétales quiservent de lest Elles sont formées parles parois non hydrolysables des cellulesvégétales Ces parois cellulaires contien-nent des chaînes carbonées qui ne sontpas attaquées par les enzymes diges-tives, ce sont par exemple la cellulose(molécules de glucose liées en 1 —' 4[î àcomparer à l'amidon où la liaison est enl — » 4 a , p 151) Le son (sous-produit demeunerie) et les graines de lin sontnches en cellulose D'autres sources vé-gétales de mucilages sont les grainesd'ivpaghui ouïes gommes de karaya Laprise de ces mucilages (en préventiond'un arrêt des selles, constipation) n'esten général associée à aucun effet secon-daire En cas d'absorption très faible deliquides et lorsqu'existe un rétrécisse-ment pathologique de l'intestin, il estpossible cependant de former un bou-chon iléal avec ces mucilages collants
Laxatifs osmotiques (C). Ce sontdes particules solubles, mais non absor-bées, qui en raison de leurs propriétésosmotiques maintiennent l'eau dans l'in-testin la pression osmotique du contenuintestinal (concentration des particules)correspond toujours à celle de l'espaceextracellulaire La muqueuse intestinalene peut pas maintenir une pression os-
motique inférieure ou supérieure a celldu contenu intestinal II se produit don6
une absorption des solutés de façon K e
osmotique, ceci signifie que l'absorptionpar ex de NaCl et de glucose est assndée a celle d'une quantité d'eau corres-'pondante Au contraire, l'eau reste dancl'intestin lorsque les molécules ne sontpas absorbées
Dans le cas du sulfate de sodiumNa^SO^, et du MgSO^ (sulfate de magnesie), les amons sulfates ne sont pasabsorbés et retiennent également les ca-tions pour maintenir l'équilibre descharges Les ions Mg peuvent en outrestimuler au niveau de la muqueuse duodenale la libération de cholécystoki-nine/pancréatozymme, qui augmenteaussi le pénstaltisme Ces laxatifs ap-pelés laxatifs salins, provoquent 1-3 haprès leur administration (si possible ensolution isotonique) des selles liquidesIls sont utilises pour la vidange de l'in-testin (par exemple avant une opération)ou pour accélérer l'élimination aprèsun empoisonnement Les contre-indica-tions sont dues au fait qu'une faiblepartie des cations est absorbée Le sul-fate de soude à cause de son contenu enNa4^ est contre-indiqué en cas d'hyper-tension, insuffisance cardiaque etœdème , le sulfate de magnésie a causede la possibilité d'une intoxication par lemagnésium dans une insuffisancerénale
Le manmtol et le sorbitol pour les-quels la membrane cellulaire ne contientaucun système de transport, au contrairedu glucose, agissent également commelaxatifs osmotiques
Le lactulose, un disacchande quin'est pas hydrolyse par les enzymes di-gestives, agit aussi comme laxatif osmo-tique La fermentation du lactulose parles bactéries du côlon, provoque une aci-dification du contenu intestinal et unealtération de la flore bactérienne Le lac-tulose est utilisé en cas d'insuffisancehépatique, pour bloquer la formation parles bactéries d'ammoniaque ainsi queson absorption (NH^ forme absorbée-> NH4+ non absorbe), de façon a éviterainsi un coma hépatique.
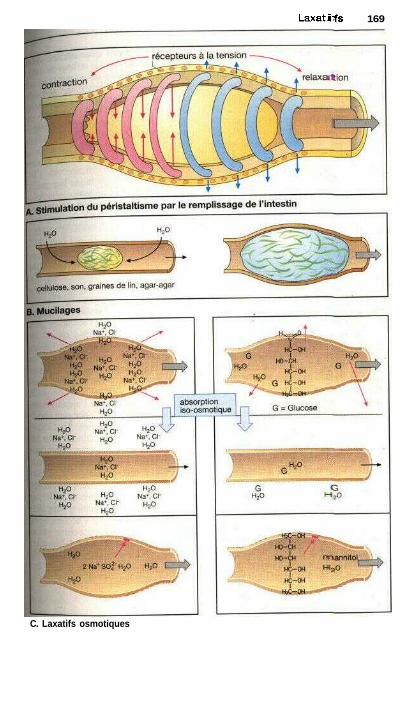
Laxati-fs 169
C. Laxatifs osmotiques

170 Laxatifs
2. Substances irritant l'intestin.Les laxatifs de ce groupe exercent uneaction irritante sur la muqueuse de l 'in-testin (A). L'absorption de fluide di-minue le péristaltisme, la sécrétion defluide et le remplissage accru de l'in-testin le stimulent ; la stimulation determinaisons sensitives entraîne parvoie réflexe une augmentation de lamotricité intestinale. Selon le site del'irritation, on distingue • l'huile dericin irritant l'intestin grêle, l'anthra-quinone irritant le gros intestin et lessubstances apparentées, dérivées du di-phénylméthane (p. 172).
Abus des laxatifs. L'idée qu'il estnécessaire d'aller à la selle au moinsune fois par jour est très répandue. Lerythme de 3 fois par semaine est cepen-dant tout à fait normal. L'idée assez ré-pandue dans l'ancien temps, que l'ab-sorption des molécules contenues dansl'intestin était néfaste pour l'organismeest sans doute à la base de ce désir deselles plus fréquentes. C'est ainsi queles purges faisaient, il y a longtemps,partie de l'arsenal thérapeutique usuel.On sait aujourd'hui, qu'un empoison-nement par les molécules du contenuIntestinal est impossible dans le casd'un fonctionnement normal du foie.Malgré tout, les laxatifs se trouvent par-fois vendus comme moyen de « purifi-cation du sang » ou bien de « nettoyagede l'organisme ».
Si l'on remplace l'absence de bal-last dans la « nourriture actuelle » parla prise de composés correspondants(fibres, mucilages), il n'y a rien à re-dire. L'utilisation de laxatifs irritantl'intestin n'est cependant pas sansdanger. Il existe le risque de ne pluspouvoir aller à la selle sans ce moyen :dépendance vis-à-vis des laxatifs. Laprise chronique de laxatifs irritants per-turbe le contenu de l'organisme en eauet électrolytes et peut provoquer dessymptômes divers (par ex. perturba-tions du rythme cardiaque liées à unehypokaliémie).
Causes d'une dépendance vi 'vis-dés laxatifs (B). Le réflexe de déf*cation est déclenché par le remplissae
du rectum. Une défécation norrn ^e
vide le gros intestin jusqu'à la branAdescendante du côlon. L'intervalle dtemps jusqu'à la prochaine défécann6
spontanée dépend donc de la vîtes n
avec laquelle ce segment de l'intestin s0
remplit de nouveau. Un laxatif irritantadministré à sa dose efficace vide l'intégralité du côlon. De façon logique »doit alors s'écouler un temps plus louajusqu'à ce qu'une nouvelle défécationspontanée soit possible. Craignant laconstipation, le sujet impatient utilise ànouveau les laxatifs, ce qui conduit àl'effet souhaité mais, à nouveau, à unevidange de la partie haute du côlonAprès l'arrêt d'un laxatif on ne doitdonc pas être alarmé par la survenued'une « pause compensatrice » ( I ) .
Dans le gros intestin, le contenufluide provenant de l'intestin grêles'épaissit par absorption d'eau et desels (passant d'environ 1 000 à 150 mlchaque jour). Si sous l'action d'unlaxatif irritant on provoque une vidangeprématurée, cela signifie aussi une perteentérale d'eau, de NaCl et de KC1.L'organisme compense l'appauvrisse-ment en eau et en NaCl par une sécré-tion accrue d'aldostérone (p. 162);cette hormone en effet stimule leur re-absorption rénale. Son effet cependants'accompagne d'une élimination rénalede KC1. Les pertes entérales et rénalesde K* s'additionnent et mènent à un ap-pauvrissement de l'organisme en K^, etune chute de la concentration sanguine(hypokaliémie). Cet état s'accompagned'une diminution du péristaltisme intes-tinal (« paresse intestinale »). Le sujetconstate une « constipation », prend denouveau des laxatifs et le cercle in-fernal est ainsi bouclé (2).
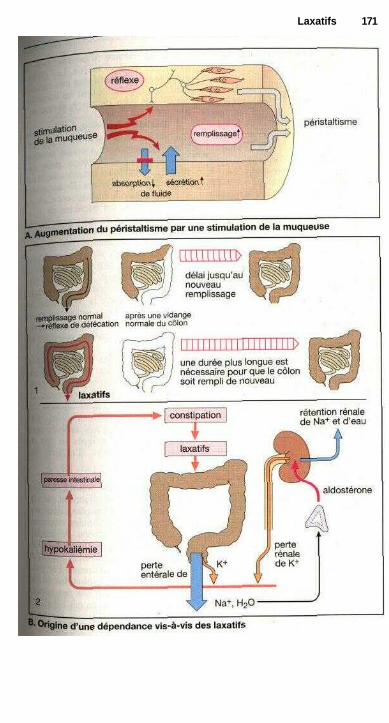
Laxatifs 171

172 Laxatifs
2. a. Laxatifs irritant l'intestingrêle : huile de ricin
L'huile de ricin provient de Ricinuscommunis (Palmier du christ, sur la fi-gure sont présentés un rameau, unegrappe de rieurs et une graine). Elle estobtenue par pression des graines (repré-sentées grandeur nature sur l'image).Après prise orale de 10-30 ml d'huilede ricin on provoque environ 1/2 à 3 hplus tard des selles liquides. Ce n'estpas l'huile de ricin qui est active maisl'acide ricinoléique. Celui-ci est forméau cours des réactions typiques de la dé-gradation des graisses : la muqueuseduodénale libère dans le sang l'hor-mone intestinale, cholécystokinine /pancréatozymine ; celle-ci stimule lacontraction de la vésicule biliaire et lalibération d'acides biliaires ainsi que lasécrétion de lipase par le pancréas(CCK/PZ stimule aussi le péristaltismeintestinal). En raison de son action radi-cale, l'huile de ricin ne convient pas autraitement d'une constipation normale.Après absorption orale d'une substancetoxique l'huile de ricin peut être utiliséepour accélérer l'élimination du poisonpar les voies naturelles et empêcher sonabsorption. Dans le cas d'une absorp-tion de poisons lipophiles, qui est faci-litée par les sels biliaires, l'huile dericin n'est pas recommandée.
2. b. Laxatifs irritant le côlon(p. 174 et suivantes)
Dérivés de l'anthraquinone (p. 175 A).Ce sont des produits végétaux. On lestrouve dans les feuilles ou les fruits duséné, l'écorce de cascara ou de bour-daine (Cortex frangulae, Cascara sa-grada), les racines de rhubarbe(Rhiwma rhei) ou bien dans les extraitsde feuilles d'aloès (p. 174). La structurede base des anthraquinones est décritedans la figure de la p. 175 A. Dans cesanthraquinones, on trouve, entre autres,deux groupements hydroxyle dont l'unest associé à un sucre (glucose, rham-nose). Après la prise du glycoside an-thraquinonique se produit avec un
temps de latence d'environ 6-8 hl'émission de fèces molles. Les glyco-sides ne sont pas actifs par eux-mêmesmais sont transformés par les bactériesintestinales pour donner la forme ac-tive.
Dérivés du diphénylméthane(p. 175 B). Ils dérivent de la phénol.phtaléine, substance à action laxativemais dont l'administration peut en-traîner dans quelques cas rares des réac-tions allergiques sévères. Le bisacodylet le picosulfate de sodium sont d'abordtransformés par les bactéries de l'in-testin en substances actives stimulantl'intestin. Après prise orale, le bisa-codyl subit l'élimination d'un groupe-ment acétyle, est absorbé, conjuguédans le foie à l'acide glucuronique (ou àl'acide sulfurique, p. 38) et enfin éli-miné avec la bile dans le duodénum.Environ 6-8 h après la prise orale, seproduit l'émission de selles molles etbien formées. Sous forme de supposi-toire, le bisacodyl produit son effet enmoins d'une heure.
Indications des laxatifs irritantle côlon : pour éviter les contractionsabdominales lors des selles : état post-opératoire, infarctus du myocarde, at-taque d'apoplexie ; pour adoucir lesdouleurs en cas de lésions anales : fis-sures, hémorroïdes. Les laxatifs sontstrictement contre-indiqués en cas dedouleurs abdominales d'origine in-connue.
3. Laxatifs lubrifiants. L'huilede paraffine n'est pratiquement pas ab-sorbée et rend les fèces plus glissantes.Elle bloque l'absorption des vitaminesliposolubles. L'absorption de goutte-lettes de paraffine peut aboutir à la for-mation dans les ganglions lymphoidesde l'intestin, de granulomes. Par pas-sage dans le tractus respiratoire, la pa-raffine peut provoquer une pneumonieinterstitielle. A cause de ces effets se-condaires, son administration n'est pa8
recommandée.
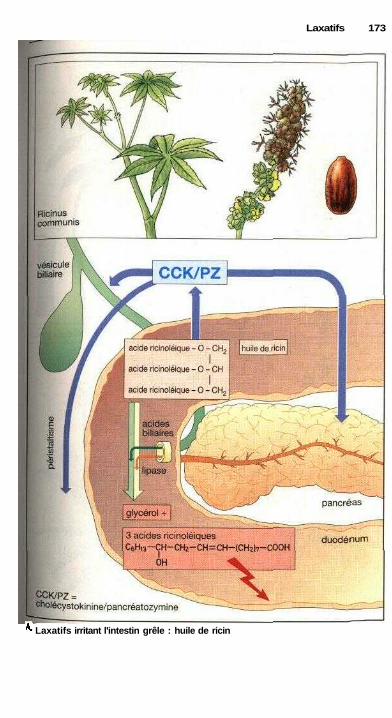
Laxatifs 173
A- Laxatifs irritant l'intestin grêle : huile de ricin
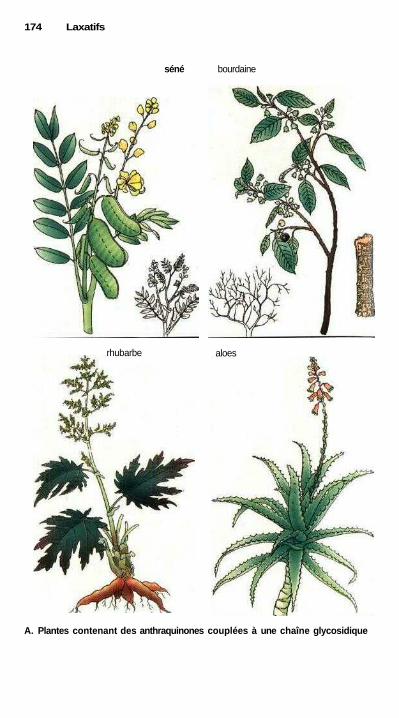
174 Laxatifs
séné bourdaine
rhubarbe aloes
A. Plantes contenant des anthraquinones couplées à une chaîne glycosidique
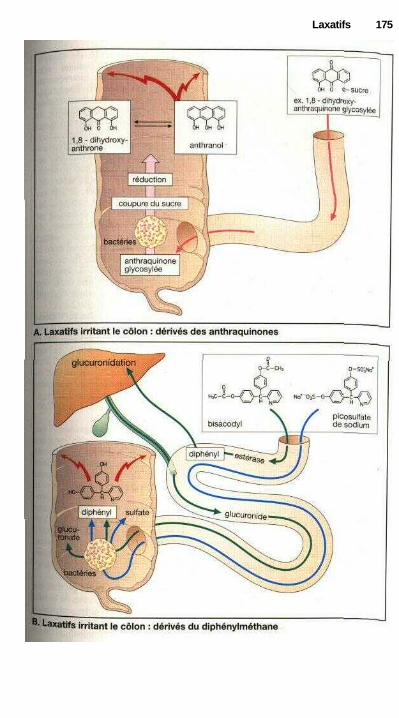
Laxatifs 175

176 Anti-diarrhéiques
Traitement d'une diarrhée
Origines d'une diarrhée (en rouge) : denombreuses bactéries (par ex. celle res-ponsable du choléra) sécrètent destoxines qui inhibent la capacité des cel-lules de l'épithélium à absorber l'eau etle NaCl, et qui augmentent les sécré-tions de liquide par la muqueuse. Lesbactéries ou les virus pénétrant dans laparoi intestinale déclenchent une in-flammation associée à une augmenta-tion de la sécrétion de fluide dans lalumière intestinale. La musculature del'intestin réagit en augmentant le péri-staltisme.
Les buts du traitement avec desanti-diarrhéiques sont les suivants :1. empêcher une perte d'eau et d'élec-trolytes par l'organisme ; 2. inter-rompre les selles fréquentes qui ne sontpas dangereuses mais gênantes. Les dif-férentes possibilités thérapeutiquesutilisées (en vert) répondent plus oumoins bien à ces buts.
Les adsorbants sont des matièresnon absorbées avec une surface consi-dérable. Les différentes molécules etentre autres les toxines se fixent surcette surface et sont donc inactivées etfinalement éliminées. Le charbon mé-dicinal est un charbon de bois ayantconservé la structure cellulaire et quiprésente une surface particulièrementimportante. La dose utilisée pour untraitement efficace des diarrhées est de4 à 8 g. Un autre adsorbant est le kaolin,un silicate d'aluminium hydraté.
Solutions orales de réhydrata-tion (pour 1 1 d'eau bouillie, 3,5 g deNaCl, 20 g de glucose, 2,5 g deNaHCOs, 1,5 g de KC1). Les solutionscontenant du glucose et des électrolytesconstituent un apport de fluide qui peutêtre absorbé après administration orale,car les toxines n'altèrent pas le trans-port simultané de glucose et de Na4' auniveau de la muqueuse intestinale (nicelui de l'eau). De cette façon, les sellesfréquentes ne sont certes pas empê-chées, mais on remédie avec succès à laperte d'électrolytes.
Opioïdes. La stimulation des ré-cepteurs opiacés des plexus nerveux de
la paroi intestinale inhibe les mouvments propulsants le contenu ve?l'avant et augmente les mouvement5
pendulaires. Cet effet anti-diarrhéiois
était autrefois obtenu par administratin6
de teinture ̂ d'opium, contenant de I11
morphine. À cause des effets centrau»(sédation, dépression respiratoirerisque de toxicomanie), on utilise es'sentiellement des dérivés ayant une ac-tion périphérique. Tandis que le diphé-noxylate peut encore provoquer dgiactions centrales marquées, le lupéra-mide à dose normale ne touche pas lesfonctions cérébrales. Le lopéramide estdonc l'opioïde anti-diarrhéique dpchoix. En raison du temps de contactplus élevé du contenu intestinal avec lamuqueuse, l'absorption de ^fluide peutégalement être améliorée. À dose tropélevée existe le risque d'une occlusionintestinale. Traitement contre-indiquéchez les enfants de moins de 2 ans.
Substances antibactériennes.C'est seulement lorsque les bactériesconstituent la cause manifeste de ladiarrhée que l'administration de cessubstances (p. 270, cotrimoxazole) a unsens. C'est rarement le cas. Il faut sesouvenir que les antibiotiques altèrentégalement la flore intestinale de l'orga-nisme, ce qui peut entraîner une diar-rhée.
Substances astringentes, parexemple les tannins (dans les remèdesde bonne femme, le thé fort) ou les selsmétalliques. Ils précipitent les protéinesde surface et peuvent ainsi provoquerune « imperméabilité » relative de lamuqueuse. La dénaturation des pro-téines ne doit pas toucher les protéinescellulaires, sinon on aboutirait à la des-truction des cellules. Les astringentspeuvent provoquer une constipation(voir sels d'aluminium, p. 164). maisleur effet dans le traitement des diar-rhées est douteux.
Mucilages, par ex. la pectine (lapomme râpée dans les recettes de bonnefemme). Ce sont des polysacchandesqui gonflent en présence d'eau. Ils soli-difient ainsi le contenu intestinal maisne possèdent aucun autre effet fav°-rable manifeste.
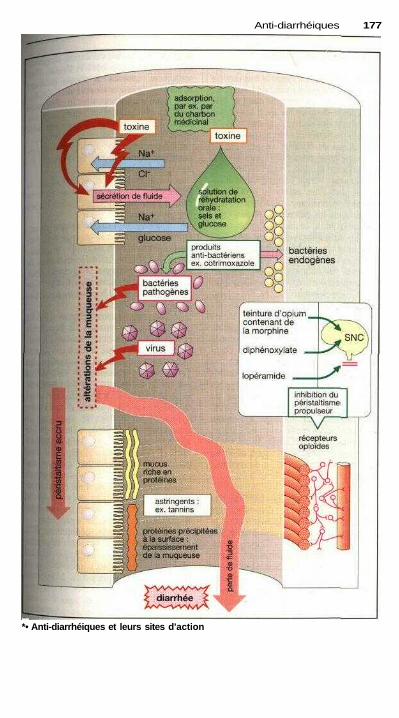
Anti-diarrhéiques 177
*• Anti-diarrhéiques et leurs sites d'action

178 Autres médicaments du tractus gastro-intestinal
Agents pour la dissolutiondes calculs biliaires (A)Le cholestérol déverse par le foie dans labile, insoluble dans l'eau par lui-même,sera maintenu en solution dans la bilesous forme de micelles avec les acidesbiliaires (et des phospholipides) S il y aplus de cholestérol déverse par le foieque les acides biliaires ne peuvent enemulsionner, il précipite et aboutit a laformation de calculs
La bile « non saturée en choles-térol » peut faire passer le cholestérolprécipite sous forme micellaire et dis-soudre ainsi les calculs de cholestérolOn utilise dans ce but une prise oraled'acide ursodésoxycholique (AUDC)ou d'acide chénodésoxycholique(ACDC) Ces deux molécules sont deuxisomères naturels des acides biliaires(groupement hydroxyle en position 7 (3AUDC , en position 7 a ACDC) Leurparticipation au contenu en acides bi-liaires de l'organisme est en tempsnormal très faible (voir le diagrammecirculaire en A), mais croît de façon no-table cependant lors d'une prise chro-nique les sels biliaires subissent uncycle enterohepatique Ils sont en parti-culier presque complètement reabsorbésdans l iléon La faible perte dans lesfèces sera compensée par une neosyn-these hépatique de sorte que le contenuen acide biliaire reste constant (3-5 g)L apport exogène élimine la nécessitéd une neosynthese hépatique d'acide bi-liaire , la proportion relative du composéadministre dans le contenu total s'ac-croît
En raison de cette modification decomposition, le pouvoir d'entraînementdu cholestérol par la bile augmente Lescalculs peuvent être dissous en l espaced un traitement de 1 — 2 ans, si cer-taines conditions sont remplies les cal-culs sont formes de cholestérol pur etsont d'une taille <15 mm , les fonctionsbiliaires sont normales et il n'y a pas demaladie hépatique , les patients sont sipossible d un poids normal L'AUDC estplus actif (dose quotidienne 8 10 mg/kg)et mieux supporte que l'ACDC(15 mg/kg/jour, avec très souvent des
diarrhées et une augmentation sanendes enzymes hépatiques) A l'issue d'1116traitement couronne de succès peiiv11'1apparaître de nouveaux calculs en*
Par comparaison avec le traitementchirurgical des calculs biliaires la disslution médicamenteuse joue un rfi?assez faible
Rappelons que l'AUDC a également une utilité en cas de cirrhose hliaire primaire
Cholérétiques : ils stimulent laformation d'une bile plus diluée mais cpprincipe n a pratiquement aucun intérêtthérapeutique
Cholagogues : ces agents stinmlent la contraction et la vidange de la vé-sicule biliaire, par exemple le jauned'œuf MgSC>4 un laxatif osmotique, laceruletide un analogue de la cholecysto-kimne (administration parenterale) Cesagents sont utilises pour tester les fonctions de la vésicule biliaire
Enzymes pancréatiques (B).Ilsproviennent des animaux de boucheneet servent de substituants en cas d insuffisance pancréatique secretoire (entreautres avec troubles de la digestion desgraisses et steatorrhee) En tempsnormal, la sécrétion d'enzymes pancreatiques est stimulée par une hormone in-testinale, cholecystokinme / pancreatozymine, libérée dans le sang par lamuqueuse intestinale au contact du bolalimentaire Lors de l'administrationorale d'enzymes pancréatiques il fautprendre en compte le fait que ces en-zymes seront partiellement mactivees enmilieu acide dans l'estomac (particulierement les lipases) Ces enzymes serontdonc administrées sous des formes gale-niques résistantes aux sucs gastriques
Lutte contre le météorisme (C).Ces agents sont utilises contre le meteonsme (accumulation excessive de gazdans le tractus digestif) Disperses sousforme de petites bulles dans le contenude l'intestin ces gaz empêchent la pro-gression du chyme Les agents « anti-mousse » de type dimeticone (dimethyl-polysiloxane) et simeticone, qui sontadministres par voie orale, stimulent laséparation des composants
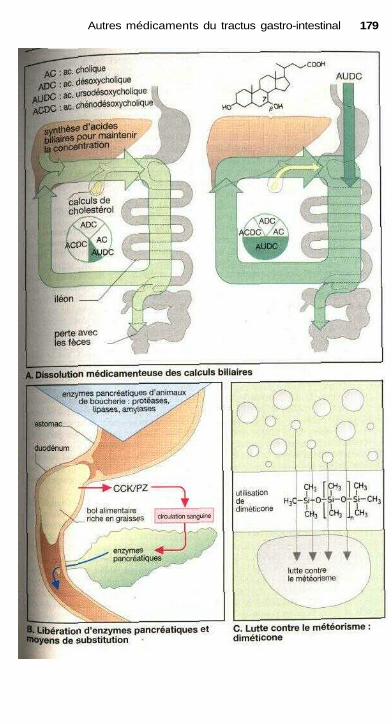
Autres médicaments du tractus gastro-intestinal 179

180 Produits agissant sur le système moteur
Substances actives sur le systèmemoteurLa plus petite unité fonctionnelle d'unmuscle squelettique est la fibre muscu-laire striée. La contraction est déclen-chée par une « impulsion » émanant deleurs nerfs moteurs. Conformément audéroulement prévu des mouvements, lecerveau envoie d'abord une impulsiondans la moelle épinière. Cet influx aboutità un motoneurone dans la corne anté-rieure de la moelle. Les prolongements dece neurone sous forme d'un faisceau defibres motrices parviennent aux cellulesmusculaires. Les mouvements réflexesélémentaires à des excitations sensitivesqui parviennent à la moelle épinière par laracine postérieure, se produisent sans in-tervention du cerveau. Pour éviter une sti-mulation trop forte des nerfs moteurs ouune contraction prolongée des muscles encas d'excitation persistante d'une termi-naison sensitive, sont intercalées dans lescircuits nerveux à travers lesquels l'influxse propage jusqu'à la moelle épinière descellules inhibitrices (encore appelées in-temeurones inhibiteurs).
La transmission neuromusculaire(B) de la stimulation du nerf moteur à lafibre musculaire se produit au niveau de laplaque motrice. L'influx nerveux libèrede l'acétylcholine (ACh) au niveau de laterminaison ; celle-ci se fixe aux récep-teurs nicotiniques de la plaque motrice.La stimulation des récepteurs provoque ladépolarisation de la plaque motrice et dé-clenche dans le sarcolemme environnantl'apparition d'un potentiel d'action qui sepropage. Le potentiel d'action entraînedans les fibres musculaires la libérationde Ça1* à partir de sites de stockage situésdans le réticulum sarcoplasmique. L'aug-mentation de la concentration de Ca^ en-traîne la contraction des myofilaments(couplage électro-mécanique). Pendant cetemps, l'ACh est dégradée par l'acétyl-cholinestérase (p. 100), l'excitation de laplaque motrice disparaît. En consé-quence, aucun potentiel d'action ne se dé-clenche et le calcium est pompé de nou-veau dans le réticulum ce qui entraîne unerelaxation des myofilaments.
Les molécules importantes sur leplan clinique agissent toutes (à l'excep-
tion du dantrolène) sur le contrôle nveux des cellules musculaires (\ np. 182 et suivantes). ' '
Myotonolytiques (A), ils ̂nuent le tonus musculaire en renforçantl'action des interneurones inhibiteurdans la moelle. Les myotonolytiques son^utilisés pour le traitement de conti iietioi»musculaires douloureuses par exeninlpassociées à des maladies spinales. Le<benwdiaiépines augmentent l'activité duneurotransmetteur inhibiteur, le GABAau niveau des récepteurs GABA(p. 224). Le baclofène stimule les récep*leurs GABAg.
Les poisons contracturants. Latoxine tétanique (cause du tétanos) et lastrychnine inhibent l'activité des inter-neurones inhibiteurs, qui utilisent unacide aminé, la glycine, comme neuro-transmetteur (A). La conséquence d'unepropagation non contrôlée des influx ner-veux jusqu'à la moelle épinière est l'ap-parition de crampes. La contracture desmuscles respiratoires constitue un dangermortel.
Toxine botulinique. C'est le pluspuissant poison connu, extrait deClostridium botulinum. La dose néces-saire pour tuer un adulte est de0,000003 mg. Elle inhibe la libéidtiond'acétylcholine par les terminaisons ner-veuses motrices (mais aussi parasympa-thiques). La mort est due à une paralysiedes muscles respiratoires.
La toxine botulinique peut être uti-lisée localement, à très faible dose. parexemple dans le cas de crampes des pau-pières (blépharospasme).
Une augmentation pathologique dela concentration des ions Mg provoqueégalement une inhibition de la transmis-sion neuromusculaire.
Le dantrolène agit dans les cellulesmusculaires au niveau du couplage élec-tromécanique, en inhibitant la libérationde calcium du réticulum. Il est employéaussi bien dans les cas douloureux despasticité chez des patients atteints de lé-sions spinales, que dans des maladiesmusculaires associées à une libérationexagérée de calcium (hyperthermie ma-ligne).
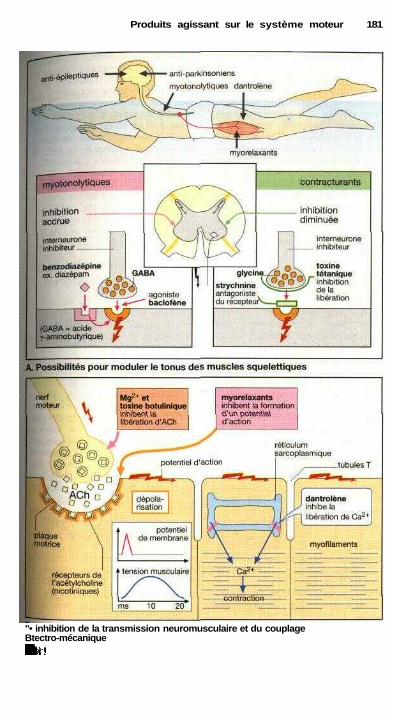
Produits agissant sur le système moteur 181
"• inhibition de la transmission neuromusculaire et du couplageBtectro-mécanique&'

182 Produits agissant sur le système moteur
Myorelaxants
Les myorelaxants entraînent un blocagedes muscles squelettiques en positionrelâchée. Ils se lient aux récepteurs del'acétylcholine de la plaque motrice etbloquent la transmission neuromuscu-laire (p. 180). Selon que la liaison auxrécepteurs de l'acétylcholine est asso-ciée à un blocage ou une stimulation dela plaque motrice, on distinguera lesmyorelaxants non dépolarisants et lesmyorelaxants dépolarisants (p. 184).Associés à un anesthésique, les myore-laxants empêchent qu'une interventionchirurgicale ne soit perturbée par Fescontractions musculaires du patient(P.214).
Myorelaxants non dépolarisantsCurare est le nom du poison végétal desflèches des indiens d'Amérique du Sud.Un être vivant qui est touché par uneflèche enduite de curare subit sousl'effet du poison qui se répand rapide-ment dans son organisme une paralysiedes muscles squelettiques et meurt carses muscles respiratoires se bloquent(paralysie respiratoire périphérique).L'animal abattu peut être consommésans danger car le poison n'est prati-quement pas absorbé au niveau dutractus intestinal. Le principe actif ducurare sur le plan médicinal est la d-tu-bocurarine. Elle possède un ammo-nium quaternaire et à l'extrémité op-posée un atome d'azote protoné à pHphysiologique. Tous les autres myore-laxants possèdent également deuxatomes d'azote chargés positivement.La présence d'une charge positive per-manente sur l'ammonium quaternaireexplique sa très mauvaise absorptionintestinale. La d-tubocurarine sera uti-lisée en injection i.v. (dose usuelle : en-viron 10 mg). Elle se fixe aux récep-teurs nicotiniques de la plaque motricesans les stimuler et agit comme un an-tagoniste compétitifàe l'ACh ; elle em-pêche la liaison de l'ACh libérée et parlà même la transmission neuromuscu-laire. La relaxation musculaire se pro-duit dans un intervalle de 4 min. La
d-tubocurarine ne pénètre pas dans leSNC. Le patient serait alors témoin enpleine conscience de la paralysie de samusculature et de son incapacité à res-pirer, sans pouvoir s'exprimer d'unefaçon quelconque. C'est pour cetteraison qu'il est nécessaire, avant d'ad-ministrer un myorelaxant, de donnerune substance provoquant la perte deconscience (narcotique). L'action d'unedose dure environ 30 minutes.
Cette durée d'action peut être rac-courcie par la prise d'un inhibiteur del'acétylcholinestérase par ex. la néo-stigmine (p. 102). Compte tenu de l'in-hibition de la dégradation de l'ACh li-bérée, sa concentration augmente dansla plaque motrice, l'ACh déplace defaçon compétitive la d-tubocurarine desrécepteurs et permet au muscle de secontracter à nouveau.
Les e f f e t s secondaires de la d-tu-bocurarine peuvent être une libérationnon allergique d'histamine par les mas-tocytes avec bronchospasme, urticaireet hypertension. On attribue cependantle plus souvent la chute de tension à unblocage des ganglions.
Le pancuronium est un composéde synthèse, souvent utilisé aujour-d'hui, ne jouant aucun rôle sur la libéra-tion d'histamine ou le blocage des gan-glions. Le pancuronium est environ5 fois plus actif que la d-tubocurarine,l'effet dure plus longtemps. On observeune augmentation de la fréquence car-diaque et de la pression artérielle liéesau blocage des récepteurs muscari-niques de l'ACh au niveau cardiaque.
Il existe d'autres myorelaxantsnon dépolarisants, ce sont : les dérivésdu pancuronium, vecuronium, rocuro-nium et pipecuronium ainsi que l'ai-curonium qui dérive d'un alcaloïde, latoxiférine. L'atracurium possède laparticularité d'être dégradé spontané-ment, sans participation d'enzymes. Ladisparition de son action est donc indé-pendante des fonctions des organesd'élimination.
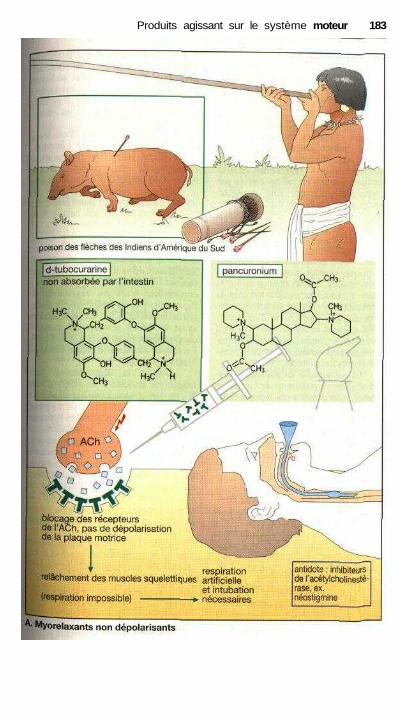
Produits agissant sur le système moteur 183

184 Produits agissant sur le système moteur
Myorelaxants dépolarisants
La succinylcholine a une utilité clinique(succinyidicholine, suxaméthonium. A).Dans son cas, il s'agit en quelque sorted'un double de l'ACh. Comme l'acétyl-choline, la succinylcholine agit au ni-veau des récepteurs nicotiniques de laplaque motrice comme un agoniste.Malgré tout, elle provoque une relaxa-tion musculaire : à l'inverse de l'ACh, lasuccinylcholine n'est pas hydrolysée parl'acétylcholinestérase. Elle constitueseulement un substrat des cholinesté-rases non spécifiques (cholinestérasessériques, p. 100). La succinylcholinesera donc dégradée plus lentement quel'ACh, persistera donc quelques minutesdans la fente synaptique et dépolariserala plaque motrice pendant un tempscorrespondant. La dépolarisation de laplaque motrice suscite d'abord dans lamembrane des cellules musculaires en-vironnantes la propagation d'un poten-tiel d'action et la contraction des fibresmusculaires : après injection intravei-neuse on peut observer des petits tres-saillements des muscles.
La naissance d'un nouveau poten-tiel d'action à proximité de la plaquemotrice n'est possible que lorsque celle-ci demeure non stimulée pendant un cer-tain laps de temps et peut se repolariser.Le potentiel d'action repose sur l'ouver-ture d'une protéine canal pour les ionsNa^ à travers laquelle se produit un fluxd'ions Na^ qui dépolarise la membrane.Après quelques millisecondes, les ca-naux sodiques se ferment automatique-ment (inactivation), le potentiel de mem-brane revient à sa valeur de repos, lepotentiel d'action se termine. Aussilongtemps que le potentiel de membranene s'est pas rapproché de sa valeur derepos, l'ouverture des canaux sodiqueset donc la formation du potentiel d'ac-tion suivant sont impossibles. Dans lecas d'une libération d'acétylcholine, larepolarisation de la plaque motrice et unretour de l'excitabilité des canaux io-niques dans les membranes environ-nantes se produisent rapidement à causede la dégradation très rapide par l'acétyl-cholinestérase. Pour la succinylcholine,
la dépolarisation de la plaque motrice e>également du domaine membranairealentour persiste. Les canaux sodiquessont maintenus dans un état inactivé etde ce fait il n'est pas possible de générerun potentiel d'action dans les mem-branes environnantes.
Comme la plupart des fibres mus-culaires sont innervées par l'intermé-diaire d'une seule plaque motricel'excitation de celle-ci permet la propa-gation d'un potentiel d'action à traversla membrane cellulaire sur plus de30 cm. Le blocage des potentiels d'ac-tion maintient la fibre musculaire dansun état relaxé.
L'action d'une dose habituelle desuccinylcholine persiste seulement unedizaine de minutes. Elle est souvent ad-ministrée au début d'une anesthésie pourfaciliter l'intubation. L'action de la suc-cinylcholine n'est pas augmentée defaçon nette par les inhibiteurs de l'acé-tylcholinestérase. Chez les rares patientsayant un déficit génétique en pseudo-cholinestérase (estérase non spécifique),l'action de la succinylcholine est nota-blement prolongée.
Comme la dépolarisation pro-longée est associée à un efflux d'ionsK^ on peut aboutir à une hyperkaliémie(avec danger d'arythmies cardiaques).C'est seulement dans quelques types demuscle (par exemple les muscles ex-ternes de l'œil) que les fibres muscu-laires sont innervées par plusieursplaques motrices. Dans ce cas, la succi-nylcholine déclenche une dépolarisationrépartie sur la totalité de la fibre. Ceci estassocié à une contraction prolongée : lapression interne de l'œil s'élève, ce quidoit être surveillé pendant une opérationde l'œil.
Pour les muscles squelettiquesdont le nerf a été sectionné, on observeque les récepteurs de l'acétylcholine serépartissent sur la totalité de la mem-brane de la fibre. Dans ce cas également,la succinylcholine produirait une dépo-larisation prolongée et une contractureainsi que le déclenchement d'une hyper-kaliémie. Ceci peut par exemple se pro-duire à la suite d'une opération chez unpatient polytraumatisé.
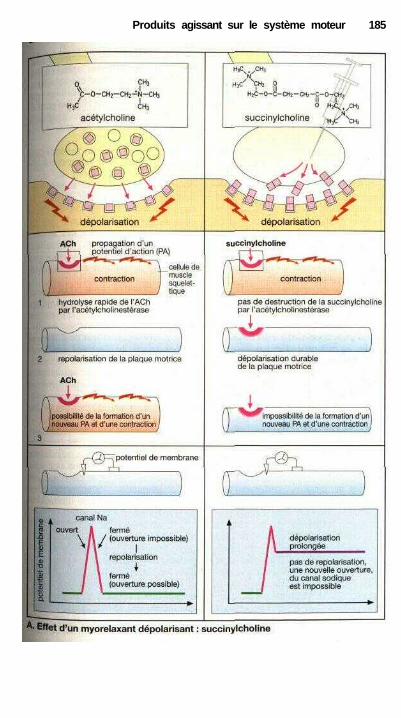
Produits agissant sur le système moteur 185

186 Produits agissant sur le système moteur
Antiparkinsoniens
La maladie de Parkinson (paralysie agi-tante) est associée à une destructiondes neurones dopaminergiques quirelient la substantiel nigra au corpusstriatum et participent au contrôle desactivités motrices extrapyramidales eninhibant l'activité des neurones choli-nergiques. La maladie repose sur un dé-ficit en dopamine (D) et un excès relatifen acétylcholine. Les principaux symp-tômes de la maladie sont : une pauvretéde mouvement (akinésie) une rigiditédes muscles (rigor) et des tremblements(tremor).
A l'aide de moyens pharmacolo-giques, on cherchera à équilibrer etcompenser la carence en D ou à dimi-nuer la prépondérance de l'activité cho-linergique.
L-DOPA. Comme il s'agit d'undéficit en D dans le système nerveuxcentral, il est nécessaire de remplacer laD dans ce système. La D, catécholaminepolaire, ne peut cependant pas traverserla barrière hémato-encéphalique. C'estpourquoi on utilisera son précuseur laL-DihydrOxyPhénylAlanine (L-DOPA)qui, en tant qu'acide aminé, sera trans-portée de façon active à travers la bar-rière hémato-encéphalique, et sera en-suite décarboxylée in situ par laDOPA-décarboxylase pour donner la D.
La L-DOPA apportée est égale-ment transformée en D à l'extérieurdu cerveau. Elle n'est cependant pas né-cessaire et produira seulement deseffets indésirables (tachycardie, alté-rations du rythme par suite d'une stimu-lation des récepteurs (3i [p. 114] etchute de tension). La formation de D enpériphérie peut être inhibée par l'admi-nistration simultanée d'inhibiteurs de laDOPA-décarboxylase (carbidopa, ben-sérazide). Ceux-ci ne traversent pas labarrière hémato-encéphalique et la dé-carboxylation dans le cerveau demeureinchangée.
Les effets secondaires d'une élé-vation cérébrale de la concentration deD peuvent être : hyperkinésie, vomisse-ments, altérations psychiques.
Agonistes dopaminergiquggPour compenser le déficit central en do-'pamine, on peut utiliser des agonistesdopaminergiques comme la bromocrin.tine (p. 114), le lisuride et le pergolideLes effets secondaires de ces agonistesne sont pas différents de ceux de laL-DOPA.
Inhibiteurs de la monoamineoxydase-B (MAO-B). La monoamineoxydase existe sous forme de deuxisoenzymes la MAO-A et la MAO-B.Le foie contient les deux formes, lecorps strié est riche en MAO-B. Cetteisoenzyme peut être bloquée par lasélégilme ; la dégradation des aminésbiogènes (noradrénaline, adrénaline,sérotonine) en périphérie ne sera pasbloquée car la capacité de la MAO-Ademeure.
Anticholinergiques. Les anta-gonistes des récepteurs muscariniquesde l'acétylcholine ayant une actioncentrale (par ex. bematropine, bipéri-dène, p. 106), permettent de diminuerl'excès relatif de l'activité choliner-gique (en particulier les tremble-ments). Les effets secondairestypiques, de type atropinique, limitentles doses utilisables. Il n'est pas pos-sible d'obtenir une disparition totaledes symptômes.
Amantadine. Au début de la ma-ladie, les symptômes peuvent être atté-nués avec l'amantadine. Le mécanismed'action de l'amantadine est vraisem-blablement un blocage du canal ioniquedes récepteurs glutamatergiquesNMDA, et finalement une diminutionde la libération d'acétylcholine.
L'administration de L-DOPA oud'un agoniste dopaminergique consti-tue le traitement le plus efficace de lamaladie de Parkinson. C'est seulement
' dans les stades précoces de la maladieet lorsque certains symptômes prédo-minent que l'amantadine ou les anti-cholinergiques seront utilisés seuls.Dans les stades plus tardifs, on devracombiner plusieurs antiparkinsomenspour contrôler les symptômes de lamaladie.
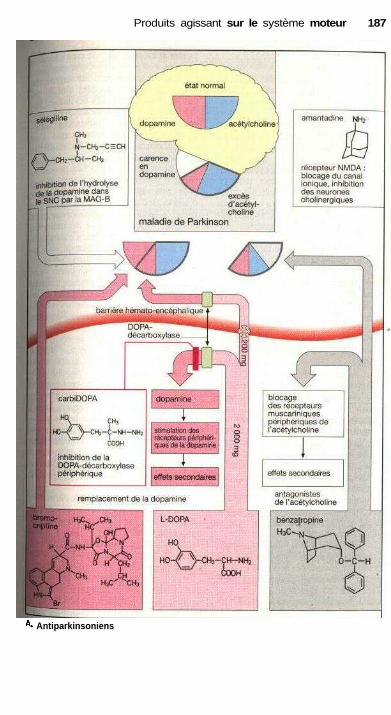
Produits agissant sur le système moteur 187
A- Antiparkinsoniens

188 Produits agissant sur le système moteur
Anticpileptiques
L'épilepsie est une maladie cérébralechronique, d'étiologie variable, caracté-risée par une excitation neuronale incon-trôlée, survenant par accès et limitée dansle temps. La décharge électrique qui peuttoucher une zone cérébrale de taille va-riable peut être mise en évidence surl'électroencéphalogramme sous formed'une activité synchronisée et peut se ma-nifester par des phénomènes moteurs,sensitifs, psychiques ou végétatifs. Etantdonné que la zone du cerveau touchée parla stimulation électrique, mais égalementl'origine de la décharge électrique peu-vent être très variables, l'épilepsie peut semanifester sous différentes formes. D'unpoint de vue thérapeutique, on distingue :- les crises généralisées ou focales ;- les crises avec ou sans perte deconscience ;- les crises avec ou sans déclencheurconnu.Compte tenu de la courte durée des accèsde convulsion un traitement médicamen-teux immédiat n'est pratiquement paspossible. Les antiépileptiques servent sur-tout à la prévention des crises et serontutilisés dans ce but de façon chronique.Ce n'est que lorsqu'un état épileptiquepersiste (succession de plusieurs accèscloniques-toniques) qu'un traitementd'urgence est appliqué, le plus souventavec une benzodiazépine, si possible eni.v., ou à défaut par voie rectale.
Les cellules entraîneuses sont indis-pensables au déclenchement de la crised'épilepsie. Elles se distinguent des autrescellules nerveuses par l'instabilité de leurpotentiel de repos, ce qui signifie qu'à lafin d'un potentiel d'action, persiste uncourant dépolarisant.
Les interventions thérapeutiques ontdonc pour but de stabiliser le potentiel demembrane des cellules nerveuses et de di-minuer l'excitabilité. Dans chaque formed'épilepsie on cherchera d'abord à at-teindre à l'aide d'un médicament une pé-riode dépourvue de crise. Dans le casd'accès généralisés, c'est en générall'acide valproi'que qui sera le premierchoix, tandis que dans le cas de crises
focales et en particulier de crises focalescomplexes on préférera la carbamazépineLa dose du médicament est augmentéejusqu'à ce qu'il n'y ait plus d'accès ouque les effets secondaires deviennent in.supportables. Ce n'est que lorsque la nio-nothérapie avec des substances diffé-rentes n'est pas suffisante, que l'onrecommandera le passage à une moléculede deuxième intention ou à une associa-tion (B), et dans ce cas il faut penser aurisque d'interactions pharmacocinétiques(voir ci-dessous). Le véritable mécanismed'action des antiépileptiques n'est pasconnu. Chacune des substances semblediminuer l'excitabilité par de multiplesmécanismes. En principe, l'excitabilitéd'un neurone peut être diminuée via l'in-hibition d'un neurone excitateur ou la sti-mulation d'un neurone inhibiteur. Lesprincipaux neurones excitateurs utilisentcomme neurotransmetteur l'acide gluta-mique, les principaux neurones inhibi-teurs utilisent l'acide -y-aminobutyrique(GABA) (191 A).
Il existe trois types de récepteursdu glutamate, dont le récepteur NMDAjoue d'un point de vue thérapeutique lerôle le plus important (le N-méthyl-D-aspartate est un agoniste de synthèse trèssélectif). Il s'agit d'un canal ioniqueactivé par un ligand, à travers lequelpénètrent après stimulation par le gluta-mate, des ions sodium mais égalementcalcium. La lamotrigine, la phénytoïneet le phénobarbital inhibent entre autresla libération de glutamate ; lefelbamatese comporte comme un antagonisteglutamatergique.
Les benzodiazépine s et \e phénobar-bital renforcent l'activation des récep-teurs GABA^ par une libération physiolo-gique de GABA (B) (p. 224). L'influxaccru de chlore s'oppose à une dépolari-sation de la membrane. Le progabide estun analogue direct du GABA. La tiaga-bine bloque l'élimination du GABA de lafente synaptique, en inhibant la recapturecellulaire. La vigabatrine inhibe la dégra-dation du GABA ; la gabapentine aug-mente la disponibilité de l'acide gluta-mique en tant que précurseur pour l2
synthèse du GABA (B).
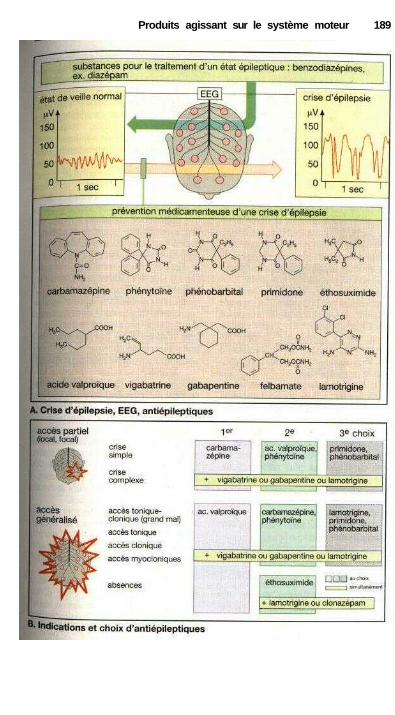
Produits agissant sur le système moteur 189

190 Produits agissant sur le système moteur
La carbamazepme 1 acide valproique et la phen\1ome bloquent descanaux sodiques dépendants du poten-tiel et inhibent la propagation de 1 exci-tation électrique
L ethosuximide bloque entre autresun canal calcique neuronal de type T(A) II occupe une position a part car iln'est actif que dans les cas d absences
Tous les antiepileptiques présen-tent des effets secondaires mais avecune intensité très variable Pratique-ment chaque traitement antiepileptiqueest accompagne d une sedatwn d unebaisse de concentration et d une dimi-nution de la motivation De plus des alterations cutanées ou de la formule san-guine peuvent imposer un changementdu produit antiepileptique Le phéno-barbital la pnmidone et la phenytomepeuvent conduire a une osteomolacie(prévention par la vitamine D) ou a uneanémie megaloblastique (préventionpar 1 acide tolique) Au cours du traite-ment par la phenytome on peut ob-server chez environ 20 % des patientsun développement des gencives (hyper-plasie gingivale)
L acide valproique est moins sé-datif que les autres anticonvulsivants,les effets secondaires les plus fréquem-ment observes sont des tremblements,des douleurs gastro intestinales et uneaugmentation de poids une chute ré-versible des cheveux est également unedes conséquences plus rare du traite-ment on observe très rarement des at-teintes hépatiques dangereuses en par-ticulier chez les enfants de moins detrois ans
Lors de 1 administration de car-bamazepine et en particulier a la suited une augmentation rapide de la dosepeuvent apparaître a cote de la seda-tion et d une sensation d engourdisse-ment des signes d intoxication nys-tagmus ataxie vision double Onobservera souvent des douleurs gastro-intestinales et des éruptions cutanéesLa carbamazepme a un effet antidiure-tique (sensibilisation du tubule collec-teur a la vasopressme -» empoisonne-ment par l'eau)
La carbamazepme est égalementutilisée pour le traitement d'une nevralgie tngemmale ou de douleurs neuropathiques
L acide valproique la carbarnazepme et d autres antiepileptiquecaugmentent le risque teratoueneCependant il est contre indique d mterrompre le traitement pendant la ç; osses'se car le risque pour 1 embryon durant une crise est encore plus grandDans ces conditions on s attacheraavec un soin particulier a utiliser la dosela plus faible ayant une action prcventive fiable et on cherchera a éviter desaltérations du tube neural par 1 administration de doses élevées d acide tolique
La carbamazepme la phenvtomeet d autres anticonvulsivants induisentdans le foie la synthèse d en^mi s participant a la dégradation des nudicaments La combinaison d anticomulsivants mais également 1 administiationsimultanée d autres médicaments peuvent conduire a des interactions ayantune repercussion clinique (surveillanceaccrue des niveaux plasmatiques)
D autres substances peuvent êtreutilisées dans les epilepsies de l'enfantsouvent difficiles a soigner ce sont parexemple le bromure 1 ACTH et ladexamethasone un glucocorticoide
II faut noter qu un ensemble demédicaments peuvent diminuer le seuild'apparition de crampes (neuroleptiques 1 isomazide un antituberculeux ou les antibiotiques P lactames aforte dose) et sont donc contre indiqueschez des patients epileptiques
Les benzodiazepines sont peu utilisees pour des traitements de longuedurée a cause de 1 apparition de phenomenés de tolérance mais ils constituentle traitement de choix pour une crised epilepsie
Le clomethlazol peut égalementêtre utilise lors de la survenue d unecrise le plus souvent il servira au traitement d états agîtes en particulier lesdélires alcooliques (ou peuvent apparaître entre autres des convulsions)
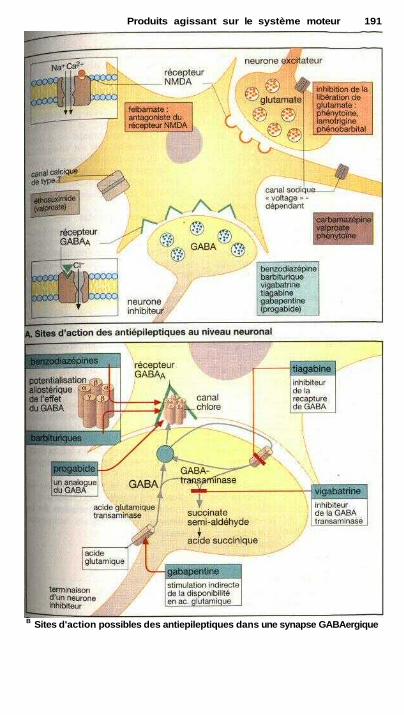
Produits agissant sur le système moteur 191
B Sites d'action possibles des antiepileptiques dans une synapse GABAergique

192 Analgésiques
Origine et conduction de la douleur
Le terme de douleur désigne un spectrede sensations dont les caractéris-tiqmes peuvent être très différentes etdoint l'intensité peut aller du désa-gréable à l'insupportable. Les stimulidoialoureux sont enregistrés par desrécepteurs physiologiques, peu diffé-reniciés sur le plan morphologique(récepteurs sensitifs) qui sont en faitdes, terminaisons nerveuses libres. Lecor-ps cellulaire du neurone bipolaire af-fénent 1 est situé dans le ganglionspiinal. La conduction de la douleurest assurée par des fibres non myélini-sée;s (fibres C, vitesse de conduction0,5i-2 m/s) et des fibres myélinisées(fibres AS, 5 à 30 m/s.). Les terminai-sorns nerveuses des fibres A8 répondentà l'a chaleur et à de fortes pressions,tanidis que les terminaisons des fibres Créâjgissent aux stimuli chimiqueslonmés à la suite d'une lésion de tissus(H^, K+, histamine, bradykinine...).Qui'elle soit provoquée par un stimuluschilmique, mécanique ou thermique, lasenisation douloureuse est fortementreniforcée en présence de prostaglan-dinies (p. 194).
Des signaux chimiques sont à labas;e des douleurs consécutives à uneinfliammation ou une ischémie (anginede poitrine, infarctus) ou encore desforttes douleurs provoquées dans la ca-vitég abdominale par une extension ouune; stimulation spastique des organesmuisculaires lisses, et qui sont entrete-nue;s par une hypoxie s'achevant entétamie (douleurs viscérales).
Les fibres A8 et C pénètrent dansla rmoelle épinière par la racine posté-riemre ; après relais sur un autre neuronela v/oie croise de l'autre côté et parvientau (cerveau par le cordon antérieur. Aucouu-s de l'évolution, les tractus néo- etspirnothalamiques se sont différenciés.La zone des noyaux thalamiques danslaqmelle aboutissent les fibres du tractus
néothalamique, envoient des irnoiiisions dans des aires définies du ayn 'post-centralis. Un stimulus empruntantcette voie sera ressenti comme pluaigu et mieux localisé. Dans le cas dpnoyaux anciens innervés par le tractupaléospinothalamique, la projectiondans le gyrus post-centralis est diffusede sorte que cette voie est considéréecomme la voie de conduction des sti-muli donnant naissance à des douleurssourdes, taraudantes, cuisantes et qui nepeuvent pas être localisées avec préci-sion par l'individu.
La conduction des impulsionsdans le tractus néo- et paléospinothala-mique est modulée par des fibres des-cendantes dont l'origine est dans la for-mation réticulée et qui se terminentdans la moelle épinière au niveau du re-lais entre les neurones 1 et 2 (systèmeantinociceptif descendant). Par la li-bération d'oligopeptides (enképha-lines), elles peuvent inhiber le passagedu neurone 1 au neurone 2.
La sensation douloureuse peutêtre influencée de la façon suivante :- interruption de la cause de la douleur,- diminution de la sensibilité des noci-
cepteurs (antipyrétiques, analgé-siques, anesthésiques locaux),
- interruption de la conduction dansles nerfs sensitifs (anesthésiques lo-caux),
- suppression du relais des influx dou-loureux dans la moelle épinière(opioïdes),
- inhibition de la perception de ladouleur (opioïdes, narcotiques) et
- modulation de l'assimilation de ladouleur (antidépresseurs comme co-analgésiques).
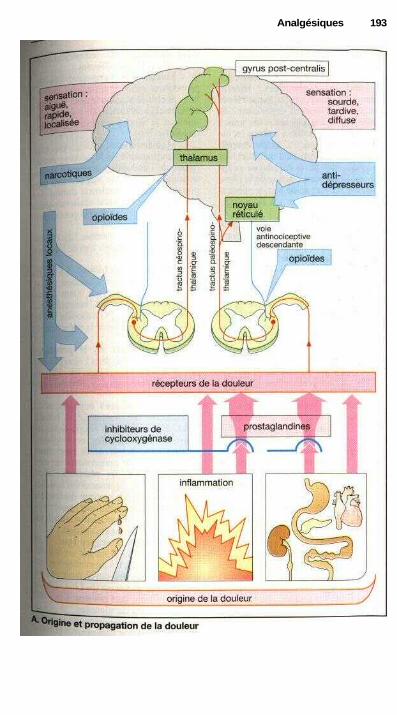
Analgésiques 193

194 Analgésiques antipyrétiques
Eicosanoïdes
Formation et métabolisme. Les eico-sanoïdes, prostaglandines, throm-boxane, prostacycline et leucotriènesproviennent dans l 'organisme del'acide arachidonique, un acide gras à20 carbones et 4 doubles liaisons (acideeicosa-tétra-énoïque). L'acide arachi-donique, un constituant habituel desphospholipides de la membrane cellu-laire, est libéré sous l'action de la phos-pholipase A;. Cet acide sert de substratà la cyclooxygénase et aux lipooxygé-nases.
La formation des prostaglandines(PG), de la prostacycline et du throm-boxane passe par l'intermédiaire d'unendoperoxyde cyclique. Dans le casdes PG, il se forme dans la chaîne car-bonée de l'acide gras un noyau cyclo-pentane. Avec la lettre suivant l'abré-viation PG (D, E, P, G, H ou I) estindiquée la différence entre les susbti-tuants hydroxyles ou cétones ; l'indicedonne une information sur le nombre dedoubles liaisons, et la lettre grecque surla position du groupe hydroxyle en C9(sur la figure est représentée la PGF^).Les PG sont en premier lieu dégradéespar l'enzyme 15-hydroxy-prostaglan-dine déshydrogénase. Dans le plasma,l'inactivation se produit très rapide-ment, en un passage à travers le pou-mon ; 90 % des prostaglandines pré-sentes dans le plasma seront dégradées.Il s'agit d'hormones locales, qui nes'accumulent en concentrations biolo-giquement actives qu'à l'endroit de leurformation.
Effets biologiques. Chacune desprostaglandines (PGE, PGF, PGI =prostacycline) présente des propriétésbiologiques distinctes.
Récepteurs de la douleur. LesPG augmentent la sensibilité aux sti-muli douleureux usuels (p. 192), cecisignifie que pour un stimulus donné, lafréquence du potentiel d'action dé-clenché au niveau des nerfs sensitifs estaugmentée.
Centre régulateur de la tempé-rature dans l'hypothalamus. Les PGaugmentent la valeur seuil du centre
thennorégulateur, la température del'organisme s'élève (fièvre).
Muscles des vaisseaux. Les PQprovoquent une vasodilatation.
Sécrétions gastriques. Les PQaccélèrent la formation de mucus et di-minuent la sécrétion d'acide chlorhy-drique (p. 166).
Menstruations. PGF;,, est sansdoute responsable de la nécrose isché-mique de l'endomètre avant les mens-truations ; en cas de saignements im-portants ou de douleurs durant lesmenstruations, il existe vraisemblable-ment un déséquilibre des proportions dechaque prostaglandine.
Musculature utérine. Les PG sti-mulent les contractions lors de l'accou-chement.
Muscles bronchiques. La PGE;provoque une bronchodilatation.
Flux sanguin rénal. Dans le casd'une réduction du flux sanguin rénalseront libérées des PG vasodilatatricesqui vont contrebalancer la réduction del'irrigation.
Thromboxane A; et prostacy-cline jouent un rôle dans l'agrégabilitédes plaquettes sanguines (p. 148) etdans la régulation du diamètre des vais-seaux.
Les leucotriènes augmentent laperméabilité des vaisseaux et consti-tuent des substances chimiotactiquespour les polynucléaires neutrophiles.Comme constituants de la slow reac-ting substance of anaphylaxis ce sontdes médiateurs des réactions aller-giques (p. 320) ; en association avec lesprostaglandines, ils peuvent déclencherl'ensemble des symptômes caractéris-tiques d'une inflammation (chaleur,rougeur, gonflement et douleur).
Utilisation thérapeutique. Lesdérivés des prostaglandines seront uti-lisés pour le déclenchement d'un ac-couchement et entre autres, pour une in-terruption de grossesse (p. 126), dans lecas d'un ulcère de l'estomac ( p. 166)ou de désordres circulatoires.
La tolérance est mauvaise car ellesne peuvent pas être administrées connuehormone locale mais doivent plutôt êtreutilisées de façon systémique.
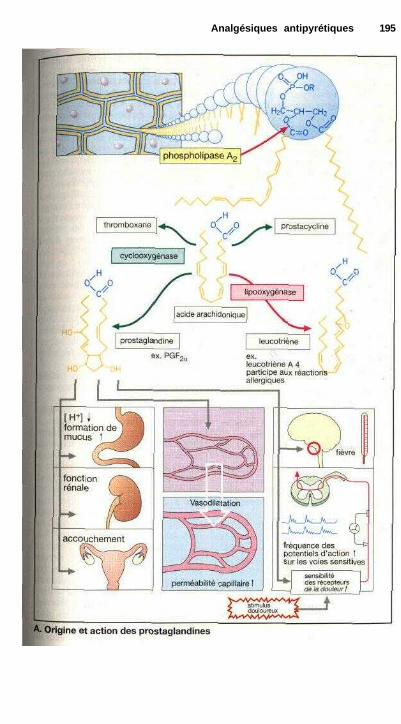
Analgésiques antipyrétiques 195

196 Analgésiques antipyrétiques
Analgésiques antipyrétiques
Le paracétamol, les acides amphiphilestels l'acide acétylsalicylique (AAS), l'ibu-profène et d'autres ainsi que les dérivésde la pyrazolone, métamizole et propyphé-nazone sont appelés analgésiques anti-pyrétiques, car ils possèdent tous, à ladifférence des analgésiques opioïdes, lapropriété de faire baisser la fièvre.
Le paracétamol a une bonne acti-vité contre les maux de tête et les maux dedents, mais il est moins actif contre lesdouleurs viscérales ou inflammatoires.Son mécanisme d'action est inconnu. Lamolécule peut être administrée per os ousous forme de suppositoire (dose indivi-duelle 0,5-1 g). L'action se manifesteaprès environ 30 minutes et dure à peuprès 3 h. Le paracétamol se conjugue dansl'organisme avec l'acide glucuronique oul'acide sulfurique au niveau du groupe-ment OH phénolique, et est éliminé par lerein sous cette forme. Aux doses théra-peutiques, une fraction plus faible seraoxydée sous forme d'une N-acétyl-p ben-zoquinonimine qui sera éliminée aprèsconjugaison au glutathion. En cas de prisede doses élevées (environ 10 g), le niveaude glutathion dans le foie ne suffit plus àassurer l'élimination et la qumonimine ré-agit avec certains composants des celluleshépatiques : les cellules meurent : nécrosehépatique. Cependant, si dans les 6-8 hsuivant l'absorption d'une dose élevée deparacétamol, on injecte par voie i.v. la N-acétylcystéine, un donneur de groupe-ments SH, il est possible d'éviter les lé-sions hépatiques. Un usage régulierpendant des années peut provoquer unealtération des fonctions rénales.
L'acide acétylsalicylique (AAS)possède à côté de son effet analgésique etantipyrétique une action antiphlogistique.Les effets peuvent être attribués à une in-hibition de la cyclooxygénase (p. 194).L'AAS peut être utilisée per os en com-primé, ou en solution en utilisant un com-primé effervescent ou encore injectéesous forme de lysinate (dose analgésiqueou antipyrétique 0,5-1 g). L'AAS est rapi-dement hydrolyse d'abord dans l'intestinet plus tard dans le sang en donnantl'acide salicylique. L'effet de l'AAS dure
plus longtemps que ne persiste la molecule dans le plasma (t,,, d'environ 20 mnutes), car la cyclooxygénase est inhibéede façon irréversible par liaison du eroupement acétyl de l'AAS et la durée d'action est donc conditionnée par la nouvel!?synthèse de l'enzyme. De plus, l'acide sa-licylique peut également contribuer al'effet. L'AAS irrite la muqueuse de l'es-tomac (p. 198). Chez des patients sensibi-lisés, il peut déclencher une bronchocons-triction (asthme aux analgésiques) etd'autres réactions pseudoallergiques)(p. 198). Comme l'AAS inhibe l'agréga-tion des plaquettes (p. 148) sanguines, ilne doit pas être utilisé chez des patientsayant des troubles de coagulation. I] fautfaire attention au syndrome de Reye chezles enfants et les adolescents ; ce syn-drome est observé lors d'une infection vi-rale associée à de la fièvre avec prised'AAS ; son pronostic est mauvais(altérations cérébrales et hépatiques).L'administration d'AAS n'est pas recom-mandée à la fin de la grossesse : diminu-tion des contractions, risque de saigne-ments chez la mère et l'enfant, fermetureprématurée du canal artériel.
Les anti-inflammatoires acides(p. 198 et suivantes) se comportentcomme l'AAS.
Le plus puissant des analgésiquesantipyrétiques est le métamizole. Ilpermet d'atténuer également les douleursviscérales. Son mécanisme d'action estinconnu. Il est absorbé de façon suffisanteaprès administration orale ou rectale etpeut, étant soluble dans l'eau, être injecté.Son métabolite actif, la 4-aminophéna-zone a une demi-vie plasmatique (t 1/2)d'environ 5 heures. La prise de métami-zole est associée à un risque rarissimemais dangereux d'agranulocytose. Chezdes personnes sensibilisées, on peut enparticulier déclencher un choc circula-toire après administration intraveineuse.Le métamizole doit être utilisé seulementdans les états douloureux qui ne peuventpas être traités par d'autres substances.Vraisemblablement, les propriétés phar-mocologiques et toxicologiques de la/w-pyphénaïone sont identiques à celles dumétamizole.
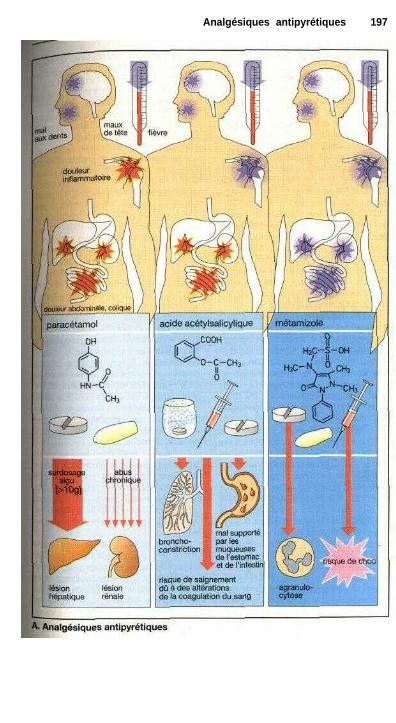
Analgésiques antipyrétiques 197

198 Analgésiques antipyrétiques
Anti-inflammatoires non stéroïdiens
A dose relativement élevée (^ 4 g/j)l'acide acétylsalicylique (AAS, p. 196)peut exercer des effets anti-inflamma-toires dans les maladies rhumatismales,par exemple la polyarthrite rhumatoide.Dans cette gamme de concentrations peu-vent cependant se manifester des signesde surdosage au niveau du SNC (bour-donnements d'oreille, vertiges, étourdis-sements, etc.). La recherche de médica-ments mieux tolérés a conduit au groupedes anti-inflammatoires non stéroïdiens(AINS). Leur caractéristique communeest leur caractère acide (anti-inflamma-toires acides). Il s'agit soit d'un acidecarboxylique (par ex. le diclofénac, l'ibu-profène, le naproxène, l'indométacine[formule p. 315]) ou bien des acides énol(par ex. azapropazone, piroxicam ainsique la phénylbutazone connue depuislongtemps mais mal tolérée).
Ces substances analgésiques, anti-pyrétiques et anti-inflammatoires agissentcomme l'AAS et bloquent la cyclooxygé-nase, mais de façon réversible contrai-rement à l'AAS. Ces substances neconviennent donc pas comme inhibiteursde l'agrégation plaquettaire. Le choixd'un traitement reposera sur les diffé-rences de propriétés pharmacocinétiqueset d'effets secondaires.
Pharmacocinétique. Les AINSsont bien absorbés par voie entérale. Leurliaison aux protéines plasmatiques estélevée (A). Ils seront éliminés avec unerapidité très variable ; comparez parexemple le diclofénac (t 1/2 = 1-2 h) et lepiroxicam (t 1/2 ~ 50 h). Cet aspect joueun rôle important en ce qui concerne lafréquence des prises et le danger d'accu-mulation, L'élimination de l'acide salicy-lique, le métabolite formé très rapidementà partir de l'AAS, présente comme parti-cularité de dépendre de la dose. Sauf dansle cas d'une urine alcaline, l'acide salicy-lique est aisément réabsorbé par le rein.Une conjugaison hépatique préalable,principalement sur la glycine (ac. salicy-lurique) ou sur l'acide glucuronique, estune condition nécessaire à une élimina-tion rapide. C'est lors de l'administrationd'une dose importante que l'on remarque
la capacité limitée des réactions de conju.gaison : l'augmentation de l'éliminationne dépend alors que de l'excrétion rénalede l'acide salicylique non métabolisé, qu,s'effectue assez lentement.
Effets secondaires caractéristiquesdu groupe (B). Ils peuvent être attribués àl'inhibition de la cyclooxygénase. Lesplus fréquents, altérations de la mu-queuse gastrique avec risque d'ulcèrepeptique, sont dus principalement (à côtéd'un effet acide direct) à l'inhibition de lasynthèse des prostaglandines (PG) proté-geant la muqueuse. La gastropathie peutêtre empêchée grâce à l'utilisation d'unanalogue des PG, le misoprostol (p. 166).Chez des patients sensibilisés, peuvent seproduire des crises d'a.\thme, vraisem-blablement par suite d'une carence enprostaglandines bronchodilatatrices etune production accrue de leucotnènes.Des réactions « pseudo-allergiques »semblables peuvent survenir avec toutesles molécules de ce groupe. Les PGjouent un rôle dans le contrôle de lacirculation rénale comme antagonistesfonctionnels de l'angiotensine II et de lanoradrénaline. Si leur libération est aug-mentée (à la suite d'une hypovolémie parex.), l'inhibition de la synthèse de PGpeut entraîner une diminution de la cin u-lation sanguine et des fonctions rénales.D'autres actions secondaires sont la for-mation d'œdèmes et l'augmentation de lapression artérielle.
On doit également faire attentionaux effets secondaires propres à chaquesubstance. Ils touchent par exemple leSNC (indométacine : maux de tête,engourdissement, confusion), la peau(piroxicam : hypersensibilité à la lumière)ou le sang (phénylbutazone : agranulo-cytose).
Perspective. Il existe deux isoen-zymes de la cyclooxygénase : Cox-1 quiest exprimé de façon constitutive (perma-nente), par exemple dans l'estomac et lesreins, et Cox-2 qui est formée au coursdes réactions inflammatoires (forme in-ductible). Les AINS dont nous disposonsaujourd'hui inhibent les deux isoen-zymes. On recherche activement des inhi-biteurs sélectifs de Cox 2 qui devraientthéoriquement être mieux supportés.
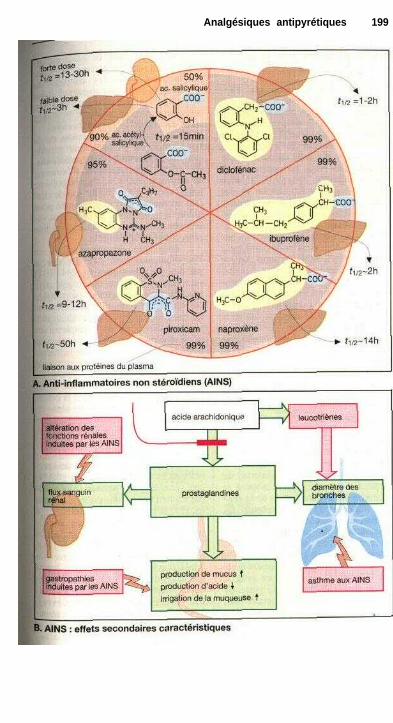
Analgésiques antipyrétiques 199

200 Analgésiques antipyrétiques
Régulation thermique du corpset antipyrétiques
La température du corps est d'en-viron 37° chez l'homme et varie à peuprès de 1 °C au cours de la journée. Aurepos, environ 25 % de la production dechaleur totale sont fournis par l'activitémétabolique du foie, 20 % par celle ducerveau, 8 % par celle du cœur et 7 %par celle des reins. La production dechaleur augmente fortement lors d'uneffort. La contribution absolue de cesorganes à la production de chaleurvarie peu lors d'une période d'activitéde l'organisme si bien que le travailmusculaire, qui au repos produit en-viron 25 % de la chaleur du corps, peutfournir Jusqu'à 90 % de cette chaleurlors d'une activité physique intense.Les vaisseaux sanguins qui irriguentla peau traversent la couche isolanteformée par le tissu adipeux et permet-tent, en fonction du diamètre des vais-seaux et de l'irrigation, de fournir àl'environnement une quantité de cha-leur très variable. L'irrigation de lapeau peut représenter, selon les besoins,à peine plus de zéro à 30 % du débitcardiaque. Le transport de chaleur parle sang, du site de production à l'inté-rieur du corps vers la surface du corps,est ainsi une voie commode d'élimina-tion de la chaleur.
À côté de la perte de chaleur parconduction et rayonnement, il est éga-lement possible d'éliminer de la cha-leur par une production accrue desueur. En effet, la sueur s'évapore etcette évaporation consomme de la cha-leur (chaleur de vaporisation). Larégulation du flux sanguin cutané et dela production de sueur par le systèmenerveux végétatif permet d'ajuster lavaleur effective de la température ducorps au seuil fourni par le centre ther-morégulateur (A). Le système sympa-thique peut, soit réduire la perte dechaleur par une vasoconstriction, soit,inversement, l'augmenter par unesécrétion accrue de sueur. Le tremble-ment des muscles est un moyen del'organisme pour augmenter la produc-tion de chaleur.
Si la production de sueur est inhibée par un empoisonnement par les na.rasympatholytiques (ex. atropine) ]„flux sanguin cutané est augmenté, si onne peut parvenir par cette voie à une éli.mination de chaleur suffisante, on aboutità une « surchauffe » (hyperthermie)
Le système de régulation de latempérature est en particulier stimulépar une hyperactivité thyroïdienneEn effet l'hypersécrétion d'hormonethyroïdienne (élévation du métabolismebasai) aboutit à une production de cha-leur accrue qui doit être éliminée pourgarder la température du corps à sa va-leur physiologique ; les patients ont unepeau chaude et transpirent.
Le centre thennorégulateur del'hypothalamus peut être déconnectépar des neuroleptiques (p. 234) (Bl)sans que d'autres centres soient déjàtouchés. De cette façon, il est possiblede refroidir le corps d'un malade sansqu'une réaction se déclenche (frissons).Ceci peut être utilisé par exemple en casde fièvre intense ou d'une opération ducœur utilisant une circulation extracor-porelle, où la température du sang peutêtre diminuée jusqu'à 10 °C.
À doses élevées, l'alcool et lesbarbituriques, inhibent aussi le centrerégulateur (Bl) et produisent ainsi unrefroidissement du corps, qui pour destempératures extérieures plus bassespeut conduire à une hypothermie mor-telle (mort de froid des ivrognes)
Les pyrogènes (par ex. des pro-duits du métabolisme bactérien) dépla-cent vers le haut, vraisemblablementpar l'intermédiaire des prostaglandines(p. 194) la valeur du thermostat dans lecentre régulateur (B2). L'organisme di-minue la perte de chaleur par une vaso-constriction des vaisseaux cutanés (sen-sation de froid) et augmente laproduction de chaleur (frissons, trem-blements) de façon à adapter la tempé-rature effective de l'organisme à la va-leur de consigne plus élevée (fièvre).Les antipyrétiques comme le paracé-tamol, l'acide acétylsalicylique et lemétamizole (p. 196) rétablissent la va-leur du thermostat (B2) et entraînentainsi une chute de la fièvre.
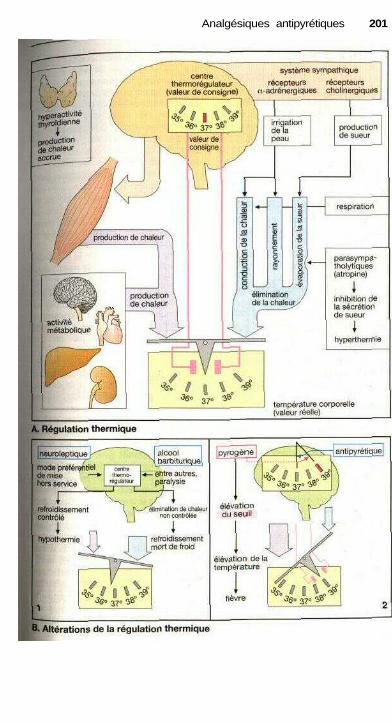
Analgésiques antipyrétiques 201

202 Anesthésiques locaux
Anesthésiques locaux
Les anesthésiques locaux inhibent de fa-çon réversible la formation et la trans-mission des stimuli dans les cellulesnerveuses. Une action de ce type est re-cherchée lorsqu'il s'agit de pratiquerune intervention douloureuse, parexemple une opération chirurgicale ouune extraction dentaire.
Mécanisme d'action. La trans-mission de l'information dans les nerfsse produit sous la forme d'un potentield'action, un changement très rapide dupotentiel de membrane, durant moinsde 1 ms. La dépolarisation a pour ori-gine un influx rapide d'ions Na versl'intérieur de l'axone (A). Cet influx seproduit à travers un canal protéique in-clus dans la membrane qui à l'état ou-vert (activé), laisse pénétrer rapidementde l'extérieur vers l'intérieur des ionssodium en suivant le gradient chimique([Na^,,,, environ 150 mM, [Na4'],,,, en-viron 7 mM). Cet influx rapide de Na'1'peut être inhibé par les anesthésiqueslocaux ; la transmission de l'excitationest bloquée (A).
Les principaux anesthésiques lo-caux existent en partie sous forme ca-tionique amphiphile (voir aussi p. 206).Cette propriété physicochimique faci-lite l'accumulation aux interfaces, do-maines frontières entre milieu polaire etapolaire. Ceux-ci se trouvent dans lesmembranes phospholipidiques et à l'in-térieur des canaux protéiques. Ceci si-gnifie que, dans certains cas, le blocaged'un canal sodique résulte de l'accumu-lation de l'anesthésique local dans lecanal protéique. Il est certain que le sited'action peut également être atteint àpartir du cytosol et que le produit doitalors traverser d'abord la membranecellulaire (p. 204).
Des substances non chargées peu-vent également exercer une action anes-thésique locale ; dans ce cas, le site deliaison doit être recherché dans le do-maine apolaire du canal ou dans lamembrane lipidique qui l'entoure.
Effets secondaires liés au moded'action. Comme l'influx de sodium
est bloqué par les anesthésiques locannon seulement dans les nerfs sensiti?'mais dans tous les tissus excitables leis
administration doit être effectuée lorlement et en prenant les précautions nécessaires pour éviter une distributiondans l'organisme (p. 204). En effet, unpassage rapide dans le sang peut provoquer des réactions secondaires systé-miques indésirables :
Par un blocage des neurones inhi-biteurs dans le système nerveuxcentral : crampes, agitation (moyen delutte contre les crampes : injection debenzodiazépine, p. 224) ; à concentra-trion plus élevée paralysie générale etblocage du centre respiratoire.
Par une inhibition de la transmis-sion de l'excitation dans le cœur : ano-malie de la conduction AV, arrêt car-diaque (moyen d'intervention : injec-tion d'adrénaline). L'inhibition, par lesanesthésiques locaux, des phénomènesd'excitation cardiaque peut être utiliséesur un plan thérapeutique en casd'arythmie (p. 134).
Types d'anesthésie locale.L'utilisation d'une anesthésie localepeut s'effectuer par infiltration dans letissu à anesthésier (infiltration), ou parinjection dans le faisceau nerveux quirassemble les fibres sensitives prove-nant de la région à endormir (anes-thésie de conduction pour les nerfs,anesthésie spinale pour la moelle épi-nière), par application de la substancesur la peau et les muqueuses (anes-thésie de contact). Dans chaque cas,l'anesthésique local doit diffuser Jus-qu'aux nerfs à anesthésier à partir d'undépôt placé sur la peau ou injecté dansle tissu.
Sensibilité élevée des nerfs sensi-tifs, sensibilité plus faible des nerfsmoteurs. La stimulation des nerfs sen-sitifs est déjà inhibée à des concentra-tions plus faibles que celles nécessairespour bloquer les nerfs moteurs. Cecipeut provenir d'une plus grande fré-quence des impulsions et d'une plusgrande durée du potentiel d'action dansle cas des nerfs sensitifs. Ou bien c'esten rapport avec le diamètre respectif
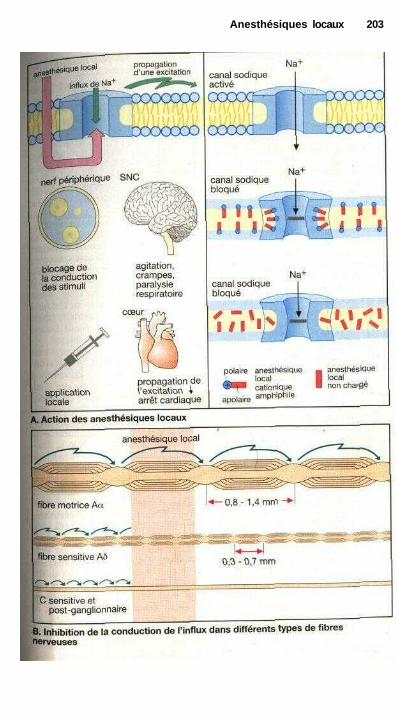
Anesthésiques locaux 203

204 Anesthésiques locaux
des nerfs sensitifs et des nerfs moteursou de l'intervalle entre les nœuds deRanvier. Dans le cas d'une conduc-tion saltatoire de l'influx, la mem-brane sera dépolarisée seulement au ni-veau des nœuds. Comme l'induction dela dépolarisation peut encore se pro-duire malgré le blocage de trois ou qua-tre nœuds,la zone dans laquelle doit êtreprésente une concentration d'anes-thésique local suffisante pour inhibercette conduction, est plus importante(p. 203 B).
Cette relation explique pourquoiles stimuli sensitifs qui passent par lesfibres myélinisées de type A8 réagis-sent à l'administration d'anesthésiqueslocaux plus tard et avec moins de sensi-bilité que les stimuli qui empruntent lesfibres C non myélinisées. Comme lesfibres végétatives post-ganglionnairesne comportent pas de couche de myé-line, elles seront également bloquéespar les anesthésiques locaux. La consé-quence de ce phénomène est une dilata-tion des vaisseaux dans la zone anesthé-siée, qui découle d'une diminution dutonus vasculaire maintenu par le sys-tème sympathique. Ce phénomène n'estpas souhaitable (voir ci-dessous).
Diffusion et action. Au cours dela diffusion à partir du site d'injection etde l'espace interstitiel du tissu conjonc-tif, vers l'axone du nerf sensitif, l'anes-thésique local doit traverser le périneu-rium. Ce périneurium est composé deplusieurs couches de cellules épithé-liales qui sont reliées les unes auxautres par des wnulae occludentes(p. 22), et qui forment ainsi une barrièrehydrophobe fermée.
Les anesthésiques locaux usuelssont des aminés tertiaires, qui dans lagamme de pH des liquides de l'orga-nisme sont en partie sous forme de baseliphophile et en partie sous forme catio-nique amphiphile, (p. 206) chargée po-sitivement. La forme non chargée peuttraverser le périneurium et parvenirdans l'espace endoneuronal où unefraction de la molécule peut, selon lepH qui règne dans cet espace, secharger à nouveau. Le même phéno-
mène se reproduit pour le passage d»anesthésiques locaux à travers la rnern5
brane de l'axone (axolemme) jusandans l'axoplasme (effet sur le canalsodique de l'intérieur de l'axoplasme^et pour la diffusion de l'espace endo^neural à travers l'endothélium non fe.nestré du capillaire jusqu'au sang.
La concentration de l'anesthé-sique local au site d'action dépendradonc de la vitesse de passage dans l'es-pace endoneural, et de la vitesse de dif-fusion vers les capillaires sanguinsPour qu'une substance puisse arriveravec une vitesse suffisante au site d'ac-tion, il faut qu'il existe un gradient deconcentration suffisamment élevé entrele dépôt injecté dans le tissu conjonctifet l'espace endoneuronal. L'injectionde solutions en concentration tropfaible reste sans effet ; par contre il fautéviter des concentrations trop élevées àcause du risque d'un passage rapidedans le sang et donc du risque associéd'un empoisonnement systémique.
Pour obtenir une action localed'une durée suffisante avec des effetssystémiques faibles, on cherchera àmaintenir l'anesthésique au site d'ac-tion et en particulier dans l'axone desnerfs sensitifs. Ceci peut être réalisé enl'utilisant associé à un agent vasocons-tricteur (l'adrénaline, plus rarement lanoradrénaline ou un dérivé de la vaso-pressine). La diffusion en dehors del'espace endoneuronal est diminuée parla réduction du flux sanguin, car le gra-dient de concentration gouvernant ladiffusion entre l'espace endoneuronalet le capillaire sanguin devient nette-ment plus faible, lorsque le flux de sangne contenant pas la molécule se réduit.L'addition d'un vasoconstricteur per-met aussi une élimination relative dusang dans la zone d'opération. L'in-convénient des vasoconstricteurs detype catécholamine est l'apparitiond'une hypérémie réactionnelle dans lazone opératoire après disparition del'effet constricteur (p. 90) ainsi quel'effet cardiostimulant, lorsque l'adré-naline passe dans le sang. On peut aussiutiliser comme adjuvant vasoconstric-
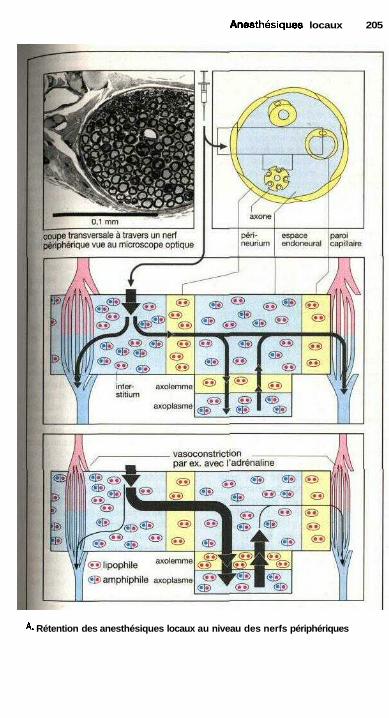
Anesthésiques locaux 205
A- Rétention des anesthésiques locaux au niveau des nerfs périphériques

206 Anesthésiques locaux
teur un dérivé de la vasopressine, lafélypressine (l'hyperémie réactionnelleest plus faible, il n'y a pas d'effet aryth-mogène mais un risque de rétrécisse-ment des artères coronaires). Les vaso-constricteurs ne doivent pas être utiliséslors d'une anesthésie locale au niveaudes extrémités (doigts, orteils).
Caractéristiques de la structurechimique. Les anesthésiques locauxsont constitués en général d'une aminésecondaire ou tertiaire, l'azote est as-socié à une chaîne latérale lipophile, leplus souvent un noyau aromatique.
L'anesthésique local, selon saconstante de dissociation (valeur depK,) et selon la valeur du pH du milieu,sera soit sous forme d'une aminé nonchargée, soit sous forme d'un cationammonium chargé. La valeur du pK,d'un anesthésique local classique varieentre 7,5 et 9. La valeur du pK, indiquela valeur du pH pour laquelle 50 % desfonctions aminés ont capturé un proton.Sous forme protonée, la molécule pos-sède aussi bien une extrémité polaire,hydrophile (azote protoné) qu'une ex-trémité apolaire lipophile (le cycle) :elle est amphiphile.
La représentation graphique de lamolécule de procaïne montre que lacharge positive n'est pas située defaçon ponctuelle sur l'azote mais est re-partie : la figure montre le potentiel àla surface de van der Waals. La formenon protonée (droite) comporte unecharge partielle négative non négli-geable dans la région du groupementester (bleu) et est par ailleurs neutre(vert). Sous la forme protonée (àgauche), la charge positive s'étend àpartir de l'azote en direction du cyclearomatique (coloration brun-rouge).
Dans les conditions de pH physio-logique et selon la valeur du pK, de 5 à50 % environ de la molécule se trou-vent sous forme lipophile non chargée.Cette propriété est importante, carl'anesthésique local traverse les bar-rières lipidiques seulement sous cetteforme (p. 26), tandis qu'il doit prendrela forme cationique amphiphile pourexprimer son activité (p. 202).
Les anesthésiques locaux les n]utilisés sont soit des esters soit damides. Des molécules possédantdes chaînes latérales constituées d, ,groupement méthylène comme par pvla chlorpromazine (p. 234) ou l'irnipramine (p. 228) agiront comme desanesthésiques locaux dans les mode'id'application correspondants. Les ânesthésiques locaux possédant une liaisonester dans la chaîne latérale seront inac-tivés par hydrolyse dès leur arrivéedans le tissu. Ceci est un avantage car lerisque d'une intoxication systémiquepar les esters est plus faible, mais c'estégalement un inconvénient car cette in-activation rapide signifie une duréed'action brève.
La procaïne ne peut pas être uti-lisée comme anesthésique de surfacecar la rapidité de son inactivation estsupérieure à la pénétration à travers lapeau ou les muqueuses.
La lidocaïne est dégradée pardésalkylation oxydative sur l'azote, enpremier lieu dans le foie.
Dans le cas de la prilocaïne et deVarticaïne, cette étape de biotransfor-mation n'est qu'à peine possible àcause de la susbtitution sur l'atome decarbone proche de l'azote. L'articaïnecomporte sur le cycle thiophène ungroupement carboxyméthyle qui peutsubir une hydrolyse, donnant naissanceà un groupement polaire -COO. De cefait, la nature amphiphile est perdue etle métabolite formé est inactif.
La benwcaïne (forme éther) est unmembre de la famille des anesthésiqueslocaux qui ne possèdent pas d'azote pro-toné dans la gamme des pH physiolo-giques. Elle sera essentiellement utiliséecomme anesthésique de contact.
De même seront utilisés commeanesthésiques de contact le pohdocanolnon chargé ainsi que la tétracaïne, ca-tion amphiphile ou la lidocaïne.
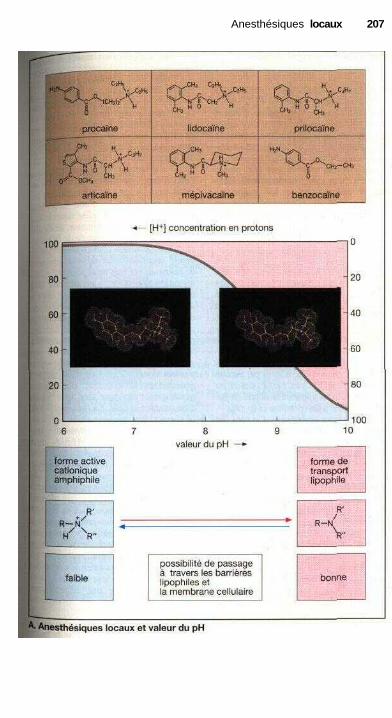
Anesthésiques locaux 207

208 Opioïdes
Analgésiques morphiniques :opioïdes
Origine des opioïdes. La morphine estun alcaloïde de l'opium (p. 4). L'opiumcontient, à côté de la morphine, d'autresalcaloïdes qui ne sont pas analgésiquestel par ex. la papavérine une substancespasmolytique. Tous les dérivés semi-synthétiques (ex. hydromorphone) oucomplètement synthétiques (ex. penta-cozine, péthydine, L-méthadone, fen-tanyï) qui possèdent les propriétésanalgésiques de la morphine, serontbaptisés opioïdes. L'effet analgésiquedes opioïdes exogènes est lié à leur affi-nité pour des récepteurs destinés à fixerles opioïdes endogènes, propres à l'or-ganisme, (enképhaline, (3 endorphine,dynorphine) (A). Les récepteurs desopioïdes sont présents à la surface descellules nerveuses. Ils sont présentsdans différentes zones du cerveau etdans la moelle épinière, mais aussi dansles plexus nerveux du tractus gastro-in-testinal et de la vessie, dont ils contrô-lent la motilité.
Il existe plusieurs types de récep-teurs des opioïdes (|i, K et ô) par l'inter-médiaire desquels sont médiés des effetsdifférents. Tous appartiennent aux récep-teurs couplés à une protéine G (p. 66).
Opioïdes endogènes. Ce sont despeptides dérivés de précurseurs, proen-képhaline, proopiomélanocortine etprodynorphine par protéolyse. Ilscontiennent tous la séquence en acidesaminés des pentapeptides Met et Leu-enkephaline (A).
Les effets des opioïdes peuventêtre complètement bloqués par des an-tagonistes (A), (ex. la naloxone) (ex-ception, la buprénorphine).
Mode d'action des opioïdes. Lamajeure partie des cellules nerveusesréagit aux opioïdes par une hyperpolari-sation (augmentation de la perméabilitéau potassium). L'influx de calcium quise produit dans la cellule nerveuse aucours d'une excitation est diminué, ré-duisant de ce fait la libération de neuro-transmetteurs excitateurs et la transmis-sion synaptique (A). Cette inhibitionpeut se manifester, selon les territoires
nerveux, aussi bien sur des effets stirnulants que sur des effets inhibiteurs (B)
Effet des opioïdes (B). L'effetanalgésique est basé sur des effets auniveau de la moelle épinière (inhibitionde la conduction douloureuse) et ducerveau (atténuation de la propagationdes influx, inhibition de la perceptionde la douleur). L'attention et la capacitéde concentration sont diminuées.
La direction vers laquelle évoluel'état d'esprit du patient dépend du ré-sultat final. A côté du soulagement as-socié à la disparition d'une forte dou-leur survient, en particulier dans le casd'une injection intraveineuse, et égale-ment lors de l'arrivée rapide de la molé-cule, une sensation de bien être et de lé-gèreté (euphorie). Le désir d'atteindrede nouveau cet état en répétant l'admi-nistration d'opioïde, peut devenir tropfort : développement d'une dépen-dance. Au moment où l'on désire ter-miner une administration régulière ap-paraissent des symptômes de sevragephysique (entre autres troubles circula-toires) ou psychique (agitation, an-goisse, dépression). Les opioïdes rem-plissent donc les critères d'un produitgénérant une toxicomanie : dépendancepsychique et physique ainsi que besoind'augmenter la dose. La prescription dela plupart des opioïdes obéit à des rè-glements particuliers (tableau des stu-péfiants). L'ordonnance précise, entreautres, les doses maximales (dose indi-viduelle, quantité maximale journa-lière, quantité maximale prescrite). Lesordonnances sont rédigées sur des car-nets à souches qui doivent être remplisselon des règles imposées. Des analgé-siques opioïdes moins actifs comme lacodéine et le tramadol peuvent êtreprescrits de façon normale car le risquede dépendance est faible.
Les différences entre les opioïdesen ce qui concerne leur activité et leurcapacité à susciter une dépendance,peuvent reposer sur des différencesd'affinité et d'activité intrinsèque vis-à-vis de chaque sous-type de récepteurs.Une substance peut ne pas agir de façonidentique comme agoniste ou antago-niste sur les différents sous-types, mais
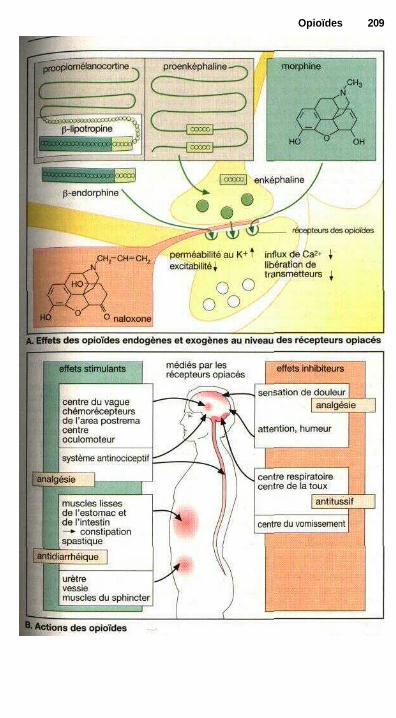
Opioïdes 209

210 Opioïdes
peut se comporter comme un agonistesur un sous-type donné et comme unagoniste partiel / antagoniste sur unautre ou encore comme un pur antago-niste (p. 212). Le danger d'une para-lysie des centres respiratoires en cas desurdosage existe pour tous les analgé-siques opioïdes puissants. L'impor-tance d'une possible inhibition ducentre respiratoire est plus faible pourdes substances agissant sur les récep-teurs des opioïdes comme des agonistespartiels / antagonistes (pentazocine,nalbuphine).
Le blocage de la toux par le biaisde l'inhibition du centre de la toux, peutêtre obtenu de façon indépendante deseffets analgésiques ou de l'action surle centre respiratoire (antitussifs :codéine, noscapine).
Les vomissements observés lorsde la première administration desopioïdes sont la suite d'une stimulationdes chémorécepteurs de l'area postrema(p. 324). L'effet émétique disparaît aucours d'un usage régulier, parce ques'établit alors un blocage direct ducentre du vomissement.
Les opiacés provoquent par unestimulation de la partie parasympa-thique des noyaux oculomoteurs (noyaude Edinger-Westphal) une dilatationdes pupilles (myosis).
Les effets périphériques touchentla motilité et le tonus des muscles lissesdu tractus digestif, les mouvements pen-dulaires de l'intestin sont renforcés, lesmouvements vers l'avant sont inhibés.Le tonus du sphincter anal est forte-ment augmenté (constipation spas-tique). L'effet anti-diarrhéique est uti-lisé sur le plan thérapeutique :lopéramide (p. 176). La vidange del'estomac est ralentie (spasme du py-lore), et l'écoulement de la bile et dusuc pancréatique est bloqué, car là aussiles sphincters sont contractés. Le fonc-tionnement de la vessie est égalementaffecté, en particulier la vidange de lavessie est bloquée par suite de l'aug-mentation du tonus des muscles dusphincter.
Utilisation : les opioïdes endo-gènes (par ex. Met-enkephaline, Leu-
enkephaline, (î endorphine) ne peuvppas être utilisés sur un plan théraoei*11
tique car, étant des peptides, ils sontd''gradés trop rapidement, ne passent n~'à travers la barrière hématoencépha8
lique et ne peuvent ainsi pas parvenir asite de leur action après administrationparentérale (A).
La morphine peut être administréeper os, par voie parentérale ou, au ni-veau de la moelle épinière, par voie éni-durale. L'héroïne et le fentanyï sont tel-lement lipophiles qu'ils parviennenttrès rapidement au SNC. Le fentanyï esten plus tellement actif, qu'il peut êtreutilisé sous forme d'un emplâtre sur lapeau. (A).
Lors d'une utilisation abusive, leproduit (en général l'héroïne = diacétyl-morphine) sera injecté (Fix) pour ob-tenir un afflux aussi rapide que possibledu produit actif dans le ceneau.Vraisemblablement, l'effet psychiquerecherché est dans ce cas particulière-ment intense. Dans ces utilisations abu-sives, des sites d'administration inhabi-tuels ont été et sont utilisés : l'opiumpeut être fumé, l'héroïne peut êtreprisée (B).
Métabolisme (C) : la morphine,comme les autres opioïdes comportantun groupement hydroxyle libre, seraéliminée par le rein sous forme conju-guée à un glucuronide. Contrairement àla glucuronidation sur l'hydroxyle 3,celle sur le groupement OH en 6 ne di-minue pas l'affinité de la molécule. Lacontribution apportée par le 6 glucuro-nide à l'effet analgésique est difficile àévaluer avec précision. L'activité de cecomposé polaire doit en tout cas êtreprise en compte lors d'une altérationdes fonctions rénales (diminution de ladose ou allongement des intervallesentre les doses).
Développement d'une tolérance.Lors d'une administration répétéed'opioides il peut apparaître pour leseffets centraux un phénomène d'accou-tumance (tolérance accrue) : au coursd'un traitement, il faudra des dosescroissantes pour parvenir à une mêmeatténuation de la douleur. Les effets pé-riphériques ne sont pas affectés par ce
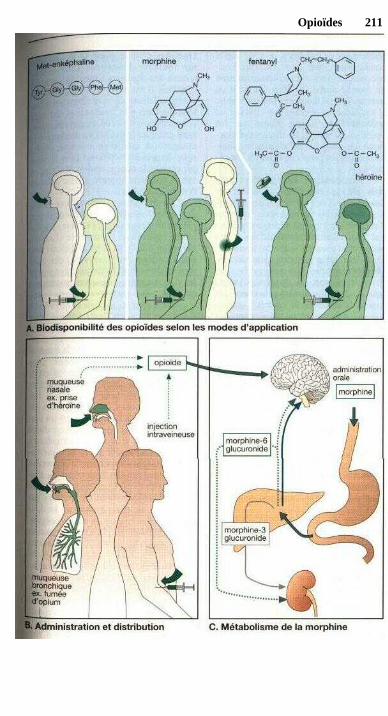
Opioïdes 211

212 Opioïdes
phénomène de tolérance, de sorte que laconstipation peut dans certaines cir-constances, lors d'une administrationde longue durée, imposer l'arrêt d'untraitement antidouleur indispensable.Il est donc nécessaire, au cours d'untraitement de longue durée par lesOpioïdes, d'utiliser à titre prophylac-tique, des moyens diététiques ou phar-macologiques pour empêcher la consti-pation.
Antagonistes ou agonistes par-tiels de la morphine. L'effet desOpioïdes peut être inhibé par des antago-nistes, naloxone ou naltrexone, indépen-damment du type de récepteur mis enjeu (A). Administrés seuls, ils n'ontaucun effet chez des individus normaux,mais leur administration peut déclen-cher les symptômes du manque chez desgens dépendants de l'opium. Comptetenu de son élimination présystémiquerapide, la naloxone ne peut être utiliséeque par voie parentérale. La naltrexoneest métaboliquement plus stable et serautilisée par prise orale. La naloxone sertd'antidote dans le cas d'une dépressionrespiratoire induite par les Opioïdes. Ilfaut noter qu'elle est éliminée plus rapi-dement que la plupart des Opioïdes etqu'il faudra donc l'utiliser le caséchéant de façon répétée. La naltrexonepeut être employée pour l'entretien d'untraitement de désintoxication.
La buprénorphine se comporteComme un agoniste partiel / antagonistesur les récepteurs . La pentazocine estun antagoniste des récepteurs et unagoniste K (A). Avec ces produits iln'est pas possible d'atteindre le mêmeeffet analgésique maximum qu'avec lamorphine ou la péthidine (B). L'effetantagoniste des antagonistes partielspeut lors du passage à un agoniste com-plet bloquer tout d'abord l'action decelui-ci.
Il n'est pas possible d'antagoniserune intoxication à la buprénorphine,car elle ne se dissocie que très lente-ment des récepteurs aux Opioïdes et, deplus, l'occupation des récepteurs parun antagoniste ne se produit pas aussirapidement que la situation cliniquel'exigerait.
Opioïdes utilisés pour le traiten,des douleurs chroniques. Lors d nt
tement des douleurs chroniques n trai'Opioïdes, il est nécessaire de maint s
continuellement la concentration î""matique dans une zone active, car as'concentration descend en dessous i?seuil critique, le patient va ressentir ri1111douleurs et considère comme née es
saire de prendre des doses plus élevJ5'par crainte de cette situation. Il s'agites
fait, à proprement parlé, d'une préven"tion de la douleur.
La morphine, comme une partides autres Opioïdes (hydromorphonepéthidine, pentazocine, codéine) est é\i.minée rapidement et sa durée d'actionest environ de 4 h. Pour maintenir uneaction analgésique constante, ces sub-stances doivent être administrées toutesles 4 h. La prise fréquente, y comprispendant la nuit, représente unecontrainte dans les maladies chroniques.En augmentant les doses individuelleson peut arriver à diminuer la fréquencedes prises mais cette pratique est asso-ciée avec un dépassement de la concen-tration thérapeutique nécessaire dansl'organisme et un risque d'effetstoxiques indésirables. Une possibilitéplus intéressante pour diminuer la fré-quence des prises, est l'utilisation deformes retard de la morphine, d'un em-plâtre de fentany] ou d'opioïdes à duréed'action plus longue (L-méthadone).Les propriétés cinétiques de la L-métha-done rendent cependant nécessairel'ajustement des doses au cours du trai-tement. Pour des doses faibles pendantles premiers jours du traitement on n'ar-rivera pas, en effet, à atténuer la dou-leur, pour des doses plus élevées cepen-dant le produit risque de s'accumulerpour atteindre des concentrationstoxiques (C). Dans des conditions parti-culières (difficulté d'une administrationorale, effets secondaires périphériquesinsupportables), les Opioïdes peuventêtre administrés continuellement soit aumoyen d'une pompe ou bien à proximitéde la colonne vertébrale sous le contrôledes patients (avantage : dosages trèsfaibles et niveau d'action constant ; in-convénient : pose d'un cathéter).
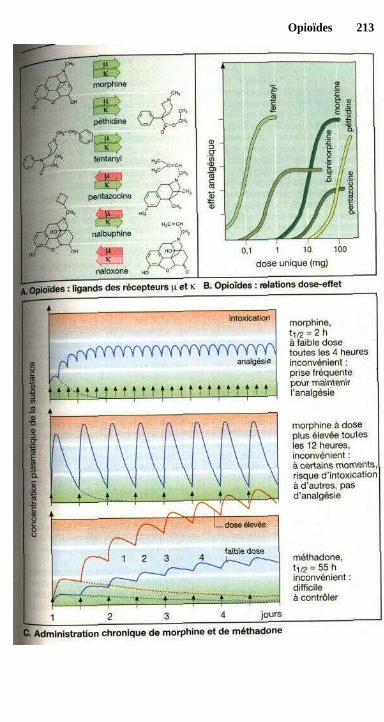
Opioïdes 213

214 Anesthésiques
Anesthésie et anesthésiques
L'anesthésie est une inhibition réver-sible des fonctions du système nerveux,provoquée par des moyens médicamen-teux, et destinée à pouvoir réaliser uneintervention chirurgicale dans un état in-conscient, en l'absence de sensationsdouloureuses, sans mouvements de reculou sans réflexes végétatifs puissants (parex. des réactions circulatoires) (A).
L'intensité de l'anesthésie va dé-pendre de l'intensité des stimuli doulou-reux, c'est-à-dire de l'importance de lastimulation du système nociceptif.L'anesthésiste va donc adapter l'anes-thésie de façon « dynamique » au dérou-lement de l'intervention.
A l'origine, l'anesthésie était prati-quée avec un seul produit (par exemplel'éther, première anesthésie ayant pourbut le déroulement d'une interventionchirurgicale par WGT Morton en 1846 àBoston). Dans une telle monoanes-thésie, la dose nécessaire pour empêcherles réflexes de retrait était plus élevéeque celle conduisant à la perte de cons-cience, et à cette concentration se pro-duisait également une inhibition defonctions vitales (par ex. régulation car-diovasculaire) (B). Dans les anesthésiesmodernes, les buts de l'anesthésie sontatteints par une combinaison de diffé-rents produits (anesthésie combinée).Ce procédé diminue le risque anesthé-sique. On a donné en C à titre d'exem-ple, quelques substances utilisées dansune anesthésie combinée, simultané-ment ou l'une après l'autre. Dans uneanesthésie par inhalation, l'ordre dépendde la propriété particulière souhaitée(voir ci-dessous). On a déjà décrit en dé-tail à d'autres emplacements des myore-laxants, des analgésiques opioïdescomme le fentanyï et de l'atropine, unparasympatholytique agissant sur lesfonctions végétatives.
Nous allons d'abord présenterquelques procédés particuliers d'anes-thésie, avant de décrire finalement lesanesthésiques.
La neuroleptanalgésie peut êtreconsidérée comme une forme particu-lière d'« anesthésie » combinée ; on
combine un analgésique opioïdg ^tion brève, tefentunyï avec un neurni'10'tique fortement sédatif et à action i?'tanciante, le droperidol. Ce procédéutilisé chez les patients à risques f86?âgés ou avec un trouble hépatique)
On désigne sous le terme de nenroleptanesthésie, l'administration cobinée d'un analgésique à action brè\wd'un anesthésique injecté, d 'un myore'laxant à courte durée de vie et d'unfaible dose d'un neuroleptique.
Dans une anesthésie régional»(anesthésie spinale) avec un anesthé-sique local (p. 202) c'est la nocicepiionqui sera interrompue ; dans ce procédé ilne s'agit plus d'une anesthésie (pas deperte de conscience).
Dans le cas des anesthésiques ausens strict on peut distinguer selon lemode d'application, les anesthésiquesinhalés et les anesthésiques injectés.
Les anesthésiques inhalés sont ad-ministrés via l'air inspiré et sont (pourune partie plus ou moins importante)également éliminés par cette voie. Ilsservent au maintien d'une anesthésie àun niveau satisfaisant. Ce groupe de sub-stance sera décrit en détail page 216.
Les anesthésiques injectée (p. 218)servent souvent à l'induction de l'anes-thésie. L'injection intraveineuse et l'ap-parition rapide de l'effet sont nettementplus agréables pour les patients que l'in-halation d'un gaz anesthésiant. L'effetdes anesthésiques injectés ne dure en gé-néral que quelques minutes.
Sous leur action on peut entre-prendre des opérations de courte durée,ou bien on débutera une anesthésie parinhalation (intubation). On chercheraalors à réguler le débit de l'anesthésiqueinhalé pour pouvoir compenser la dimi-nution de l'effet de l'anesthésique in-jecté.
Au cours d'anesthésies combinéesde longue durée on utilise en proportionscroissantes des anesthésiques injectés àla place des inhalations (AnesthésieIntraveineuse Totale, AIVT).
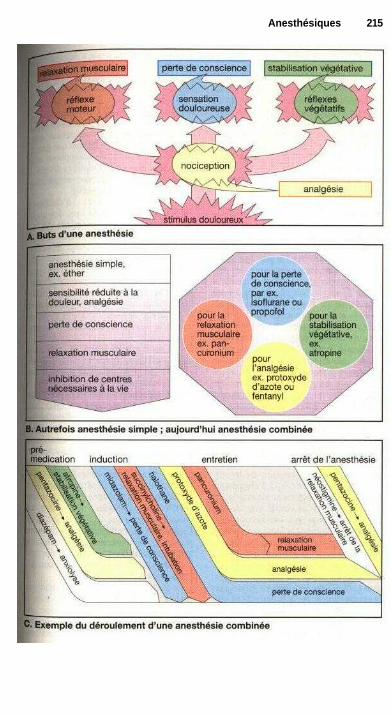
Anesthésiques 215

216 Anesthésiques
Anesthésiques inhalés
Le mécanisme d'action des anesthé-siques inhalés est inconnu. Compte tenude la multiplicité des structures chimiquesagissant comme anesthésique (gaz raresxénon, hydrocarbures, hydrocarbures ha-logènes), il semble exclu d'envisager uneinteraction avec des récepteurs spéci-fiques. Parmi les hypothèses, on envisageune insertion du produit dans la coucheinterne lipophile de la double membranephospholipidique des cellules nerveuses,ce qui bloquerait l'excitabilité électriqueet la propagation de la stimulation dans lecerveau. Cette proposition permettraitd'expliquer la corrélation entre l'inten-sité de l ' e f f e t anesthésique et la lipophiliedes anesthésiques (A). On peut égalementpenser à une interaction avec les do-maines lipophiles de protéines membra-naires. L'activité narcotique sera donnéesous forme de CAM (concentration al-véolaire minimale du narcotique) ; à cettevaleur 50 % des patients ne présentent au-cune réaction de recul face à un stimulusdouloureux défini (coupure cutanée).Tandis que le protoxyde d'azote (N;0)faiblement lipophile doit être respiré enforte concentration (> 70 % de l'air ins-piré doit être remplacé), il est nécessaired'utiliser des concentrations beaucoupplus faibles d'halothane, un composé li-pophile (< 5 %).
La vitesse avec laquelle l'actiond'un anesthésique inhalé s'installe et dis-paraît est très variable et dépend égale-ment du caractère lipophile de la sub-stance. Dans le cas du N;0, l'éliminations'effectue très rapidement lorsque le pa-tient est de nouveau ventilé avec de l'airpur : compte tenu de la pression partielleélevée dans le sang, la force poussant aupassage dans l'air (expire) est importante,et l'organisme peut être rapidement« purgé » du N;0 à cause de la faible cap-ture dans les tissus. Au contraire, la pres-sion partielle dans le sang est faible dansle cas de l'halothane et la quantité de pro-duit répartie dans l'organisme est impor-tante, si bien que l'élimination a lieu net-tement plus lentement.
Le protoxyde d'awte seul (gaz hila-
rant N;0) ne permet pas d'atteindre unprofondeur d'anesthésie suffisante noipratiquer une opération chirurgicalr
même lorsqu'il représente 80 % en volume de l'air inspiré (il est nécesaired'avoir 20 % d'oxygène en volume). M r>possède une bonne action anesthésiquequi sera utilisée en combinaison avecd'autres anesthésiques. En tant que gaz leprotoxyde d'azote peut être appliqué sansque l'on ait à s'occuper d'autres détails, ildemeure inchangé et est inspiré quantita-tivement par les poumons (B).
L'halothane (point d'ébullition50 °C), doit être vaporisé avec des appa-reils spéciaux, comme l'enflurane (56 °C)et l'isoflurane (48 °c). Une partie de l'ha-lothane administré peut donner naissanceà des métabolites hépatotoxiques. (B).Lors d'une anesthésie à l'halothane il peutse produire une lésion hépatique, événe-ment rare et pratiquement imprévisible.Le risque de cette atteinte augmente avecla fréquence des anesthésies et le faibleintervalle entre deux anesthésies succes-sives.
Lors d'une anesthésie à l'enfluraneou à l'isoflurane (fraction biotransformée< 2 %) les produits de dégradation nejouent pratiquement aucun rôle.
L'halothane a un effet hypotenseurmarqué, auquel participe un effet ino-trope négatif. L'enflurane et l'isofluraneexercent une action dépressive plusfaible sur l'activité cardiovasculaire.L'halothane sensibilise le muscle car-diaque contre les catécholamines (atten-tion : tachyarythmies sévères, fibrillationventriculaire en cas d'administration decatécholamines comme antihyperten-seurs ou tocolytiques). Cet effet estmoindre dans le cas de l'enflurane et del'isoflurane. L'enflurane et l'isofluranepossèdent au contraire de l'halothaneune action relaxante sur les muscles, quis'ajoute à celle des myorelaxants non dé-polarisants.
Le desflurane a une structureproche de celle de l'isoflurane, mais ilest moins lipophile. On obtient donc uneinduction et une élimination particulière-ment rapide ainsi qu'une bonne maniabi-lité de l'anesthésie.
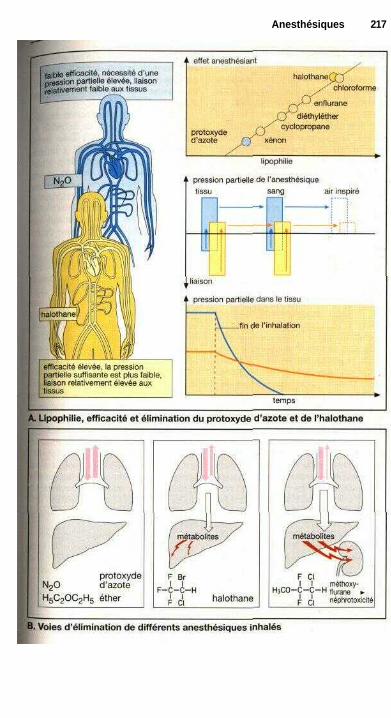
Anesthésiques 217

218 Anesthésiques
Anesthésiques injectés
Des substances appartenant à différentsgroupes chimiques peuvent après admi-nistration intraveineuse inhiber laconscience et servir d'anesthesiques(A) A la différence des anesthesiquesinhales, la plupart d'entre elles cepen-dant n agissent que sur l'état deconscience et n'ont aucun effet analgé-sique (exception ketamine) L'expli-cation de cette action est, de façon in-discutable (sauf peut être pour lepropofol) une insertion non spécifiquedans la membrane des neurones
La plupart des anesthesiques in-jectes se caractérisent par une duréed'action brève La diminution rapidede 1 effet repose essentiellement surune distribution après injection in-traveineuse s'établit rapidement dansle cerveau bien irrigue une concentra-tion élevée, l'anesthesie débute Avecle temps, le produit va se repartir defaçon égale dans l'organisme ce qui si-gnifie que la concentration a la péri-phérie augmente tandis que celle dansle cerveau diminue distribution etdissipation de l'effet anesthesique (A)L'effet s'estompe sans que le produitne quitte 1 organisme Une deuxièmeinjection de la même dose immédiate-ment après dissipation de l'effet de ladose précédente peut pour cette raisonprovoquer une action plus longue etplus intense Dans la plupart des casces produits ne seront donc injectésqu'une seule fois Le propofol et l'eto-midate seront cependant perfuses éga-lement pendant une durée plus longue,pour provoquer une perte deconscience Si lors d'une anesthesieaucun anesthesique inhale n'est utilisé,on parle d'une anefthem intravei-neuse totale (AIVT)
Le thiopental ainsi que le meto-hexital font partie des barbituriques,qui, en fonction de la dose, auront uneaction sédative, hypnotique ou anesthe-sique Les barbituriques diminuent le
seuil de la douleur et peuvent ainsiciter des mouvements de recul ils . u s
bent les centres respiratoires Les bdrhtunques servent souvent a l'induch 1
d'une anesthesie t1
La ketaimne a une action analgesique qui selon l'état de la perte ri'conscience, dure jusqu a une heureaprès injection La capacité a induireune perte de connaissance dure seulement un quart d'heure environ Apresson réveil, le patient peut éprouver unedissociation entre le monde exteneur etses sensations intérieures (anesthesiedl'iwilalive) On observe souvent uneperte des souvenirs de la phase de ré-veil, cependant les gens se plaignent enparticulier au réveil, d'expériences pé-nibles Celles ci peuvent être évitéespar 1 administration de benzodiaze-pines (ex midazolam) L'action cen-trale de la ketamine réside dans uneinterférence avec le glutamate unneurotransmetteur excitateur La keta-mine bloque un pore cationique au ni-veau d'un canal active par le glutamateappelé récepteur NMDA Le NMDAou N-Methyl-D-Aspartate, est une mo-lécule exogène, qui est un agoniste spé-cifique de ce récepteur La ketaminepeut augmenter le rythme cardiaque etla pression artérielle par l'intermédiaired'une libération de catecholammes
Le propofol est une substance remarquablement simple a synthétiser,son action débute rapidement et s es-tompe facilement, d'une façon trèsagréable pour le malade L'intensité de1 effet est aisément modulable lorsd'une administration de plus longuedurée
L'etomidate affecte a peine lesfonctions végétatives II inhibe la syn-thèse de cortisol, ce qui peut être utiliselors d'une hyperactivite des glandessurrénales (maladie de Cushing)
Le midawlam est une benzodiaze-pine a dégradation très rapide (p 226)qui peut donc être utilisée pour l'induc-tion d'une anesthesie
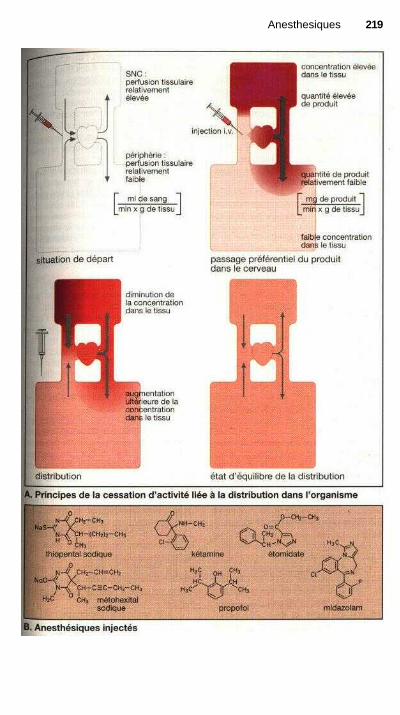
Anesthesiques 219

220 Hypnotiques
Somnifères, hypnotiques
Le sommeil est une phase de repos pen-dant laquelle se produisent plusieursphases d'activité cérébrale, répétées denombreuses fois, et qui peuvent être dis-tinguées les unes des autres sur un électro-
' encéphalogramme. Les phases de som-meil se succèdent 4 à 5 fois par nuit,chacun des cycles étant interrompu parune phase de sommeil dite REM (RapidEye Movements, sommeil « paradoxal »avec mouvements rapides de yeux) (A).Les périodes REM sont reconnaissablespar un tracé EEG comparable à celui d'unétat de veille, par des mouvements rapidesdes yeux, des rêves animés et des tres-saillements occasionnels de certainsmuscles squelettiques, par ailleurs atones.En temps normal, une phase REM ne peutêtre atteinte qu'après une phase préalableNREM (No Rapid Eye Movements, som-meil orthodoxe). En cas d'interruptionsfréquentes du sommeil nocturne, la pro-portion de sommeil paradoxal diminue.Une diminution de la durée du sommeilREM (normalement environ 25 % de ladurée totale du sommeil) provoque pen-dant la journée une agitation et une exci-tabilité accrue. Dans une période de reposnocturne non perturbé, un déficit en som-meil paradoxal sera compensé pendant lesnuits suivantes par un allongement dusommeil REM (B).
Peuvent servir de somnifère les ben-zodiazépines (par ex. triazolam, téma-zépam, clotiazépam, mtrazépam), lesbarbituriques (ex. hexobarbital, pento-barbital), l'hydrate de chloral, et les anti-histamimques H, à action sédative. Lesbenzodiazépines possèdent des récepteursspécifiques (p. 224). Le site et les méca-nismes d'action des barbituriques, del'hydrate de chloral et des antihistami-niques sont peu clairs.
Tous les somnifères raccourcissentles phases de sommeil paradoxal (B). Lorsde l'absorption régulière de somnifèrespendant une longue pénode, on observeque le rapport entre les stades de sommeilse normalise maigre la prise de somnifère.A l'arrêt du somnifère se produit une régu-lation en sens contraire, la proportion dusommeil paradoxal augmente et se norma-
lise après quelques jours (B). Corninpphase REM est associée à des rêves a» -un sommeil où la proportion de c es'phase est accrue sera ressenti cornn10
moins reposant. Lorsque l'on essaye drêter la pnse régulière d'un somnifère ar'phénomène donne l'impression qu'il «e
nécessaire pour un sommeil reposa8»d'utiliser un somnifère et favonse la possiblité d'une dépendance.
Selon la concentration dans le sanples benzodiazépines et les barbituriquesagiront comme des calmants et des séda-tifs, les benzodiazépines étant aussi desanxiolytiques ; à plus forte concentra-tion, ils auront une action sur le sommeilagité et finalement sur l'endormisse-ment (C). A dose plus faible c'est l'ac-tion anoxiolytique de benzodiazépinesqui prédomine.
Au contraire des barbituriques, lesdérivés des benzodiazépines n'ont pasd'action narcotique par voie orale, ilsn'inhibent pas de façon génénque l'acti-vité du cerveau (la paralysie respiratoireest pratiquement impossible) et ils n'af-fectent pas les fonctions autonomes tellesla pression artérielle, la fréquence car-diaque ou la température corporelle Lafenêtre thérapeutique des benzodiazé-pines est également nettement plus largeque celle des barbituriques.
Le zolpidem (dont la structure estcelle d'une imidazo-pyridme) et le zopi-clone (une cyclopyrrolone) sont des hyp-notiques qui en dépit de leur structure chi-mique distincte peuvent stimuler lerécepteur des benzodiazépines (p. 224)
Les barbituriques à cause de leurfenêtre thérapeutique étroite (risque d'uti-lisation dans des suicides) et des risquesde dépendance ne sont plus utiliséscomme somnifère ou seulement rarement.La dépendance peut prendre tous lessignes d'une toxicomanie (p. 208).
L'hydrate de chloral n'est utili-sable comme hypnotique que pour debrèves périodes à cause d'une toléranced'installation rapide.
Les antihistammiques (par ex. di-phénhydramine, doxylamine, p. 114) sontutilisés comme somnifère sans ordon-nance, dans ce cas leurs effets secon-daires servent comme action principale
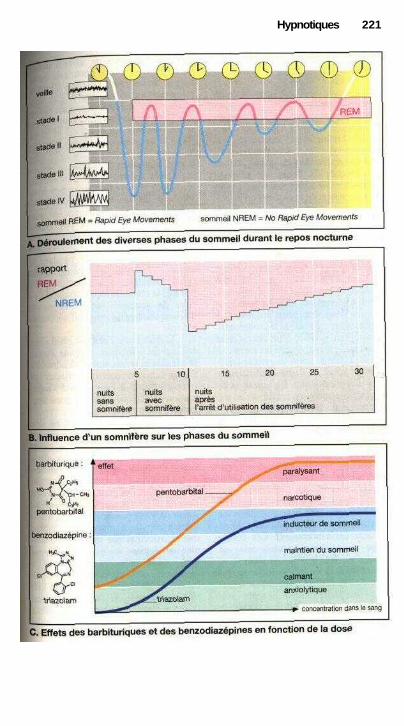
Hypnotiques 221

222 Hypnotiques
Rythmes d'éveil et de sommeilet somnifères
Les mécanismes physiologiques de ré-gulation du rythme d'éveil et de som-meil ne sont pas bien connus. Ce qui estdémontré, c'est que l'activité des neu-rones histaminergiques, cholinergiques,glutamatergiques et adrénergiques estplus élevée pendant la phase d'éveilque pendant la phase de sommeilNREM. Les neurones que nous venonsde citer, partent du tronc cérébral princi-palement en direction du thalamus où ilsstimulent les voies thalamo-corticales etinhibent les neurones GABAergiques.Pendant le sommeil, l'activité électriqueen provenance du tronc cérébral est ré-duite ce qui provoque en même tempsune diminution de l'activation thalamo-corticale et une désinhibition de l'acti-vité GABAergique. (A). La modifica-tion de l'équilibre entre les neuronesexcitateurs (rouge) et les neurones inhi-biteurs (vert) conduit à un changementcircadien de la préparation au sommeil :elle est faible le matin, augmente lente-ment au début de l'après-midi (sieste),pour diminuer ensuite à nouveau et at-teindre finalement son maximum au mi-lieu de la nuit (Bl).
Traitement des troubles dusommeil. Les moyens pharmacolo-giques ne sont indiqués que lorsque letraitement causal est sans effet. Lescauses des troubles du sommeil peu-vent être des chocs émotionnels (peur,stress, chagrin), des problèmes phy-siques (toux, douleurs) et la prise demédicaments (boissons contenant dela caféine, sympathomimétiques oucertains antidépresseurs). Ces condi-tions conduisent (comme cela estmontré en B2 dans le cas d'un chocémotionnel) à un déséquilibre en fa-veur de l'activité excitatrice. Le tempsde latence entre la mise au repos et leseuil de sommeil s'allonge, la duréemoyenne diminue et le sommeil peutêtre interrompu par plusieurs périodesd'éveil.
Le traitement médicamenteux destroubles du sommeil s'effectue à l'aide
de benzodiazépines (p. 224) à actionbrève (t 1/2 = 4-6 h, triazolam, broti-zolam) ou moyenne (t 1/2 == lO-ls ),'lormétazépam, témazépam). Ces suhstances raccourcissent la période précé-dant l'endormissement, allongeant ladurée moyenne du sommeil et dimi-nuent la fréquence des réveils au coursde la nuit. Ils renforcent l'activité inhi-bitrice. Même lors de la prise de benzo-diazépines à durée de vie plus longuele patient se réveille, après 6-8 h desommeil, car, pendant les heures de lamatinée, l'activité excitatrice est plusimportante que la somme des inhibi-tions physiologiques et pharmacolo-giques (B3). L'effet du somnifère peutcependant se faire sentir pendant lajournée, lorsque le patient prendd'autres substances à action sédative(alcool) et réagit de façon inhabituelle :effet de synergie (altération de laconcentration et des possibilités de ré-action).
L'écart entre l'activité excitatriceet l'activité inhibitrice diminue durantla vieillesse, tandis que la tendance àl'apparition de courtes périodes desommeil durant la journée et à l'inter-ruption plus fréquente du sommeil noc-turne augmente (C).
La prise d'un somnifère ne doitpas dépasser une durée de 4 semainescar une accoutumance peut se déve-lopper. Le risque d'une nouvelle dimi-nution de la disponibilité au sommeilà l'arrêt du traitement peut être évitépar une réduction graduelle des doses.Lors de la prescription d'un somnifèreil faut toujours penser au danger d'uti-lisation en vue d'un suicide. Commeune intoxication par une benzodiazé-pine ne devient vraiment dangereuseque lorsque d'autres substances àinhibition centrale sont prises enmême temps (alcool) et comme ellepeut être traitée de façon spécifique(antagonistes des benzodiazépines),les benzodiazépines doivent être utili-sées préférentiellement comme som-nifères plutôt que les barbituriquesadministrés autrefois comme hypno-tiques.
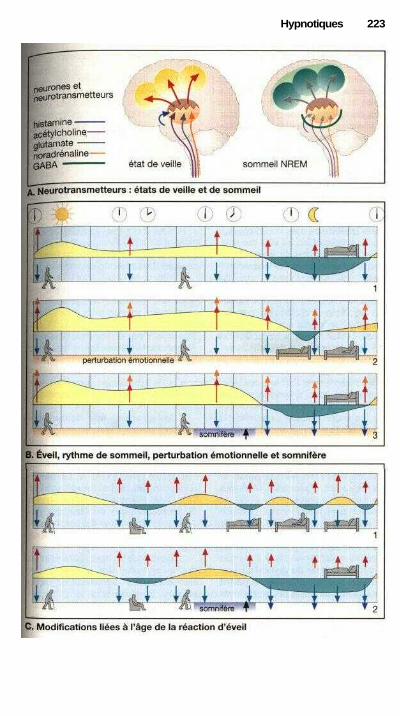
Hypnotiques 223

224 Médicaments du psychisme
Benzodiazépines
Les benzodiazépines provoquent unchangement des réactions émotion-nelles aux perceptions, en particulierelles donnent un certain flegme en facede situations angoissantes : effet anxio-lytique. Les benzodiazépines ont despropriétés calmantes (sédatives), ellesempêchent la tendance aux crampes(effet anticonvulsivant), et diminuentle tonus des muscles squelettiques (effetmyotonolytique). Toutes ces actionsreposent sur le fait que les benzodiazé-pines renforcent l'influence de neu-rones inhibiteurs dans le cerveau et lamoelle épinière. Ceci est dû à une inter-action avec des sites de liaison spéci-fiques, les récepteurs des benzodiazé-pines, qui forment une partie desrécepteurs GABA^, canaux ioniques sti-mulés par un ligand. Le neurotransmet-teur inhibiteur GABA (acide •y amino-butyrique) provoque une ouverture decanaux chlore : la perméabilité auchlore de la membrane des cellules ner-veuses augmente, ce qui atténue l'actionde stimuli dépolarisants. Les benzodia-zépines augmentent l'affinité du GABApour ses récepteurs si bien que pour unemême concentration de GABA, on auraune liaison plus élevée au récepteur etun effet plus fort. L'excitabilité de lacellule nerveuse est diminuée.
Cette action des benzodiazépinespeut être utilisée sur le plan thérapeu-tique dans les névroses d'angoisse, lesphobies et les dépressions anxieuses.Les benzodiazépines ne résolvent ce-pendant aucun problème mais atténuentles réactions face au problème et allè-gent la psychothérapie indispensable.Elles sont indiquées pour diminuer unestimulation cardiaque liée à l'angoisseen cas d'infarctus du myocarde, pourdiminuer les troubles du sommeil,dans la préparation des opérations,pour le traitement des crampes oul'abaissement du tonus des musclessquelettiques (myotonolyse en cas detensions spastiques).
Les synapses GABAergiques sontprésentes uniquement dans le SNC et
les benzodiazépines n'affectent que [pfonctions contrôlées par les synapse»;GABAergiques. Les centres qui régnlent la pression artérielle, la fréquencecardiaque et la température du corpsn'en font pas partie. La fenêtre théra-peutique, évaluée par l'écart entre ladose nécessaire pour obtenir les effetssouhaités et la dose toxique (dépressionrespiratoire) est pour les benzodiazé-pines > 100, soit plus de 10 fois supé-rieure à celle des barbituriques et desautres sédatifs. Dans le cas d'une in-toxication, il y a la possibilité d'utiliserun antidote (voir ci-dessous).
Sous l'emprise des benzodiazé-pines, il n'est plus possible de réagir ra-pidement aux stimuli extérieurs (par ex.conduite d'un véhicule automobile).
Malgré la bonne tolérance aiguëdes benzodiazépines, il ne faut pas né-gliger les possibles changements depersonnalité (inertie) et la dépendanceassociée à une prise régulière. Cettedépendance repose vraisemblablementsur une accoutumance qui se manifesteà l'arrêt du traitement par des symp-tômes de manque : angoisse et agita-tion. Ces symptômes favorisent uneutilisation prolongée de benzodiazé-pines.
Antagonistes des benzodiazé-pines. Certaines molécules comme lefluma-iénil possèdent une affinité pourles récepteurs des benzodiazépines etoccupent ceux-ci sans modifier la fonc-tion des récepteurs GABA. Le fluma-zénil pourra être utilisé comme antidotelors d'une absorption trop importantede benzodiazépines ou chez des pa-tients sous sédation par des benzodiazé-pines pour les réveiller après une opéra-tion.
Tandis que les benzodiazépines entant qu'agonistes des récepteurs desbenzodiazépines augmentent de façonindirecte la perméabilité au chlore, ilexiste des agonistes inverses qui pro-voquent une diminution de cette per-méabilité. Ces substances pour les-quelles on ne connaît aucune indicationthérapeutique provoquent agitation,excitation, angoisse et crampes.
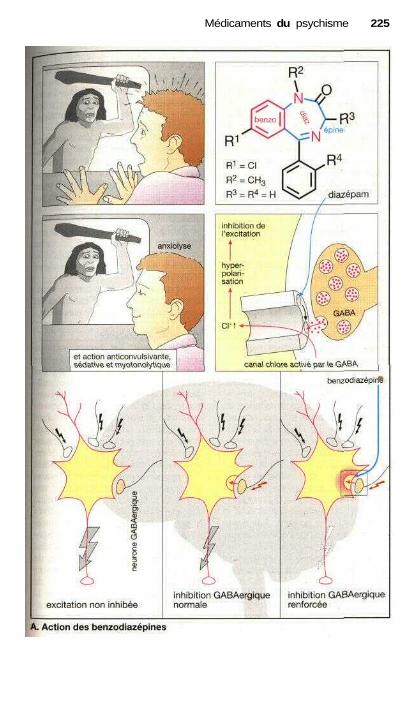
Médicaments du psychisme 225

226 Médicaments du psychisme
Pharmacocinétiquedes benzodiazépines
Toutes les benzodiazépines exercentleur action au niveau des récepteurs desbenzodiazépines (p. 224). Le choixd'une substance pour les différentes in-dications dépend uniquement de la rapi-dité de l'effet, de son intensité et de sadurée et donc des propriétés physico-chimiques et pharmacocinétiques desmolécules. Chaque benzodiazépine sé-journe un temps différent dans l'orga-nisme, et sera éliminée principalementpar biotransformation. L'inactivationpeut être accomplie en une seule réac-tion chimique ou au contraire en plu-sieurs étapes (ex. diazépam), avantqu'un métabolite inactif et propre àl'élimination rénale ne soit formé.Comme les sous-produits sont en partieactifs et en partie éliminés, mais beau-coup plus lentement que les moléculesinitiales correspondantes, ils peuvents'accumuler au cours d'une administra-tion régulière et finalement participerde façon importante à l'action sou-haitée. Ce sont les substituants sur lecycle diazépine (diazépam : déalkyla-tion sur l'azote en position 1, 11/2 ~30 h ; midai.olam : hydroxylation dugroupement méthyle sur le noyau imi-dazole, t 1/2 ~ 2 h), ou le cycle diazé-pine lui-même qui seront d'abord tou-chés par les transformations chimiques.Le midawlam hydroxylé sera éliminétrès rapidement par le rein après conju-gaison avec l'acide glucuronique. Lediazépam déméthylé sur l'azote (nor-diazépam) est biologiquement actif etsera hydroxylé en position 3 du noyaudiazépine avec un 11/2 de 50-90 heures.Le métabolite hydroxylé (oxazépam)est également pharmacologiquementactif. Le diazépam est déjà éliminé len-tement et s'accumule donc au coursd'une administration régulière maiscette accumulation est encore plus fortepour son métabolite le nordiazépam.L'oxazépam est conjugué à un acide
glucuronique sur le groupement hvdroxyle avec un t 1/2 d'environ 8 h eiéliminé par le rein (A). En (B) sont re-présentées pour différentes benzodiazé^pines ou leurs métabolites actifs lesvaleurs des demi-vies d'éliminationfigurées par des surfaces grises.
Les substances avec des demi-viestrès brèves et qui ne donnent pas nais-sance dans l'organisme à des méta-bolites actifs pourront être utiliséescomme inducteurs de sommeil ou pourmaintenir le sommeil (désigné en B pardes surfaces bleu clair), tandis que lessubstances avec des demi-vies pluslongues doivent être réservées pour destraitements anxiolytiques à long ternie(surfaces vert clair). Elles permettent demaintenir un niveau plasmatique élevéet régulier.
Le midawlam sert comme anes-thésique injecté pour l'induction etl'entretien d'une anesthésie combinée.
Risque de dépendanceL'usage régulier des benzodiazépinespeut entraîner le développement d'unedépendance. Cette relation n'est pasaussi évidente qu'avec les autres sub-stances pouvant entraîner une toxico-manie, car l'effet des premières benzo-diazépines mises sur le marché duretrès longtemps, de sorte que les symp-tômes de manque (le signe éclatantd'une dépendance installée) ne se déve-loppent que très tardivement. Pendantcette période de manque se manifestentagitation, nervosité, excitabilité et an-goisse. Ces symptômes peuvent à peineêtre distingués de ceux considéréscomme les indications des benzodiazé-pines. L'administration d'un antago-niste des benzodiazépines entraînel'apparition brutale de symptômes decarence. Il faut noter que les substancesavec une demi-vie d'élimination dedurée intermédiaire, seront utilisées leplus souvent de façon abusive et mon-trent également le risque de dépendancele plus élevé (surface violette en B).
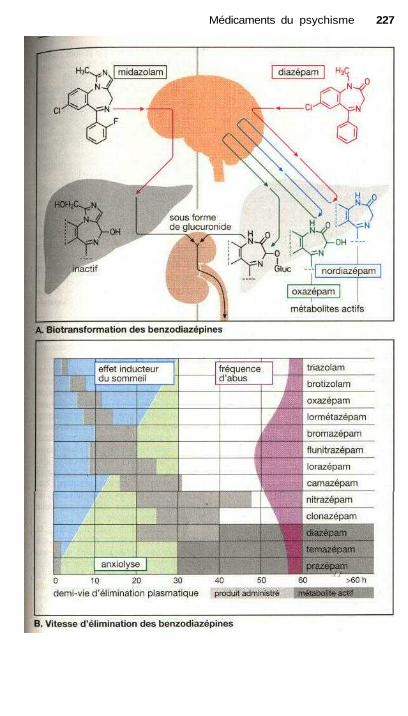
Médicaments du psychisme 227

228 Médicaments du psychisme
Traitement de la cyclothymie
Sous la dénomination de cyclothymie, ondésigne une maladie de l'esprit (psychoseaffective), dans laquelle survient par pé-riode et sans cause extérieure une altéra-tion pathologique de l'humeur. Dans lecas d'une humeur dépressive, il s'agitd'une dépression endogène (mélancolie).L'état pathologique inverse est la manie(p. 232). Ces épisodes surviennent defaçon périodique, et sont entrecoupés dephases où l'humeur est normale. Selon lespatients, l'humeur peut aller d'une direc-tion à l'autre ou bien toujours dans lamême direction (unipolaire).I. Traitement de la dépression endogèneAu cours d'une dépression endogène, lepatient se trouve dans un état de souf-france profonde (insupportable) ; à caused'un sentiment de culpabilité, il se fait desreproches amers. Le déclenchement d'ac-tivités ou d'actions est inhibé. Il existe unrisque de suicide, mais le passage à l'acteest peu probable à cause de l'affaiblisse-ment de la volonté d'entreprendre. Deplus surviennent diverses altérations so-matiques (troubles du sommeil, perted'appétit, constipation, sensations car-diaques, impuissance). La dépression en-dogène est figurée en (A) par une largebande de couleurs sombres, la volontéd'entreprendre symbolisée par une lignesinusoïdale est fortement diminuée.
Les traitements peuvent être divisésen deux groupes :- thymoleptiques pour lesquels l'actionantidépressive et l'amélioration de l'hu-meur est nettement marquée : antidépres-seurs tricycliques ;- thymérétiques pour lesquels se mani-festent surtout une action désinhibitrice etune augmentation de l'esprit d'entreprise,par ex. les inhibiteurs de monoamine oxy-dase.Ce serait une erreur de traiter un patientavec des psychostimulants tels les amphé-tamines qui n'accroissent que l'espritd'entreprise. En effet, son humeur nechange pas mais l'inhibition de sa volontéd'entreprendre s'estompe (A), ce qui aug-mente le risque de suicide.
Les antidépresseurs tricycliquessont depuis longtemps utilisés pour le
traitement de la dépression et sont encnraujourd'hui les médicaments les plus mportants, par ex. imipramine.
Le cycle central à sept côtés de cessubstances entraîne le fait que les deuxcycles de part et d'autre forment entre euxun angle de 120°. C'est une différencestructurale nette par rapport aux neurolep-tiques de type phénothiazines (p. 237) n -ont une structure cyclique plane. L'azotede la chaîne latérale est essentiellementprotoné à pH physiologique.
Ces substances présentent une a f f i -nité pour les récepteurs et les systèmes detransport des neurotransmetteurs et fonc-tionnent comme des inhibiteurs. C'estainsi que la recapture de la noradrénaline(p. 82) et de la sérotonine (p. 116) seraempêchée et leurs effets renforcés. Les ré-cepteurs muscariniques et les récepteursadrénergiques ainsi que les récepteurs del'histamine seront bloqués ; il y a peud'interférence avec le système dopami-nergique.
On ne sait pas exactement de quellefaçon l'effet antidépresseur dérive decette perturbation des neurotransmetteurs.D'abord, ce n'est qu'après une adminis-tration de longue durée, de l'ordre de se-maines, que se met en place l'effet anti-psychotique proprement dit : améliorationde l'état d'esprit et du tonus. L'effet surles neurotransmetteurs est déjà pratique-ment obtenu dès le début du traitement.Ce sont vraisemblablement des modifica-tions adaptatives qui se produisent lente-ment en réaction à ces interférences, quiseraient la véritable origine de cette actionantipsychotique. Chez des gens dont l'hu-meur est normale, les antidépresseurs de
'façon remarquable n'altèrent pas cettehumeur (aucune euphorie).
A côté des effets antipsychotiques, seproduisent également des effets aigus, quise manifestent également chez des gensnormaux. Ils sont plus ou moins marquésselon la substance, ce qui permet uneadministration thérapeutique adaptée(p. 230). Ceci est dû à un éventail d'inter-férences variables avec les systèmes deneurotransmetteurs. L'amitriptyline a uneaction sédative, anxiolytique et tempère letonus psychomoteur. Elle sert à traiter lespatients dépressifs anxieux et surexcités.

Médicaments du psychisme 229

230 Médicaments du psychisme
La désipramine au contraire agiten stimulant l'activité psycho-motrice.L'imipramine occupe une position inter-médiaire. Il faut noter que la désipramine(déméthyl-imipramine) se forme égale-ment dans l'organisme à partir de l'imi-pramine. Le dérivé déméthylé de l'ami-tnptyline (nortriptyline) est par ailleursmoins inhibiteur que la substance mère,
L'action anxiolytique et sédativepeut être utilisée chez des malades où lessouffrances organiques sont très mar-quées par le psychisme, de façon à obtenirun « découplage psychosomatique ». Onpeut également noter son utilisationcomme co-analgésique (p. 192).
Les effets secondaires indésirablesdes antidépresseurs tricycliques reposenten grande partie sur leur antagonisme vis-à-vis de divers neurotransmetteurs. Ceseffets débutent également immédiatementaprès le début du traitement. Le blocagedes récepteurs muscariniques de l'acétyl-choline provoquent des effets de typeatropinique : tachycardie, inhibition dessécrétions glandulaires (sécheresse de labouche), constipation, troubles de la mic-tion et troubles de la vision.
Les modifications du systèmeadrénergique sont complexes. L'inhi-bition de la recapture des catécholaminespeut provoquer des effets sympathomi-métiques indirects. Les patients sont éga-lement hypersensibles aux catéchola-mines (par exemple, addition d'adrénalineà une anesthésie locale). D'un autre côté,le blocage des récepteurs a, peut provo-quer une hypotension orthostatique.
En fonction de leur nature chimique,certaines de ces molécules vont agircomme des substances cationiques am-phiphiles en stabilisant les membranes, cequi peut entraîner des altérations de laconduction avec des arythmies et une di-minution de la contractilité cardiaque.Tous les antidépresseurs tricycliques fa-vorisent la tendance aux crampes. La sti-mulation de l'appétit peut entramer uneaugmentation du poids.
La maprotiline qui est au sens strictun antidépresseur tétracyclique ne pré-sente, en ce qui concerne ses propriétés cli-niques et pharmacologiques, pratiquementpas de différence avec les substances tricy-
cliques. La miansérine, également unesubstance tétracyclique au sens strictdistingue dans la mesure où elle au»ment^la concentration de noradrénaline dans 1fente synaptique via un blocage des récenleurs a, présynaptiques et non par l'inhibi-tion de la recapture. Les effets atropinique<ide la miansérine sont moins marqués
La fluoxétine est un exemple d'anti-dépresseurs nouvellement développés quisont « atypiques » en ce qui concerne leurstructure et leur action. Ce n'est pas un tri-cyclique et son action sur les neurotrans-metteurs est sélective : elle n'inhibe que larecapture de la sérotonine. La fluoxétine aune composante excitatrice, et son activitéantidépressive semble être moins forte quecelle des antidépresseurs tricycliques.L'avantage, c'est qu'elle ne présenteaucun effet atropinique et souvent aucuneffet de stabilisation de membrane sui lescellules cardiaques. La fluoxétine pro-voque une réduction de l'appétit et uneperte de poids. Les effets secondaires sontnervosité, tremblement, perte de sommeilet angoisse. Dans l'ensemble, le tableaudes effets de la fluoxétine semble plutôtêtre celui d'un thymérétique.
Laparoxétine et bfluvoxamme sontd'autres inhibiteurs de la recapture de lasérotonine.
Le moclobémide est un nouveau re-présentant du groupe des IMAO. Comptetenu de l'inhibition de la dégradationintraneuronale de sérotonine et de nora-drénaline, leur concentration augmentedans la fente synaptique. Dans le cas desinhibiteurs de la MAO, c'est l'action th\-mérétique, stimulant le désir d'entre-prendre, qui est à la base de leur effetL'autre substance de ce groupe, la tranyl-cypromine, inhibe de façon irréversibleles deux enzymes, MAO-A et MAO-B. Lacapacité du foie à éliminer de façon pré-systémique les aminés biogènes, commela tyramme, apportées par l'alimentation(fromages, chianti) est donc diminuéePour éviter une élévation de la tension ar-térielle, il faut associer un traitement par latranylcypromine à des prescriptions diété-tiques strictes. Dans le cas du moclobé-mide, le danger est beaucoup plus faible,car seule la forme A de la MAO est in-hibée et de plus cet effet est réversible.
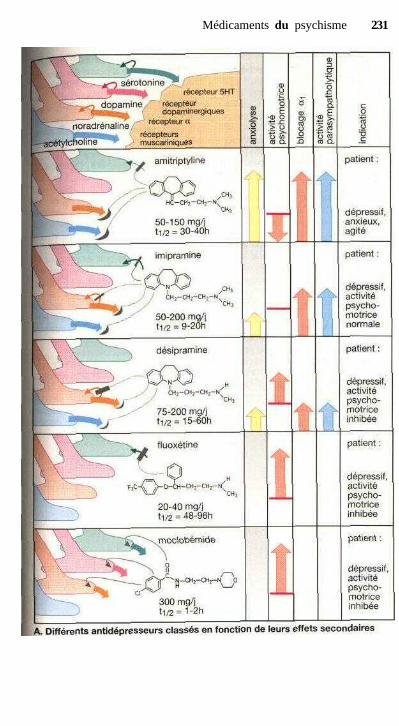
Médicaments du psychisme 231

232 Médicaments du psychisme
II. Traitement de la manieLa phase maniaque est reconnais-sable par une bonne humeur exa-gérée, un flot d'idées et un esprit d'en-treprise anormalement élevé. Ceci estmontré en (A) par un tableau coloreavec des structures brisées et des cou-leurs agressives. Les patients se suresti-ment, montrent une activité sans repos,expriment un flot d'idées bizarres, agis-sent de façon irresponsable (sur le plansexuel ou financier).
Ions lithium. Le traitement de laphase maniaque utilise des sels de li-thium par ex. sous forme d'acétate oude carbonate. L'effet se manifeste en-viron 10 jours après le début du traite-ment. En raison de l'étroitesse de lafenêtre thérapeutique, il est nécessaired'effectuer des contrôles sanguins, laconcentration sérique doit être prochede 0,8-1 mM le matin à jeun. Pour desvaleurs plus élevées, apparaissent déjàdes e f f e t s secondaires : les troubles duSNC se manifestent par un tremblementléger des extrémités, mais aussi par desaltérations des mouvements (ataxie) oudes crampes. L'action de l'ADH au ni-veau du rein peut être inhibée ce qui estvisible par une polyurie et une soif(p. 162). La fonction thyroïdienne estinhibée (p. 244) tandis qu'apparaît unehypertrophie compensatrice.
On n'a aucune certitude concernant le mécanisme d'action des ions ]ithium. Chimiquement, le lithium appartient au groupe des métaux alcalinsparmi lesquels le sodium et le potas-sium occupent dans l'organisme uneplace considérable. On peut admettreque les ions lithium interfèrent, au ni-veau de sites non définis, avec les per-méabilités ioniques des membranespour les ions Na4^ et K^ ou avec lespompes ioniques et qu'il s'ensuit desconséquences significatives pour lefonctionnement des cellules du cerveaudébouchant sur une action positive surla cyclothymie. Une diminution ducontenu membranaire en phosphatidylinositol diphosphate peut aussi être im-portante, car ce phospholipide joue unrôle important dans la transduction dessignaux (p. 66).
Il faut remarquer que les neuro-leptiques peuvent également être uti-lisés pour calmer un état maniaque(voir ci-dessous).
III. Prévention de la cyclothymieAprès 6-12 mois de traitement, les ionslithium empêchent l'apparition de nou-velles phases maniaques. Les phases.dépressives sont également prévenues.Les sels de lithium stabilisent le carac-tère dans un état à peu près normal.
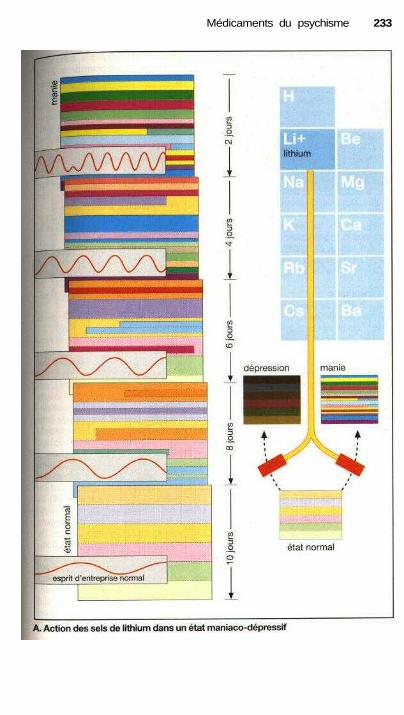
Médicaments du psychisme 233

234 Médicaments du psychisme
Traitement de la schizophrénie
La schizophrénie est une psychose endo-gène qui se développe par poussées suc-cessives Les symptômes de base sont desaltérations de la pensée (par ex incohé-rence pensées illogiques et décousues ,blocage du raisonnement perte soudainedu cours des idées, repliement del'esprit le malade prétend que ses pen-sées lui sont retirées de l'extérieur) del'affectivité (état d'esprit sans relationavec la situation) et de la volonté d'entre-prendre Des symptômes supplémentairessont par ex des délires (délires de persé-cution), ou des hallucinations auditivesfréquentes et angoissantes L'incohérencede l'état mental est symbolisé en haut àgauche (A) (comparez avec l'état normalde l'esprit en bas de la page 233)
NeuroleptiquesAprès le début du traitement, on observed'abord une sédation seule Les visions etles hallucinations qui tourmentent les pa-tients schizophrènes perdent de leur force(A, affaiblissement des couleurs vives), lecomportement psychotique persiste ce-pendant Au cours des semaines suivantes,les phénomènes psychiques se normali-sent lentement (A), les à-coup psycho-tiques s'estompent Une normalisationcomplète ne peut souvent pas être ob-tenue Mais même lorsque l'on ne peutpas parvenir à une guenson, les modifica-tions que nous venons de décrire ont ce-pendant une conséquence Pour les ma-lades en effet, le supplice que constitue lechangement de leur moi sera atténué, lasurveillance sera plus souple et laconfiance que lui accorde la communautéreviendra plus rapidement II existe deuxtypes de composés avec des structureschimiques différentes permettant un trai-tement neuroleptique, antipsychotique1. les phénothiazines dérivées d'un anti-histamimque, la prométhazme (dont lasubstance de référence est la chlorproma-zme) et leurs analogues (par ex thioxan-thènes) et 2 les butyrophénones (sub-stance de référence, l'halopéndol) Lesphénothiazines et les thioxanthènes peu-vent être séparés selon la structurechimique de la chaîne latérale en
- composés aliphatiques substitués(chlorpromazme, tnflupromazine, p 2371- piperazmes substituées (tnflupérazine'fluphénazme, flupentixol, p 237)
L'effet antipsychotique repose vrai-semblablement sur une action antaromste au niveau des, récepteur', dopami-nergiques A côté de l'effet principalanti-psychotique, les neuroleptiques pré-sentent simultanément des effets antago-nistes vis-à-vis de- l'acétykholme, au niveau des récep-teurs muscanmques -» effets de typeatropmique ,- la noradrenahne, au niveau des récep-teurs a -» troubles de la régulation de lapression artérielle,- la dopamme, au niveau des récepteursdopammergiques de la substance noire -»troubles extrapyramidaux, de l'area post-rema -> action anti-émétique (p 324), del'hypophyse — augmentation de la sécré-tion de la prolactme (p 240) ,- l'hittamme —* origine probable de lasédationCes effets complémentaires peuvent éga-lement se produire chez des individuspsychiquement sains, leur intensité estvariable d'un produit à l'autre
Indications complémentaires. L'ad-ministration de neuroleptiques provoquede façon aiguë une tedation et une anxwlyse Ces effets peuvent être utilisés dansdes maladies ayant une forte composantepsychique pour induire un découplagepsychosomatique L'effet distanciant aégalement une utilité lors de l'associationd'un neuroleptique (le dropéndol, un dé-rivé butyrophénone) à un opiolde pour in-duire une neuroleptanalgesie (p 214) ouencore pour calmer un panent wexciléou pour le traitement du delmum tremens(halopéndol) L'administration dans lecas d'une manie a été signalée précédem-ment (p 232)
Les neuroleptiques n'ont pas d'ac-tion anticonvulsivante A cause de leuraction mhibitnce sur les centres thermoré-gulateurs, les neuroleptiques peuvent êtreutilisés pour refroidir le corps de façoncontrôlée au cours d'une opération (hiber-nation artificielle, p 200)

Médicaments du psychisme 235

236 Médicaments du psychisme
Effets secondaires. Les effets se-condaires les plus fréquents et qui limi-tent fréquemment le traitement sont lestroubles moteurs extrapyramidaux ; ilsproviennent du blocage des récepteursdopaminergiques. Une dyskinésie pré-coce peut être notée immédiatementaprès le début du traitement sous formede mouvements involontaires et anor-maux, surtout au niveau de la tête, ducou et des épaules. Après des semainesou des mois de traitement on peut ob-server des symptômes analogues à ceuxde la maladie de Parkinson (tremble-ment, raideur, lenteur des mouvements)ou bien une akathisie (agitation mo-trice). Toutes ces altérations peuventêtre soignées par des antiparkmsoniensappartenant au groupe des anticholiner-giques (ex. bipéridène). Ces symptômesdisparaissent toujours à l'arrêt des neu-roleptiques. Une dyskinésie tardivepeut être notée, en particulier au mo-ment de l'arrêt du traitement, après uneadministration de plusieurs années. Elleest due à une hypersensibilité du sys-tème des récepteurs dopaminergiques ets'aggrave après administration d'anti-cholinergiques.
Au cours d'une administrationchronique de neuroleptique peuvent seproduire de rares lésions hépatiquesavec cholestase. Un effet secondairerarissime mais dramatique est le syn-drome malin des neuroleptiques (hy-perthermie, raideur des muscles sque-lettiques, stupeur), qui peut être fatal enl'absence d'un traitement médical in-tensif (entre autre dantrolène).
Différences entre neurolep-tiques. Il est clair en ce qui concerne letraitement, qu'il existe des dérivés desphénothiazines ou des analogues dontles propriétés se distinguent nettementde celles de la chlorpromazine, la sub-stance de référence, et s'apparententplutôt à celles de la butyrophénone.
.Cela touche l'activité antipsychotique(symbolisée par la flèche), l'importancede la sédation et la possibilité de provo-quer des troubles extrapyramidaux.
Le déclenchement variable dggtroubles extrapyramidaux peut être attribué à un rapport variable entre les ap'tivités antagonistes vis-à-vis de la dona'mine ou de l'acétylcholine (p. 186) i«risque de troubles extrapyramidaux estplus important dans le cas des dérivésbutyrophénones que dans celui des phé-nothiazines. Ils ne possèdent en effetaucune action anticholinergique etl'équilibre entre l'activité des neuronesdopaminergiques et cholinergiques estplus profondément altéré.
Les dérivés substitués par une pi-pérazine (ex. trifluopérazme, fluphéna-zine) ont une activité antipsychotiqueplus élevée, à doses comparables, queles dérivés substitués par une chaînealiphatique, (ex. chlorpromazine, triflu-promazine), mais la qualité des effetsantipsychotiques n'est pas modifiée.
Les thioxanthènes sont des ana-logues structurels des phénothiazines(ex. flupentixol, chorprothixène) pourlesquels l'azote présent dans le cyclecentral est remplacé par un atome decarbone relié à la chaîne latérale par unedouble liaison. Ils se distinguent desphénothiazines par une composanteadditionnelle thymoleptique.
La clowpine est un neuroleptiqueà la structure atypique, censé ne provo-quer aucun trouble extrapyramidal.Cette propriété est vraisemblablementdue au fait qu'il bloque parmi les ré-cepteurs dopaminergiques plus particu-lièrement le type D4. Mais les récep-teurs 5HT,, H, et muscanniques sontégalement touchés. La clozapine peutêtre utilisée lorsque les autres neuro-leptiques ne peuvent plus être prescritsà cause de leurs effets extrapyrami-daux. La clozapine peut provoquer uneagranulocytose et ne doit donc être uti-lisée qu'avec une surveillance régu-lière de la formule sanguine. Elle a uneforte action sédative.
La fluphénazine ainsi que l'halo-péridol peuvent être administrés sousforme de dépôt intramusculaire aprèsestérification par un acide gras.
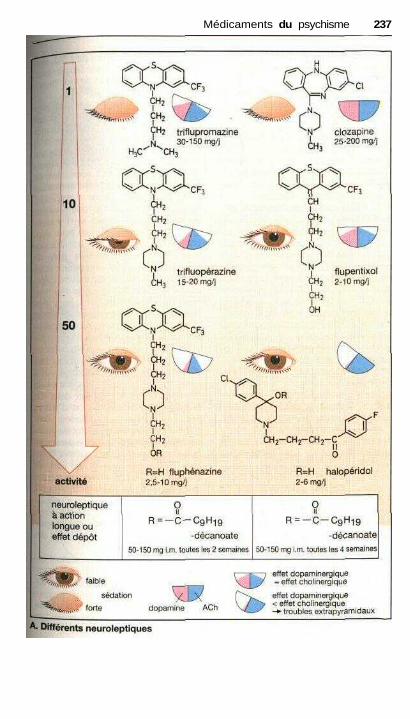
Médicaments du psychisme 237

238 Médicaments du psychisme
Psychomimétiques (substanceshallucinogènes ou psychédéliques)
Les psychomimétiques ont la faculté dedéclencher des modifications psy-chiques analogues à celles qui peuventse manifester au cours d'une psychose :visions, illusions et hallucinations.Cette expérience peut donc posséder uncaractère fantastique, la transformationémotionnelle et du raisonnement provo-quée par ce phénomène paraît follepour celui qui est à l'extérieur.
Une action psychomimétique peutêtre illustrée par l'exemple des portraitsexécutés par un peintre sous l'influencede l'acide lysergique diéthylamide(LSD). Il raconte que sous l'effet de lagriserie du LSD, arrivant par vagues, levisage du modèle devient de plus enplus grimaçant, phosphorescent dansdes coloris bleu violet et s'agrandit ouse rétrécit comme à travers l'objectifd'un zoom. Les changements confus deproportions entraînent donc une succes-sion de mouvements bizarres. La cari-cature diabolique apparaît menaçante.
Les illusions se font égalementsentir dans le domaine de l'audition et del'odorat : les sons sont vécus comme despoutres suspendues et les impressionsoptiques comme des odeurs (par ex.d'ozone). Sous l'emprise du LSD, l'indi-vidu se voit par moment de l'extérieur etanalyse son état. En outre, les frontièresentre son être et l'environnement s'effa-cent. Un sentiment exaltant de fusionavec les autres et le cosmos s'installe. Lanotion de durée n'existe plus, il n'y aplus ni avant ni après. Des objets sontvus qui n'existent pas. Des expériencessont faites qui ne sont pas explicables.C'est pourquoi on parlera d'un effet dedilatation de la conscience à propos duLSD (révélations psychédéliques).
Le contenu de ces hallucinationspeut de temps à autre être extrêmementmenaçant (bad trip), l'individu se voitéventuellement poussé à une action vio-lente ou au suicide.
Après la « griserie » du LSD, sur-vient une phase de grande fatigue avecun sentiment de honte et de vide humi-liant.
Le mécanisme de l'action psy-chomimétique est inconnu. Commeune partie des substances hallucino-gènes telles le LSD, la psilocine et lapsilocyhine (tirées d'un champignonmexicain, le psilocybe) la bufoténine(tirée entre autres des sécrétions dela peau d'un crapaud), la mescaline(extraite d'un cactus mexicain,Arihalonium lewinii, peyotl) présentedes analogies structurales avec la séro-tonine et l'adrénaline, on peut sup-poser l'existence d'une interférenceavec ces aminés biogènes dans leSNC. La structure d'autres moléculescomme le tétrahydro-cannahinol (dé-rivées du cannabis indica, le chanvre,haschich, marihuana), le muscimul(extrait d'un champignon amanitemuscaria) ou ïaphencyclidine, synthé-tisée comme anesthésique injectablene montre pas ces caractères com-muns. Des hallucinations peuvent êtreassociées, comme effet secondaire, àla prise d'autres substances, par ex. lascopolamine (au Moyen Age dans « lepeuplier des sorcières ») ou d'autresparasympatholytiques d'action cen-trale. Des substances hallucinogènesnaturelles ont été utilisées dans cer-taines religions par des prêtres (cha-mans) pour parvenir à un état detranses. Le LSD a été consomméassez fréquemment dans les années 60,en particulier par des artistes : art psy-chédélique qui consiste à représenterd'une façon qui ne peut pas être appré-hendée par la raison, des rêves ou dessignes hallucinatoires.
Comme il n'est pas possible d'ex-clure le développement d'une dépen-dance ou d'altérations psychiques du-rables après la prise d'un psycho-mimétique, leur production et leur com-merce sont interdits (stupéfiants non encirculation).
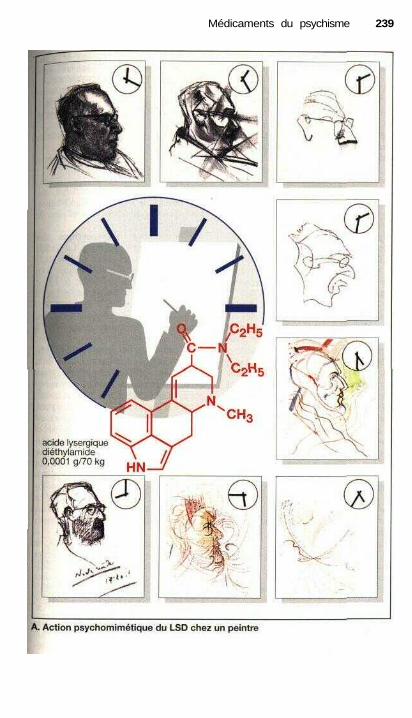
Médicaments du psychisme 239

240 Hormones
Hormones hypothalamiqueset hypophysaires
Le système endocrinien est contrôlé parle SNC. Les cellules nerveuses de l'hy-pothalamus synthétisent et libèrent desmédiateurs qui vont gouverner dans l'hy-pophyse antérieure la sécrétion d'hor-mones ou qui seront, elles-mêmes, distri-buées dans l'organisme comme hormones.
Enfin, il y a les hormones de l'hy-pophyse postérieure : les prolongementsdes neurones hypothalamiques projettentdans la post-hypophyse (neurohypo-physe), stockent à cet emplacement unnonapeptide (ADH : hormone antidiuré-tique) et Yocytocine, et les libèrent dans lesang en cas de besoin. Les traitements uti-lisant ces hormones peptidiques (ADH,p. 162, ocytocine, p. 126) seront réaliséspar voie parentérale ou en nébulisationnasale.
Les médiateurs hypothalamiquessont des peptides qui atteignent leurs cel-lules cibles dans l'adénohypophyse par unsystème porte, c'est-à-dire par deux zonescapillaires disposées l'une à la suite del'autre. La première est située dans la tigede l'hypophyse ; à ce niveau, les hor-mones libérées par les terminaisons ner-veuses des neurones hypothalamiquesdiffusent dans le sang. La seconde com-prend les capillaires de l'anté-hypophyse.A ce niveau, les hormones diffusent dusang vers les cellules cibles qu'ellescontrôlent. Les hormones libérées par lescellules de l'anté-hypophyse parviennentdans le sang et sont ensuite distribuéesdans l'organisme (1).
Dénomination des médiateurs /hormones de libération. RH : releasinghormone, hormone de libération. RIH :release inhibiting hormone, hormonebloquant la libération.
GnRH : gonadotrophine-RH = go-nadoreline (gonadolibérine) ; stimule laproduction de FSH (hormone folliculo-stimulante) et de LH (hormone lutéini-sante).
TRH : protiréline, stimule la sécré-tion de TSH (hormone thyréostimulante).
CRH : corticotropine RH (CRF) ;stimule la sécrétion d'ACTH (hormoneadrénocorticotrope = corticotropine).
GRH : growth hormone-RH ; ai.mule la libération de GH (growth hor-mone = STH = hormone somatotrope).
GRIH : somatostatine, inhibe la sé-crétion de STH (et aussi d'autres hor-mones peptidiques produites par ex. par lepancréas et l'intestin).
PRH : prolactine-RH, son existenceest hypothétique.
PRIH : inhibe la sécrétion de pro-lactine, ce pourrait être la dopamine.
Les hormones hypothalamiquessont essentiellement administrées parvoie parentérale à des fins diagnostiquespour tester la fonction des cellules del'adénohypophyse.
Modulation thérapeutique descellules de l'anté-hypophyse. La GnRHsera utilisée dans les cas de stérilité de lafemme d'origine hypothalamique pourstimuler la sécrétion de FSH et de LH etdéclencher une ovulation. Dans ce but, ilfaut imiter le rythme de la sécrétion phy-siologique (pulsatile, environ toutes les90 min) (administration parentérale àl'aide d'une pompe spéciale).
Analogues des gonadorélines, su-peragonistes : ce sont des analogues de laGnRH dont l'affinité pour les récepteursde la GnRH sur les cellules de l'hypo-physe est beaucoup plus élevée. La consé-quence d'une stimulation ininterrompueet non physiologique des récepteurs estune interruption de la sécrétion de FSH etde LH après une phase initiale de stimula-tion. Buséréline, leuproréline, gosérélinesont utilisées chez des patients souffrantd'un carcinome prostatique pour dimi-nuer la production de testostérone qui fa-vorise le développement de la tumeur. Leniveau de testostérone chute autantqu'après l'ablation chirurgicale des testi-cules (2).
L'agoniste dopaminergique D;, labromocriptine (p. 114) inhibe les cellulesà prolactine de l'anté-hypophyse (indica-tions : sevrage, tumeurs hypophysairessécrétant de la prolactine). Une produc-tion anormale de STH peut être égalementréduite (indication : acromégalie) (3).
L'octréotide est un analogue de lasomatostatine. Il sera par ex. utilisé dansle cas de tumeurs de l'hypophyse qui sé-crètent de la GH.
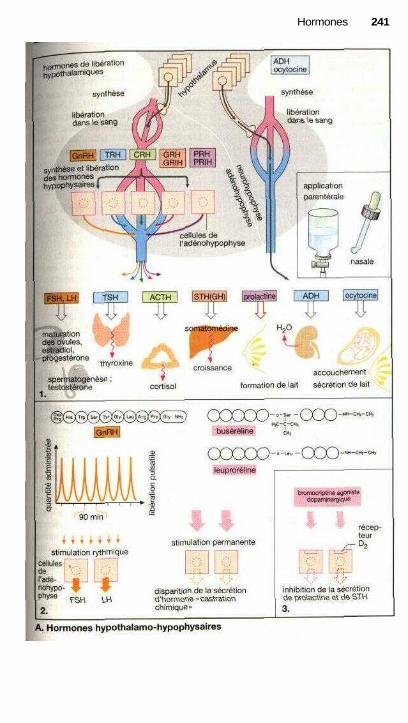
Hormones 241

242 Hormones
Traitements par les hormonesthyroïdiennes ,
Les hormones thyroïdiennes agissent en .stimulant le métabolisme. Leur libéra-
tion (A) est gouvernée par une glyco-protéine hypophysaire, la TSH, dontla libération est, de son côté, sous lecontrôle d'un tripeptide hypothala-mique TRH. La sécrétion de TSHdécroît lorsque la concentration d'hor-mone thyroïdienne dans le sang aug-mente ; à l'aide de ce mécanisme derétrocontrôle négatif s'établit automati-quement une production d'hormonesadaptée aux besoins.
La glande thyroïde produit essen-tiellement la thyroxine CT^). La formeactive semble cependant être la triiodo-thyronine (T,). T^ est en partie trans-formée en T, dans l'organisme et les ré-cepteurs des cellules cibles ont uneaffinité 10 fois supérieure pour T^.L'action de T^ se produit plus rapide-ment et dure un peu moins longtempsque celle de T^. Le 11/2 d'éliminationplasmatique atteint environ 7 jours pourÏ4 mais seulement 1,5 jour pour T.,. Ladégradation de T^ et T, libère de l'iode.Il y a 100 |xg d'iode contenus dans150 |Jig de T4.
Pour l'utilisation thérapeutique,on préférera ^.^. T, est certes la formeactive et est mieux absorbée par l'in-testin mais avec T,, on atteint pourtantun niveau plasmatique équivalent car sadégradation est très lente. Puisque l'ab-sorption de Ï4 est maximale à jeun, ellesera administrée environ 1/2 h avant lepetit déjeuner.
Traitement de substitution dansle cas d'une hypothyroïdie. Un hypo-fonctionnement de la thyroïde, qu'ilsoit primaire et lié à une maladie de lathyroïde ou secondaire via une carenceen TSH, sera soigné par l'administra-tion orale de thyroxine. Pour com-mencer, la dose de Ï4 sera en généralchoisie assez faible, car on peutcraindre un changement trop rapide dumétabolisme avec le risque d'une sur-charge cardiaque (angine de poitrine,,infarctus), et augmentée graduellement.
La dose finale permettant l'installationd'une euthyroïdie dépend des besoinsindividuels (environ 100 p,g/j).
Traitement suppressif en cas degoitre euthyroïdien (B). L'origined'un goitre est principalement un ap-port d'iode alimentaire insuffisant.L'augmentation de l'action de la TSHpousse la thyroïde à utiliser de façontellement intensive la faible quantitéd'iode disponible qu'une hypothyroïdiene produit pas mais que la thyroïdegrossit.
En raison de la régulation de lafonction thyroïdienne selon le principedu rétrocontrôle négatif, on peut aboutiren administrant T^ à une dose (100 -150 |Jig/j) équivalente à celle de la pro-duction quotidienne d'hormone endo-gène, à un arrêt de la stimulation thyroï-dienne. La glande inactive et maintenueau repos diminue de taille.
Dans le cas d'un goitre euthyroï-dien par carence en iode, installé depuisun temps court, il est possible de dimi-nuer la taille de la thyroïde par une aug-mentation de l'apport en iode (com-primés d'iodure de potassium).
Chez des patients âgés, présentantun goitre avec carence en iode, existe ledanger de déclencher une hyperthy-roïdie par l'augmentation de l'apportd'iode (p. 245) : après des années de sti-mulation maximale, le tissu thyroïdienpeut devenir indépendant de la stimula-tion par la TSH (« tissu autonome »).Lors de l'augmentation de l'apportd'iode, la production d'hormone thyroï-dienne s'accroît et, à cause des rétro-contrôles négatifs, la sécrétion de TSHdiminue. L'activité du tissu autonomedemeure cependant élevée, l'hormonethyroïdienne est libérée en excès, unehyperthyroïdie induite par l'iode s'estdonc installée.
Prévention par les sels d'iode. Legoitre avec une carence en iode est lar-gement répandu. Par l'administrationde sel de cuisine iodé, on peut assureraisément les besoins en iode (150-300 |Jig/j d'iode) et éviter le goitreeuthyroïdien.
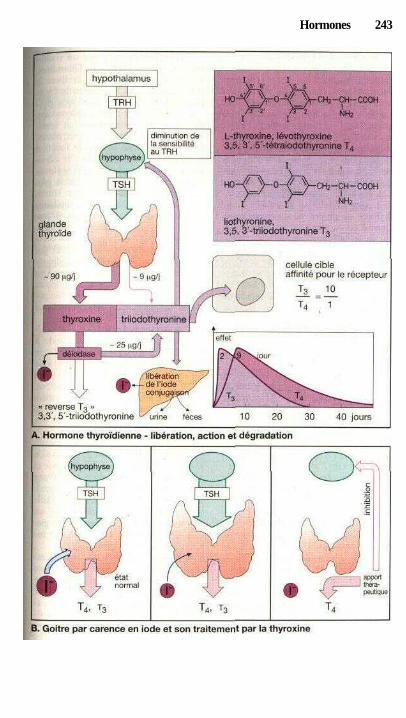
Hormones 243

244 Hormones
Hyperthyroïdie et thyréostatiques
Hyperthyroïdie. Le fonctionnementanormalement élevé de la glande thy-roïde dans la maladie de Basedow (A)est dû à la formation d'anticorps (IgG)qui se fixent aux récepteurs de la TSH etles stimulent. La conséquence de cettestimulation est une surproduction d'hor-mones (avec disparition de la sécrétionde TSH). La maladie de Basedow peutdisparaître spontanément en 1 ou 2 ans.Son traitement réside donc en premierlieu dans l'inhibition réversible de lathyroïde au moyen de thyréostatiques.Dans d'autres formes d'hyperthyroïdie,par ex. les adénomes thyroïdiens produi-sant de l'hormone (morphologiquementbénins), l'option thérapeutique de choix
^ est l'ablation du tissu soit par voie chirur-gicale, soit par administration d'iode i31!en quantité suffisante. L'iode radioactifest capté par la thyroïde et détruit le tissudans une zone de quelques millimètrespar le rayonnement (3 (électrons) émislors de la décroissance radioactive.
Pour les hyperthyroïdies induitespar l'iode, voir p. 242.
Thyréostatiques. Ils inhibent lafonction thyroïdienne. Avant sécrétionde l'hormone thyroïdienne se déroulentles événements suivants (C). L'iodesous forme d'iodure est activementcaptée par une « pompe » dans les cel-lules thyroïdiennes. Il se produit alorsune réduction en iode, une liaison sur lachaîne latérale d'une tyrosine de la thy-réoglobuline, une association de deuxgroupements tyrosine iodés avec forma-tion des résidus T^ et 13. Cette réactionest catalysée par l'enzyme peroxydase.Dans l'intérieur du follicule thyroïdien,la thyréoglobuline portant la 14 eststockée sous forme de colloïde. En casde besoin, l'hormone thyroïdienne seralibérée à partir du colloïde, après endo-cytose et hydrolyse par les enzymes ly-sosomiales. Un effet thyréostatique peutavoir lieu par une inhibition de la syn-thèse ou de la libération de l'hormone.En cas d'interruption de la synthèse, lecolloïde est encore utilisable et l'effetthyréostatique est alors d'apparitionlente.
Thyréostatiques pour un traite-ment de longue durée (C). Thiamides'dérivés de la thiourée. Ils inhibent laperoxydase et donc la synthèse de l'hor-mone. Deux thérapeutiques sont pos-sibles pour rétablir un état euthyroi'diendans la maladie de Basedow : a) admi-nistration de thiamide seule avec réduc-tion progressive de la dose en fonctionde la régression de la maladie ; b) admi-nistration de thiamide à dose plus élevéeet ajustement de l'inhibition de la pro-duction d'hormones par apport simul-tané de thyroxine. Les effets secondairesdes thiamides sont rares, mais il fautfaire attention à la possibilité d'une agra-nulocytose.
Les perchlorates administrés parvoie orale sous forme de sels sodiquesinhibent le transport actif d'iodure. Desanémies aplasiques peuvent se produirecomme effet secondaire. En compa-raison des thiamides, leur importancethérapeutique est faible.
Substances destinées à une inhi-bition de courte durée (C). L'iode àdose élevée ( > 6 000 p.g/j) agit de façontransitoire comme un thyréostatiquedans les états hyperthyroïdiens, mais engénéral pas dans les états euthyroïdiens.Comme ce traitement inhibe égalementla libération de l'hormone, son effet sefera sentir plus rapidement que celui desthiamides.
Indications '. mise au repos préopé-ratoire avant une ablation de la thyroïdeavec une solution de lugol (5 % d'iode et10 % d'iodure de potassium, 50-100 mgd'iode/j pour un maximum de 10 jours).Dans les crises thyréotoxiques, on utili-sera l'iode avec les thiamides et unB-bloquant. Effets secondaires : allergie.Contre-indication : thyréotoxicose in-duite par l'iode.
Les ions lithium inhibent la libéra-tion de l'hormone. Les sels de lithiumpeuvent être utilisés à la place de l'iodeen cas de thyréotoxicose induite parl'iode pour obtenir une suppression ra-pide de la fonction thyroïdienne. L'uti-lisation des sels de lithium dans les psy-choses maniaco-dépressives endogènesest décrite page 232.
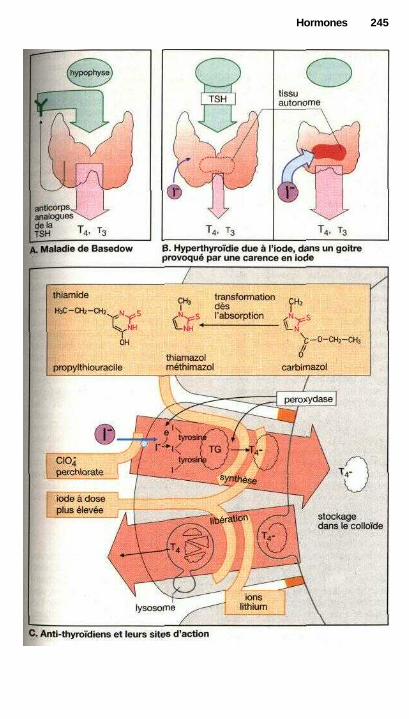
Hormones 245

246 Hormones
Utilisations thérapeutiquesdes glucocorticoïdesI. Traitements de substitution. Lecortex surrenalien produit un glucocorti-coide, le cortisol (hydrocortisone) et unminéralocorticoide, l'aldostérone. Cesdeux hormones stéroïdes sont d'un intérêtvital pour l'adaptation à des situations dif-ficiles comme par exemple une maladieou une opération. Le stimulus pour la sé-crétion de cortisol est l'ACTH hypophy-saire et pour l'aldostérone principalementl'angiotensine II (p. 124). En cas de dys-fonctionnement du cortex surrenalien {in-suffisance primaire de la glande surré-nale, maladie d'Addison) ce sont lecortisol et l'aldostérone qu'il faut rem-placer, dans le cas d'une production insuf-fisante d'ACTH par l'hypophyse (insuffi-sance surrénalienne secondaire) seul lecortisol doit être remplacé. Le cortisol estactif par voie orale (30 mg/j 2/3 le matinet 1/3 l'après-midi). Dans des situationsdifficiles, la dose sera augmentée d'en-viron 5 à 10 fois. L'aldostérone, peu ac-tive par voie orale, sera remplacée par lefludrocortisone(0,l mg/j).II. Traitement pharmacodynamiquepar les glucocorticoïdes (A). A concen-trations élevées, supraphysiologiques, lecortisol et les autres glucocorticoïdes sup-priment toutes les phases (exsudation,prolifération, cicatrisation) de la reactioninflammatoire, c'est-à-dire du mode dedéfense de l'organisme contre les corpsétrangers ou irritants. Cette action reposesur une multitude de composantes dont lacaractéristique commune est une modula-tion de la transcription de gènes (p. 64).C'est ainsi que sera stimulée la synthèsed'une protéine, la lipocortine (annexine),qui inhibe la phospholipase A;, réduisantainsi la libération d'acide arachidonique àpartir des phospholipides membranaireset donc la formation des médiateurs lipi-diques de l'inflammation, prostaglan-dines et leucotriènes (p. 194). Les gluco-corticoïdes diminuent également lasynthèse d'une série de protéines impor-tantes pour les phénomènes inflamma-toires, par ex. les interieukines (p. 296) etd'autres cytokines, la phospholipase A;(p. 194), la cyclooxygénase 2 (p. 198).
A très fortes concentrations peuvent aan, •apparaître des effets non gmomiques. l
Effet souhaité. Les glucocorncoïdes sont remarquablement ^.rcomme agents anti-allergiques iminitngsuppresseurs et antiphloghtiques dan-, le»réactions inflammatoires exagérées oiichroniques telles l'allergie la polyarthritprhumatoide ou d'autres.
Effets indésirables. Lors d'une ad-ministration brève, les glucocorticoïdesmême à doses élevées, ne présentent pra."tiquement aucun e f f e t secondaire.
Au cours d'une administration àlong terme, ils entraînent des altérationsqui sont proches de celles observées dansle syndrome de Cushing (surproductionendogène de cortisol). Conséquence despropriétés anti-inflammatoires : tendanceà l'infection, altération des processus decicatrisation. Conséquence de l'activitéglucocorticoïde exagérée : a) augmenta-tion de la néoglucogenèse et de la libéra-tion de glucose, sous l'action de l'insulinetransformation du glucose en triglycérides(dépôt adipeux : visage lunaire, épaississe-ment du tronc, « cou de buffle »), en casd'augmentation insuffisante de la sécré-tion d'insuline « diabète stéroïdien » ;b) dégradation accrue des protéines (cata-bolisme protéique) avec atrophie desmuscles squelettiques (finesse des extré-mités), ostéoporose, troubles de crois-sance chez l'enfant, atrophie cutanée.Conséquence de l'activité minéralocorti-coïde du cortisol, faible en temps normalmais maintenant augmentée : rétentiond'eau et de NaCl, augmentation de la pres-sion artérielle, formation d'œdème ; pertede KC1 avec risque d'hypokaliémie.Moyens d'atténuer ou d'éviterle syndrome de Cushingd'origine médicamenteuse
a) Remplacement du cortisol par desdérivés ayant une activité minéralocorti-coïde faible (prednisolone) ou nulle (triam-cinolone, dexaméthasone) (tableau desactivités relatives, figure A p. 247). Lespropriétés glucocorticoi'des, anti-inflam-matoires et le blocage de la production en-dogène (voir p. 248) vont cependant depair, et la partie glucocorticoïde des symp-tômes de Cushing ne peut être évitée.
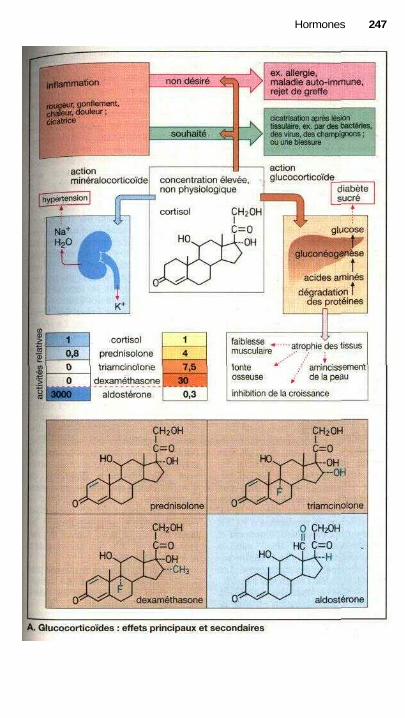
Hormones 247

248 Hormones
b) Administration locale. Atten-tion aux effets secondaires locaux, parexemple une atrophie de la peau ouune colonisation des muqueuses pardes candida. Pour maintenir, lorsd'une inhalation, les effets systé-miques aussi faibles que possible, ondoit utiliser des composés avec uneélimination présystémique impor-tante : dipropionate de béclométha-sone, flunisolide, budésonide, propio-nate de fluticason (p. 14).
c) Dose aussi faible que possible.Pour des traitements de longue durée,il faut essayer de donner la dose justesuffisante. Il faut cependant penser quela prise de glucocorticoides exogènesaboutit, via la boucle de contrôle, à unediminution de la production endogènede cortisol. Une dose faible peut decette façon être « amortie », de sorteque l'effet anti-inflammatoire ne se ma-nifeste pas.
Effet d'un traitement par lesglucocorticoïdes sur la productionde cortisol par le cortex surrénalien.La libération de cortisol est sous la dé-pendance de l'ACTH hypophysaire,tandis que la sécrétion d'ACTH estelle-même régulée par un facteur hy-pothalamique (CRF = CRH). Dansl'hypophyse et l'hypothalamus, ilexiste des récepteurs du cortisol dontl'occupation par le cortisol inhibe lasécrétion d'ACTH ou de CRF. Lescentres supérieurs contrôlent si lesconcentrations réelles de cortisol (ni-veau effectif) correspondent auxconcentrations souhaitées (valeur affi-chée). Si le niveau réel dépasse la va-leur affichée, la production d'ACTHet donc celle de cortisol diminuent etinversement. C'est ainsi que laconcentration de cortisol oscille au-tour du « point de consigne ». Lescentres supérieurs réagissent à un glu-cocorticoïde synthétique comme aucortisol. L'administration exogène de
cortisol ou d'un autre glucocorticoïdeimpose, pour que le niveau réel resteidentique au seuil affiché, une diminu-tion de la production endogène de cor-tisol. La libération de CRF et d'ACTHs'effondre (« inhibition des centressupérieurs par les glucocorticoïdesexogènes ») entraînant une diminutionde la sécrétion de cortisol. L'admi-nistration de doses élevées de cortisolpendant des semaines entraîne uneatrophie du cortex surrénalien. Lacapacité de synthèse d'aldostéronepersiste cependant. Lors d'un arrêtsoudain du traitement par les gluco-corticoïdes, le cortex surrénalien atro-phié ne peut fournir une quantité suffi-sante de cortisol -* carence en cortisoldangereuse. C'est pour cette raisonqu'un traitement par les glucocorti-coïdes doit toujours se terminer parune diminution lente des doses.
Méthodes pour éviter une a'tro-phie du cortex surrénalien. La sécré-tion de cortisol est élevée le matin etfaible le soir (rythme circadien). Lesoir, la sensibilité des centres supé-rieurs au cortisol est élevée.
a) Administration circadiennela dose journalière de glucocorticoïdesera donnée le matin. Le cortex surré-nalien a déjà commencé sa propre pro-duction, la possibilité d'inhibition descentres supérieurs est relativementfaible ; dès les premières heures dumatin suivant se produiront de nou-veau une libération de CRF etd'ACTH et une stimulation du cortexsurrénalien.
b) Traitement alterné : une doubledose quotidienne sera administrée lematin un jour sur deux. Durant le joursans traitement, il y aura une synthèseendogène de cortisol.
Les deux conduites ne mettent pasà l'abri d'une réapparition des symp-tômes de la maladie durant les inter-valles entre les traitements.
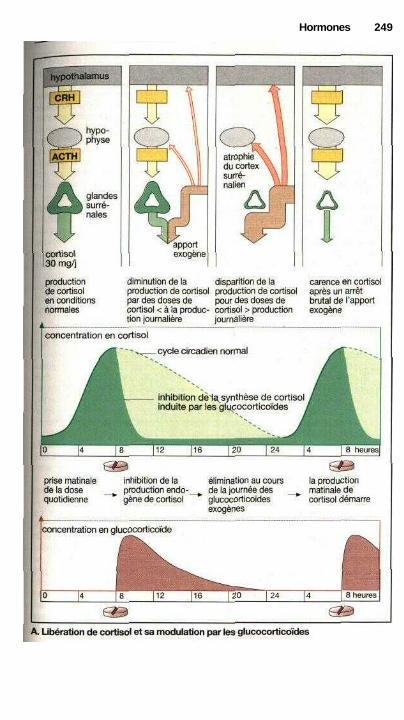
Hormones 249
24 4

250 Hormones
Androgènes, anabolisants,antiandrogènes
Les androgènes sont les molécules « fai-sant l'homme ». L'hormone sexuellepropre à l'homme est le stéroïde testosté-rone (T) provenant des cellules deLeydig, cellules interstitielles des testi-cules. La sécrétion de T est stimulée par laLH hypophysaire (hormone lutéinisante)dont la libération est elle-même activéepar la sécrétion pulsatile de la GnRH hy-pothalamique (gonadoréline, p. 240). LaT inhibe en un rétrocontrôle négatif leshormones hypothalamo-hypophysaires.Dans quelques tissus, par ex. la prostate,la T sera réduite en dihydrotestostéronequi se lie avec une affinité plus élevée auxrécepteurs des androgènes. La dégrada-tion se produit rapidement dans le foie(t 1/2 plasmatique environ 15 min) pourdonner entre autres l'androstérone, éli-minée par le rein sous forme de produitsconjugués (17 céto-stéroides). En raisondu métabolisme hépatique très rapide, T neconvient pas pour une prise orale ; elle seraitcertes absorbée mais aussi éliminée presquecomplètement de façon présystémique.
Dérivés de la testostérone à usagethérapeutique. Les esters pour dépôt i.m.sont le proprionate et Vheptanoate (énan-tate) de testostérone. Ces esters sont in-jectés par voie intramusculaire en solutiondans l'huile. Après diffusion, les estéraseslibèrent rapidement la forme acide — T.En même temps que le caractère lipo-phile, la tendance de l'ester à persister auniveau du dépôt s'accroît ; la durée d'ac-tion augmente. L' undécanoate de T peutêtre utilisé par voie orale. En raison de lanature d'acide gras de l'acide undéca-noïque, l'ester se retrouve dans la lympheaprès l'absorption et de là dans la circula-tion en passant par le canal thoracique eten évitant le foie. La 17-a-méthyltestosté-rone est active par voie orale grâce à unestabilité métabolique accrue. En raison dela toxicité hépatique des androgènes al-kylés en 17 (cholestase, tumeur) leur ad-ministration doit cependant être évitée.
La mestérolone, active par voie ora-le, est la 1-a-méthyldihydrotestostérone.
Indication : substitution en cas d'in-suffisance de la production endogène deT : esters de T en injection dépôt.
Pour stimuler la spermatogenèselors d'une carence en gonadotrophine(LH, FSH), on utilise des injections avecHMG et HCG. HMG, gonadotrophine defemme ménopausée, provient de l'urinede femmes après l'entrée en ménopausequi est riche en FSH. L'HCG, la gonado-trophine chorionique humaine, est ex-traite de l'urine de femmes enceintes etagit comme la LH.
Anabolisants. Ce sont des dérivésde la testostérone (ex. clostebol, météno-lone, nandrolone, stanozolol) qui sont uti-lisés chez des malades gravement atteintsen raison de leurs effets bénéfiques sur lasynthèse protéique (possibilité d'usageabusif chez les sportifs). Ils agissent via lastimulation des récepteurs des androgèneset possèdent donc également des effetsandrogéniques (ex. virilisation de l'aspectchez la femme).
L'antiandrogène cyprotérone estun antagoniste compétitif de la T. Il agiten plus comme un progestatif, dans la me-sure où il diminue la sécrétion de gona-dotrophines (p. 254), Indication : chezl'homme, calme la libido en cas de sexua-lité exacerbée, carcinome de la prostate.Chez la femme : traitement de manifesta-tions virilisantes, avec le cas échéant utili-sation de l'effet contraceptif important.
Le flutamide est un antagoniste durécepteur des androgènes à la structuredifférente et qui ne présente aucune acti-vité contraceptive.
Le finastéride inhibe la 5 a réduc-tase qui catalyse la formation de la dihy-drotestostérone (DHT) à partir de la T. Lastimulation androgénique sera donc ré-duite dans les tissus où la DHT est laforme active (ex. la prostate). Les tissuset les fonctions contrôlées par la T neseront pas ou à peine affectés, par ex. lesmuscles squelettiques, le rétrocontrôle né-gatif de la sécrétion de gonadotrophines etla libido. Le finastéride pourra être utilisédans un cas d'hyperplasie bénigne, pourréduire la taille de la glande et faciliter lamiction.
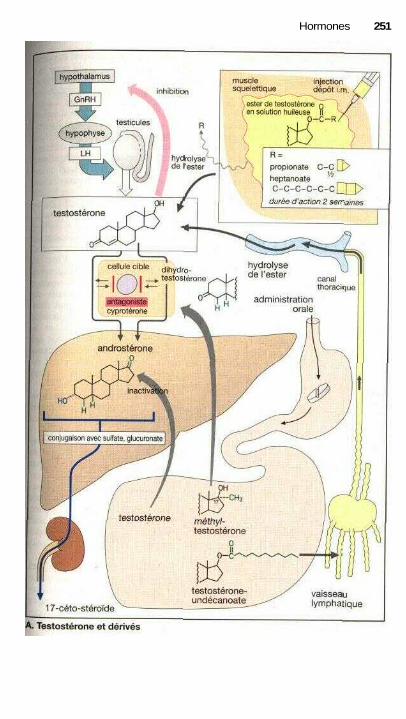
Hormones 251

252 Hormones
Maturation des ovules et ovulation,formation des œstrogèneset des progestogènes
La maturation et la ponte des ovulesainsi que la formation associée des hor-mones sexuelles féminines se produisentsous l'effet des gonadotrophines hypo-physaires FSH (hormone follicule-sti-mulante) et LH (hormone lutémisante).Dans la première moitié du cycle, laFSH induit la maturation de l'ovocyte enfollicule tertiaire qui commence à syn-thétiser de l'estradiol. L'estradiol favo-rise la prolifération de la muqueuse del'endomètre et augmente la perméabilitédu mucus cervical aux spermatozoïdes.La libération de FSH sera inhibée parun mécanisme de rétrocontrôle négatiflorsque le niveau d'estradiol dans lesang se rapproche d'un seuil établi dansles centres supérieurs. Compte tenu duparallélisme entre la maturation del'ovocyte et la libération d'estradiol,l'hypothalamus et l'hypophyse peuvent« suivre » le développement du phéno-mène de maturation par la déterminationdu niveau d'estradiol. Après l'ovulation,le follicule tertiaire donne naissance aucorps jaune (corpus luteum) qui libèrede la progestérone sous l'action de laLH. La progestérone provoque la phasede sécrétion de l'endomètre et diminuela possibilité de pénétrer à travers lemucus cervical. Les follicules restantdans l'ovaire continuent à produire desœstrogènes sous l'action de la FSH.Après deux semaines, la synthèse deprogestérone et d'œstrogènes s'effondrece qui a pour conséquence l'évacuationde la muqueuse sécrétoire de l'endo-mètre (menstruation).
Les hormones naturelles ne con-viennent pas pour une administrationorale car le foie provoque une élimina-tion présystémique après leur absorp-tion. L'estradiol sera transformé en es-trone et estriol, tous les trois peuventêtre éliminés par le rein après une conju-gaison les rendant plus polaires. Dans lecas de la progestérone, le métaboliteprincipal est le pregnandiol, qui est luiaussi éliminé par les reins après conju-gaison.
Forme médicamenteuse des œs-trogènes. Préparations dépôt pour in.jection i.m. Ce sont des esters de l'estra-diol sur les groupements hydroxyle en3 ou 17, en solution dans l'huile La vi-tesse de libération ou la durée d'actionvarient en fonction du caractère hydro-phobe des chaînes d'acides p. 250. Lesesters libérés seront hydrolyses, donnantnaissance à l'estradiol. Préparationsorales : 1'' éthinylestradiol (EE) est méta-boliquement stable, après administrationorale il traverse le foie et agit sur les ré-cepteurs des œstrogènes comme l'estra-diol. Le mestranol lui-même est inactifaprès hydrolyse du groupement méthylporté par l'oxygène en C, on obtient denouveau l'EE comme forme activeDans les contraceptifs oraux, l'une deces molécules constitue la composanteœstrogénique (p. 254). Les œstrogènesconjugués (sulfates) sont extraits del'urine de jument et sont présents dansles formes utilisées pour le traitementdes troubles de la ménopause et laprévention de l'ostéoporose. Dans lespréparations pour l'application trans-dermique, on utilise un emplâtre, l'es-tradiol passe dans l'organisme à traversla peau.
Formes médicamenteuses des pro-gestogènes. Les préparations-dépôtpour l'application i.m. sont le caproatede 17-a-hydroxyprogestérone et \'acé-tate de médroxyprogestérone. Les pré-parations orales sont des dérivés del'éthinyltestostérone ou éthistérone (parex. noréthistérone, lynestrénol, désoges-trel, gestodène) ou de l'acétate de 17-a-hydroxyprogestérone (par ex. l'acétatede chlonnadmone ou de cyprotérone)Les substances citées ci-dessus sontprincipalement utilisées comme compo-sant progestatif dans les contraceptifsoraux.
Indications. Pour les œstrogèneset les progestogènes ce sont : la contra-ception hormonale (p. 254), les traite-ments de substitution en cas de carencehormonale (prévention de l'ostéopo-rose), le saignement et les troubles ducycle. Pour les effets secondaires, voirp.254.
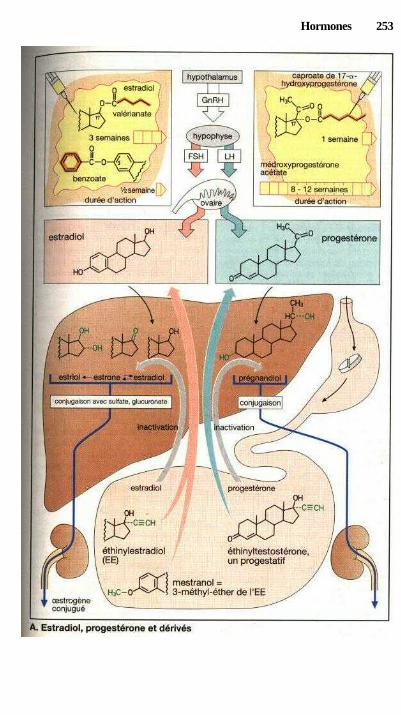
Hormones 253

254 Hormones
Contraceptifs oraux - Pilule
Inhibiteurs d'ovulation. Il est possibled'inhiber la maturation des ovocytes etl'ovulation en mettant à profit le rétro-contrôle négatif de la libération des gona-dotrophines. L'apport d'œstrogènesexogènes (éthinylestradiol ou mestranol)durant la première moitié du cycle en-traîne une diminution de la productionde FSH (obtenue également en présencede progestogènes). En raison de la dimi-nution de l'effet stimulant de FSH sur lefollicule tertiaire, on aboutit à la perturba-tion de la maturation d'ovocytes et donc àun blocage de l'ovulation. Par l'adminis-tration d'œstrogènes, on fait en quelquesorte croire aux centres supérieurs que lamaturation du follicule tertiaire se pour-suit normalement et qu'une stimulationsupplémentaire de la FSH n'est pas néces-saire. En cas d'apport d'œstrogènes seulsdans la première moitié du cycle, leschangements de la muqueuse utérine et dumucus cervical ainsi que les autres effetsdans l'organisme vont se dérouler norma-lement. Avec l'apport supplémentaired'un progestatif (p. 252) dans la deuxièmemoitié du cycle, la phase sécrétoire del'endomètre ainsi que les autres effetspourront être déclenchés normalement.Après l'arrêt de la prise hormonale, lesmenstruations apparaîtront.
Le cours physiologique de la libéra-tion d'œstrogène et de progestérone serareproduit par les préparations dites bi-phasiques (séquentielles) voir (A). Dansles préparations simultanées (phaseunique), les œstrogènes et la progesté-rone sont combinés pendant toute la duréede la période d'administration. L'admi-nistration précoce de progestéronecontribue à une inhibition des centres su-périeurs, empêche au niveau de l'endo-mètre une prolifération normale et la pré-paration à la nidation et diminue laperméabilité du mucus cervical aux sper-matozoïdes. Ces deux derniers effetscontribuent également ' à l'effet anti-conceptionnel. Selon l'échelonnement dela dose de progestérone, on peut différen-cier (A) : les préparations à un, deux ou
trois degrés. Dans le cas des préparationssimultanées (pilule placebo), l'arrêt del'apport hormonal déclenche égalementun saignement « de privation ».
E f f e t s indésirables : le risque accrude thrombose et d'embolie est lié enparticulier au composant œstrogéniqueHypertension, rétention de fluide, choles-tase, tumeurs bénignes du foie, nauséedouleurs de la poitrine peuvent se pro-duire. Le risque de tumeur maligne n'estglobalement pas augmenté de façon signi-ficative.
Minipilule. L'administration inin-terrompue d'un progestogène faiblementdosé peut aussi empêcher la grossesse. Engénéral, l'ovulation n'est pas bloquée,l'action repose sur les changements pro-voqués par le progestatif sur le canal cer-vical et l'endomètre. Ces formules serontrarement utilisées en raison de la néces-sité d'une prise régulière au même mo-ment de la journée, d'une efficacitécontraceptive plus faible et de saigne-ments plus fréquemment irréguliers.
« Pilule du lendemain ». Elle cor-respond à l'administration à forte dosed'œstrogène et de progestérone jusqu'à48 h après le coït. L'action de l'hormonedéclenche un saignement menstruel quirend peu probable la nidation de l'œuffécondé dans l'utérus (en temps normal,7 jours après fécondation, p. 74).
La mifépristone, un antagonistedes récepteurs de la progestérone, em-pêche l'entretien de la muqueuse utérineau début de la grossesse. Cette substancepeut être utilisée pour une interruption degrossesse en association avec des prosta-glandines.
Inducteurs d'ovulation. Une aug-mentation de la sécrétion des gonado-trophines peut être induite par une admi-nistration pulsatile de GnRH (p. 240). Unantagoniste des œstrogènes, le clomifène,bloque dans les centres supérieurs les ré-cepteurs qui sont impliqués dans le phé-nomène de rétrocontrôle négatif et« désinhibent » la libération de gonado-trophines. Un apport de gonadotro-phines s'effectue par l'administration deHMG et HCG (p. 250).
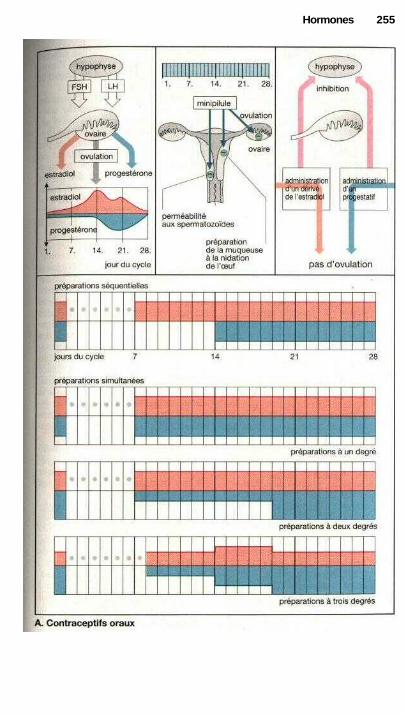
Hormones 255

256 Hormones
Traitement par l'insuline
L'insuline est produite par les cellules (ides îlots de Langerhans dans le pan-créas. C'est une protéine (poids molécu-laire 5 800) qui est formée de deuxchaînes peptidiques reliées entre ellespar deux ponts disulfure : la chaîne Aavec 21 acides aminés et la chaîne Bavec 30 acides aminés. L'insuline estl'hormone diminuant le sucre dans lesang. Lors d'un apport d'hydrates decarbone par la nourriture, elle sera sé-crétée et empêchera une élévation plusimportante de la concentration de glu-cose dans le sang en stimulant la captureet l'utilisation du glucose par le foie, lescellules musculaires et le tissu adipeux.
Sur le plan thérapeutique, l'insu-line sera utilisée en traitement de sub-stitution en cas de sécrétion insuffisantepar le pancréas et également dans le dia-bète sucré (diabètes mellitus).
Origine de l'insuline utilisée surle plan thérapeutique (A). L'insulinepeut être extraite du pancréas des ani-maux de boucherie. L'insuline de porcse distingue de l'hormone humaine parun seul acide aminé sur la chaîne B, \'in-suline de bœuf par deux acides aminéssur la chaîne A et 1 sur la chaîne B.Grâce à ces faibles différences, lesinsulines animales ont la même activitébiologique que l'hormone humaine.L'antigénicité est dans le cas de l'insu-line de porc à peine plus élevée et pourl'insuline de bœuf un peu plus forte quecelle de l'insuline humaine.
L'insuline humaine peut être ob-tenue de deux façons : par hémisynthèse,en remplaçant dans l'insuline de porcl'alanine (en position 30 de la chaîne B)par une thréonine ; par génie génétique :les bactéries Eschenchia coli peuventêtre amenées à produire de l'insuline hu-maine après introduction dans leur gé-nome de l'ADN correspondant.
Formes d'administration (B).Étant un peptide, l'insuline ne convientpas pour une forme orale (dégradationpar les protéases dans l'estomac et l'in-testin) et doit être administrée de façonparentérale. Le plus souvent, les prépa-rations d'insuline sont injectées par voie
sous-cutanée. La durée d'action dépendalors de la vitesse avec laquelle l'insu-line peut diffuser du site d'injection à lacirculation sanguine.
Solution d'insuline. L'insulineen solution est baptisée insuline nor-male ou insuline rapide. En cas d'ur-gence, par exemple un coma hypergly-cémique, elle peut être administrée parvoie intraveineuse (essentiellement enperfusion, car l'action d'une injectionintraveineuse est de courte durée). Encas d'administration par voie sous-cutanée, la plus courante, l'action sefait sentir en 15-20 minutes, atteint unmaximum après environ 3 h et dure àpeu près 6 h.
Suspensions d'insuline. On in-jecte une suspension de particules conte-nant de l'insuline, qui ne se dissolventque lentement dans le tissu sous-cutanéet libèrent l'insuline (insuline-retard).Les particules peuvent être constituéesde complexes apolaires et peu solublesdans l'eau entre l'insuline chargée néga-tivement et un partenaire comportant descharges positives par exemple la prota-mine, une protéine polycationique oubien l'aminoquinuride. En présenced'ions zinc, l'insuline forme des cris-taux ; la taille des cristaux conditionne lavitesse de dissolution. Les insulines in-termédiaires agissent pendant une duréemoyenne, les insulines ultra-lentes jus-qu'à 24 h et plus.
Insulines combinées. Elles contien-nent de l'insuline ordinaire et des sus-pensions d'insuline, le pic plasmatiqueest la somme des courbes propres auxdeux composants.
Effets indésirables. Une hypogly-cémie peut être la conséquence d'un sur-dosage absolu ou relatif (p. 258). Lesréactions allergiques sont rares : loca-lisées (au site d'injection rougeur ouégalement atrophie du tissu adipeux :lipodystrophie) ou généralisées (ana-phylaxie, exanthème). Une résistance àl'insuline peut provenir de la liaison àl'insuline d'anticorps responsables deson inactivation. Il peut se produire ausite d'injection une hypertrophie dutissu cutané qui peut être évitée en chan-geant la place de l'injection.
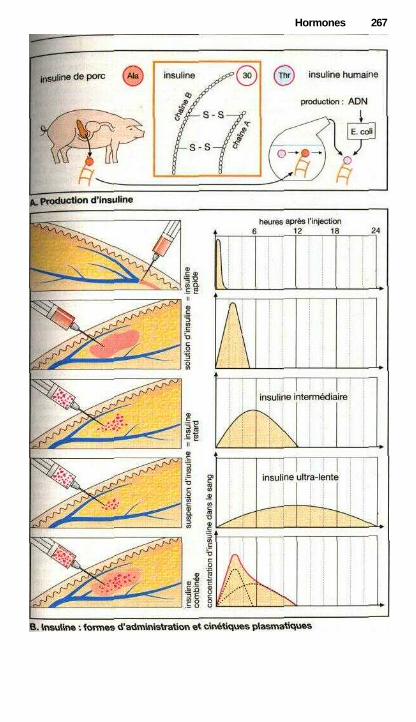
Hormones 267

258 Hormones
Traitement du diabète sucréavec carence en insuline
La « maladie sucrée » survenant chezl'enfant ou le jeune adulte (diabète juvé-nile ou de type I) est la conséquence d'undéclin des cellules (3 du pancréas produi-sant l'insuline. L'insuline doit être substi-tuée (dose journalière, environ 40 unitéscorrespondant à 1,6 mg).
Buts du traitement : 1. Empêcherle coma hyperglycémique (diabétique)qui constitue un danger potentiellementmortel. 2. Empêcher les altérations pa-thologiques dues au diabète (lésions vas-culaires avec oblitération, infarctus dumyocarde, insuffisance rénale) ; ilconvient pour cela d'éviter également lesaugmentations pathologiques de courtedurée de la concentration de glucose dansle sang (pics de sucre) par une bonne« équilibration » du malade. 3. Eviter unsurdosage d'insuline avec le danger graved'une chute de sucre (choc hypoglycé-mique : altérations du SNC liées à unecarence en glucose).
Principes thérapeutiques. Chez lesbien portants, la quantité d'insuline li-bérée est ajustée « automatiquement » àl'apport en hydrates de carbone ou à laconcentration de glucose dans le sang. Lestimulus essentiel de la sécrétion est uneaugmentation de la concentration de glu-cose dans le sang. La prise de nourritureet l'activité physique (déversement accrude glucose dans les muscles, diminutiondu besoin en insuline) vont de pair avecdes changements correspondants de la sé-crétion d'insuline (A), colonne de gauche.
Chez le diabétique, l'insuline pour-rait en principe être administrée commeelle est libérée chez l'individu en bonnesanté : au moment des principaux repas,de l'insuline ordinaire en injection sous-cutanée, le soir, administration d'insulineretard pour éviter un manque durant lanuit, avec ajustement des doses aux chan-gements des besoins. Une telle pratiqueexige des malades bien formés, prêts àcollaborer et capables de s'y conformer.Assez fréquemment, il sera nécessaired'appliquer un protocole fixe. Parexemple, une injection d'insuline com-binée matin et soir en dose pratiquement
constante (A). Pour éviter une hypo- ouune hyperglycémie, l'apport en hydratesde carbone de la nourriture doit corres-pondre à la période de libération de l'in.suline à partir du dépôt sous-cutané : ré-gime ! La nourriture (environ 50 % descalories sous forme d'hydrates de car-bone, 30 % en graisse, 20 % en protéine)doit être répartie en petits repas pouratteindre une répartition constante desapports : en-cas, souper pour la nuit. Lessucres rapidement absorbés (sucreriesgâteaux) doivent être évités (pics san-guins) et remplacés par des hydrates decarbone à digestion lente.
L'acarbose (un inhibiteur de l'a-glucosidase) inhibe dans l'intestin la libé-ration de glucose à partir de disaccha-rides.
Chaque modification des habitudesalimentaires ou du mode de vie peut per-turber l'organisation du traitement : sauterun repas conduit à l'hypoglycémie, unapport accru d'hydrocarbones à l'hyper-glycémie, un effort physique inhabituel àl'hypoglycémie.
Une hypoglycémie est caractériséepar des symptômes annonciateurs : tachy-cardie, agitation, tremblements, sueur, pâ-leur. Certains de ces symptômes sont liésà la libération d'adrénaline, une hormonemobilisant le glucose. Moyen de lutte :apport de glucose, hydrates de carbone àabsorption rapide, ou en cas de perte deconscience 10-20 g de glucose i.v. ; danscertains cas, injection de glucagon, l'hor-mone pancréatique augmentant le glucosesanguin.
Malgré un bon équilibre du traite-ment, l'administration sous-cutanéed'insuline ne peut pas imiter complè-tement la situation physiologique. Chezle bien portant, le glucose absorbé etl'insuline libérée par le pancréas attei-gnent le foie ensemble et en concen-tration élevée, ceci permet une réelle
' élimination présystémique du glucose etde l'insuline. Chez le diabétique, l'insu-line injectée en s.c. se distribue égale-ment dans tout l'organisme. Le foien'est pas perfusé par une concentrationélevée d'insuline et il y aura moins deglucose retire du sang de la veine porte.Une quantité de glucose plus importanteparvient à l'organisme.
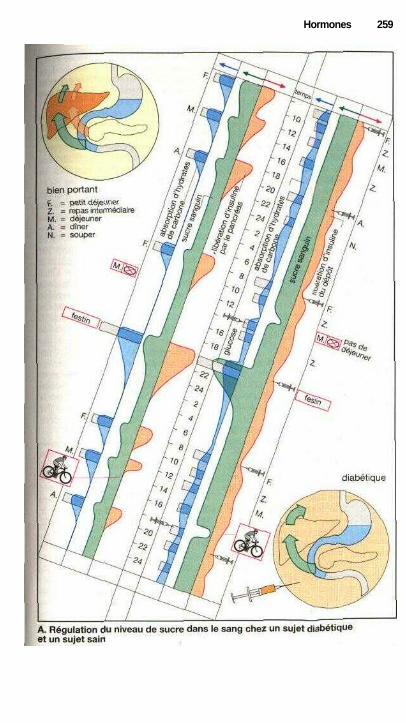
Hormones 259

260 Mormones
Traitement du diabète de l'adulte
Lorsque chez un adulte en excédent depoids, s'installe une situation métabo-lique de type diabétique (diabète de typeII, diabète de l'adulte), il existe la plu-part du temps une carence relative eninsuline : un besoin accru en insulineest associé à une diminution de la sécré-tion. L'origine du besoin accru en in-suline est une diminution des récep-teurs de l'insuline par ex. dans le tissuadipeux. La conséquence en est une di-minution de la sensibilité à l'insulinedes cellules (A). Chez un obèse, laliaison maximale de l'insuline (plateaude la courbe) est diminuée de façon cor-respondante à la réduction du nombrede récepteurs. Pour de faibles concen-trations d'insuline, il y aura égalementune liaison plus faible que chez les indi-vidus de poids normal. Pour obtenir uneffet métabolique donné (par exemplela transformation des hydrates de car-bone contenus dans une part de tarte), ilfaut qu'un nombre donné de récepteurssoient occupés, et qu'une liaisondonnée d'insuline soit atteinte. A partirdes courbes de liaison, on peutconstater que ces conditions peuventaussi être réunies dans le cas d'une di-minution des récepteurs, mais pour uneconcentration d'insuline plus élevée.
Développement d'un diabète detype II (B). En comparaison avec un in-dividu de poids normal, l'obèse a be-soin en permanence d'une libérationd'insuline plus élevée (courbe orange)pour empêcher lors d'une surcharge englucose, une élévation trop importantedu glucose sanguin (courbe verte). Si lacapacité du pancréas à libérer del'insuline s'épuise, cela se remarquerad'abord par une élévation de la concen-tration de glucose (diabète latent) encas de surcharge. Ensuite, il ne sera pluspossible de maintenir la valeur de laconcentration plasmatique de sucre, àjeun (diabète avéré).
Traitement. Un régime pour at-teindre un poids normal s'accompagned'une augmentation de la densité de ré-cepteurs et de la sensibilité à l'insuline.Maintenant, la quantité d'insuline qui
peut être libérée est de nouveau suffi-sante pour une situation métaboliquenormale. Le traitement de premierchoix est la réduction de poids et nonla prise de médicaments !
Si le diabète ne disparaît pas, i]faut en premier lieu penser à une substi-tution par l'insuline (p. 258). Les anti-diabétiques oraux de type sulfony-lurée stimulent la libération d'insulineà partir des cellules p du pancréas. Ilsinhibent les canaux potassiques ATP-dépendants et favorisent ainsi une dé-polarisation de la membrane. En tempsnormal les canaux sont fermés, lorsquela concentration intracellulaire en glu-cose et donc en ATP augmente. A cegroupe de composés appartiennent parex. le tolbutamide (500-2000 mg/j) et laglibenclamide (1,75-10,5 mg/j). Chezquelques patients, l'augmentation de lasécrétion d'insuline n'est pas possibledès le début, chez d'autres s'installeplus tard une résistance au traitement. Ilest nécessaire d'adapter l'apport denourriture (régime) au traitement parles antidiabétiques oraux. L'effet se-condaire le plus important est une hy-poglycémie. Il peut y avoir renforce-ment de l'action due à une interactionmédicamenteuse : déplacement de laliaison aux protéines plasmatiques parex. par les sulfamides ou l'acide acétyl-salicylique.
La metformine, un dérivé bigua-nide, permet de normaliser un niveaude glucose trop élevé, en présence d'in-suline. La metformine ne stimule pas lalibération d'insuline, mais augmentel'utilisation du glucose périphérique etdiminue la libération du glucose par lefoie. Le danger d'hypoglycémie n'estsouvent pas accru. Les effets secon-daires assez fréquents sont une perted'appétit, des diarrhées et des nausées.-Un effet secondaire rare mais dange-reux est une surproduction d'acide lac-tique (lactacidose). La metformine peutêtre utilisée seule ou en associationavec les sulfonylurées. Elle est contre-indiquée en particulier chez les per-sonnes ayant une insuffisance rénale etne doit donc pas être utilisée chez lespatients âgés.
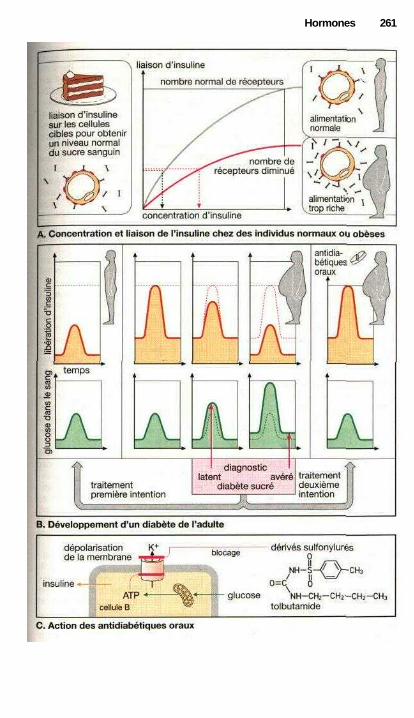
Hormones 261

262 Hormones
Substances utilisées pour maintenirl'homéostasie du calcium
Au repos, la concentration intracellu-laire d'ions calcium (Ca^) est maintenueà 0,1 u-M (mécanismes impliqués, p. 128).Lors d'une stimulation, une élévation jus-qu'à 10 U.M provoque la contraction descellules musculaires (couplage électromé-canique) ou, dans les cellules glandu-laires, la vidange des vésicules (couplageélectrosécrétoire). Le contenu cellulairede calcium est en équilibre avec laconcentration extracellulaire de calcium(environ 1 000 p-M) ; de même que lafraction de calcium liée aux protéinesdans le sang. Le calcium peut cristalliseren présence de phosphate, sous formed'hydroxyapatite, constituant minéral desos. Les ostéoclastes sont des cellules« gloutonnes » qui libèrent du calcium àpartir de la dégradation des os. Des chan-gements très faibles de la concentrationextracellulaire de calcium peuvent modi-fier les fonctions de l'organisme, c'estainsi que l'excitabilité des muscles sque-lettiques augmente de façon importanteavec une diminution du Ça2* (par exemplelors d'une tétanie résultant d'une hyper-ventilation). Trois hormones ont pourfonction dans l'organisme de maintenirconstante la concentration extracellulairedeCa2".
Hormone dérivée de la vita-mine D. Elle provient de la vitamine D(cholécalciférol). La vitamine D peut éga-lement être formée dans l'organisme : àpartir du 7-déhydrocholestérol, dans lapeau, sous l'action de la lumière ultravio-lette. En cas d'ensoleillement insuffisant,l'apport dans la nourriture est suffisant ;l'huile de foie de morue est riche en vita-mine D. L'hormone dérivée de la vita-mine D, métaboliquement active, seforme par deux hydroxylations : dans lefoie en position 25 ( -> calcifédiol), puisdans le rein en position 1 ( -» calcitriol,hormone-vitamine D). L'hydroxylationen 1 dépend de l'état de l'homéostasie ducalcium et sera stimulée par la parathor-mone ainsi que par la chute des concen-trations de phosphate et de Ca^ dansle sang. L'hormone-vitamine D [1,25(OH)^] stimule l'absorption de Ça2* et
de phosphate au niveau de l'intestin ainsique leur réabsorption par le rein. Par suitede l'élévation des concentrations de Ca^et de phosphate, la tendance à la cristalli-sation dans l'os sous forme d'hydroxy-apatite augmente. En cas de carence en vi-tamine D, la minéralisation osseuse estinsuffisante (rachitisme, ostéomalacie),L'utilisation thérapeutique est un traite-ment de substitution. En général, ondonne de la vitamine D, on peut aussi uti-liser le calcifédiol chez des sujets ayantdes troubles hépatiques ou le calcitriolchez les malades du rein. L'activité ainsique la rapidité d'apparition de l'action oude sa disparition augmentent dans l'ordrevit. D, 25-OH-vit. D, 1,25 (OH);-vit. D.En cas de surdosage, se produit une hy-percalcémie avec dépôt de sels de Ça dansles tissus (principalement les reins et lesvaisseaux) : calcinose.
Le polypeptide parathormone estsécrété par les glandes parathyroïdes lorsd'une baisse de la concentration sanguinede Ca^. Elle active les ostéoclastes et fa-vorise la dégradation osseuse ; Dans lerein, elle stimule la réabsorption du Çamais augmente en revanche l'excrétion dephosphate. La diminution de la concentra-tion de phosphate dans le sang diminue latendance du Ca^ à être incorporé dans latrame osseuse. En cas de carence en para-thormone, on utilisera en remplacement lavitamine D, qui au contraire de la para-thormone est active par voie orale.
Le polypeptide calcitonine est libérépar les cellules C de la thyroïde en casd'hypercalcémie. Il diminue le Ca^ en in-hibant l'activité des ostéoclastes. On l'uti-lise en outre en cas d'hypercalcémie etd'ostéoporose. De façon remarquable, onobserve qu'une injection de calcitoninepeut avoir un e f f e t analgésique persistanten cas de douleurs osseuses sévères.
Une hypercalcémie peut être" soi-gnée par : 1. une solution de NaCl à 0.9 %et le cas échéant du furosémide -* î e Mïé-tion rénale de Ça2*, 2. par la calcitonine quiinhibe les ostéoclastes, la plicamycine oule clodronate (un diphosphonate) -» <L mo-
bilisation osseuse du calcium, 3. pss un
complexant du calcium, EDTA sodiqii6 ou
citrate sodique, ainsi que, le cas échléant,4. par les glucocorticoïdes.
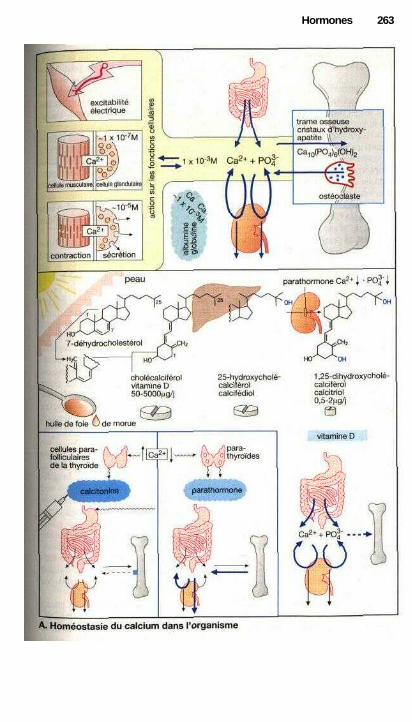
Hormones 263

264 Substances antibactériennes
Médicaments contreles infections bactériennes
Si les bactéries traversent les bar-rières cutanées ou muqueuses et pénè-trent dans l'organisme, on obtient alorsune infection bactérienne. L'organismeest souvent capable d'éliminer les bac-téries par l'intermédiaire d'une réactiondu système immunitaire, sans que dessymptômes de maladie se manifestent.Lorsque les bactéries se multiplientplus vite que les défenses de l'orga-nisme ne peuvent les détruire, se dé-clenche une maladie infectieuse accom-pagnée de signes inflammatoires, parex. infection purulente d'une écorchure,ou infection des voies urinaires. Pour letraitement de ces infections, il faut dessusbtances qui affectent les bactéries etempêchent donc leur multiplication ul-térieure mais qui cependant ne touchentpas les cellules de l'organisme (1).
En termes de nomenclature, lesantibiotiques sont produits par desmicro-organismes (bactéries, champi-gnons) et sont dirigés « contre la vie »des bactéries mais aussi des champi-gnons ou des cellules humaines. Lesagents chémothérapeutiques provien-nent d'une synthèse chimique. Cettedistinction n'est aujourd'hui plus uti-lisée dans le langage courant.
Une atteinte spécifique des bacté-ries sera en général possible lorsqu'unesubstance agit sur une voie métaboliquecaractéristique des bactéries, quin'existe pas dans les cellules humaines.Ceci est particulièrement net pour lesinhibiteurs de synthèse de la paroi, carles cellules humaines ne possèdent pasde paroi. Les points d'impact des sub-stances antibactériennes sont pré-sentés en (2), dans une cellule bacté-rienne très simplifiée.
Dans les pages suivantes, il nesera pas fait mention de la polymyxineet de la tyrothricine. Ces antibiotiquespolypeptidiques augmentent la perméa-bilité de la membrane cellulaire. Enraison de leur mauvaise tolérance, ilsseront utilisés chez l'homme unique-ment en application locale.
Le résultat de l'action des sub-stances antibactériennes peut êtreétudié m vitro (3). Les bactéries se mul-tiplient dans des conditions contrôléessur un milieu nutritif. Si ce milieu nu-tritif contient une substance anti-bacté-rienne, il faut distinguer deux effets :1. les bactéries sont tuées : effet bacté-ricide ; 2. les bactéries survivent maisne se multiplient plus, effet bactério-statique. Même si des variations peu-vent se produire dans les conditionsthérapeutiques, les différentes sub-stances peuvent être classées selon leurprincipe d'action (soulignés en couleurdans la figure 2).
Si la multiplication bactériennepersiste sous l'action d'une substanceantibactérienne, on a affaire à unphénomène de résistance des bacté-ries. Ces phénomènes peuvent repo-ser sur le fait qu'une souche de bacté-ries en raison de son métabolismepropre, est naturellement insensible àla substance (résistance naturelle).Selon qu'une substance est capabled'atteindre seulement un petit nom-bre ou bien de très nombreuses es-pèces bactériennes, on parlera doncd'un antibiotique à spectre étroit (parex. pénicilline G) ou bien à spectrelarge (ex. tétracycline). Des souchesbactériennes sensibles au début peu-vent devenir résistantes sous l'in-fluence de substances antibiotiques(résistance acquise). Une modifica-tion au hasard du patrimoine hérédi-taire (mutation) donne naissance à unebactérie résistante. Sous l'influence dela molécule, les autres bactéries meu-rent, tandis que le mutant qui n'estpas touché se multiplie. L'apparitiond'une souche bactérienne à la résis-tance acquise augmente avec la fré-quence d'utilisation d'un antibiotique(par ex. germes résistants dans lescliniques).
La résistance est également trans-missible dans la mesure où l'ADN,dans lequel la résistance est inscrite(encore appelée plasmide de résis-tance), peut être transféré à d'autresbactéries.
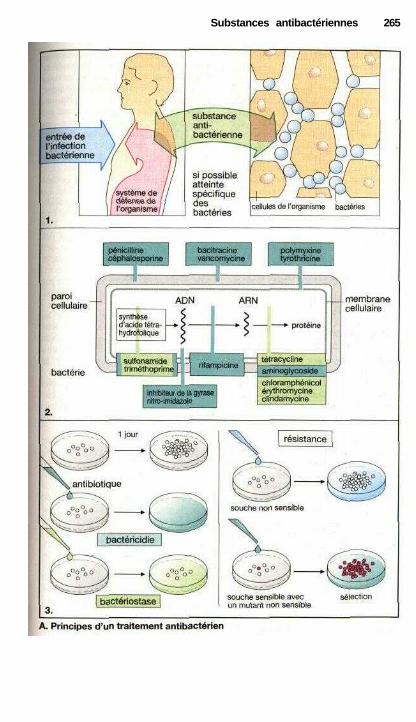
Substances antibactériennes 265

266 Substances antibactériennes
Inhibiteurs de synthèse de la paroibactérienne
Dans la plupart des cas, une paroi cellu-laire entoure les bactéries comme uneécorce rigide ; elle les protège des agres-sions extérieures et empêche unerupture de la membrane cellulaire sousl'influence d'une pression interne (osmo-tique) élevée. La solidité de la paroi cellu-laire repose avant tout sur la structure dela muréine (peptidoglycane). Elle secompose d'éléments de base rassemblésen une énorme macromolécule formantun réseau. Ces éléments contiennent lesdeux sucres aminés N-acétylgiucosamineet acide N-acétylmuramique enchaînésles uns aux autres. Ce dernier comporteune chaîne peptidique. Les « briques »sont synthétisées dans la bactérie, trans-portées vers l'extérieur à travers la mem-brane cellulaire et assemblées selon leschéma ci-contre. Ensuite, l'enzymetranspeptidase relie les chaînes pepti-diques de deux polymères voisins desucres aminés.
Inhibiteurs de synthèse de laparoi. Ils conviennent comme substanceantibactérienne car les cellules humainesne possèdent pas de paroi. Ils sont bacté-ricides pour les germes qui poussent et semultiplient. C'est de cette façon qu'agis-sent les antibiotiques (3-lactames, cépha-losporines et pénicillines ainsi que baci-tracine et vancomycine.
La pénicilline (A). La substanceoriginelle de ce groupe est la pénicil-line G (benzylpénicilline). Elle a été ob-tenue à partir de cultures de moisissures,initialement de Pénicillium notatum. Lapénicilline G contient l'élément de basecommun à toutes les pénicillines, l'acide6-aminopénicillanique (6AAP, p. 269),avec un cycle P-lactame à 4 côtés. Le6AAP lui-même n'a pas d'action antibac-térienne. Les pénicillines interrompent lasynthèse de la paroi en inhibant la trans-peptidase. Si les bactéries se trouventdans une phase de croissance et de multi-plication, les pénicillines provoquent lamort cellulaire (bactéricidie) ; en raisondu défaut de la paroi, les bactéries gon-flent puis éclatent.
Les pénicillines sont bien suppor-tées chez l'homme. La dose journalièrepeut aller pour la pénicilline G d'environ0,6 g i.m. ( = 106 unités internationales1 Mega UI) jusqu'à 60 g en perfusion!L'effet secondaire le plus fréquent estune réaction allergique (fréquence jus-qu'à 5 % des malades traités), dont lesmanifestations peuvent aller de manifes-tations cutanées jusqu'à des chocs ana-phylactiques (moins de 0,05 % des cas).Ces molécules sont contre-indiquées chezdes sujets allergiques à la pénicilline. Enparticulier à cause du danger de sensibili-sation, les pénicillines ne doivent pas êtreutilisées en applications locales. Dese f f e t s neurotoxiques, principalement descrampes, peuvent survenir lorsque desconcentrations très élevées agissent sur leSNC, par ex. en cas d'administration ra-pide de doses élevées en i.v. ou bien paradministration directe dans le liquide cé-phalo-rachidien.
La pénicilline G est éliminée auniveau des reins, essentiellement sousforme inchangée et de façon très rapide(demi-vie plasmatique environ 1/2 heure).
Il est possible de prolonger ladurée d'action :
1. Administration à dose plus éle-vée, pour une demi-vie plasmatique iden-tique la concentration persiste plus long-temps au-dessus de la valeur seuilnécessaire à l'action antibactérienne.
2. Association avec le probénécide.L'élimination rénale de la pénicilline Gs'effectue en grande partie par le sys-tème de sécrétion des anions (acides)dans le tubule proximal (fonction acidedu 6AAP !). Le probénécide, un acide(p. 310), entre en concurrence avec cettevoie d'élimination et ralentit ainsi l'élimi-nation de la pénicilline G.
3. Injection intramusculaire endépôt. La pénicilline G sous forme anio-nique (-COO-) forme avec des substancescontenant des groupements aminéschargés, des sels peu solubles dans l'eau(procaïne, p. 206, clémizole un anti-his-taminique, benzathine une substance di-cationique). Selon la substance, la libéra-tion de la pénicilline G à partir du dépôts'étend sur un intervalle de temps variable.
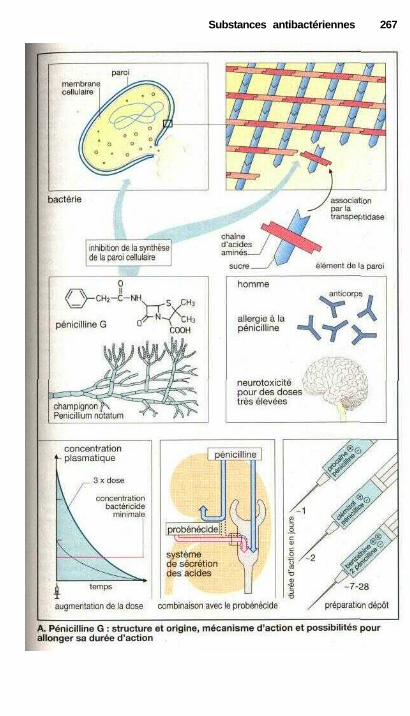
Substances antibactériennes 267

268 Substances antibactériennes
La pénicilline G est très bien sup-portée mais présente cependant des in-convénients (A) qui restreignent son uti-lité thérapeutique : 1. L'acidité gastriquehydrolyse le cycle p-lactame et inactive lapénicilline G qui doit donc être injectée.2. Le cycle p-lactame peut égalementêtre dégradé par une enzyme bactérienne(P-lactamase) qui peut être produite enparticulier par certaines souches de sta-phylocoque, ce qui les rend résistantes àla pénicilline G. 3. Le spectre antibacté-rien est étroit. Il englobe beaucoup debactéries Granit ainsi que des cocciGram- et l'agent de la syphilis mais n'af-fecte pas beaucoup de germes Gram-.
Les dérivés comportant un autresubstituant sur l'acide 6-aminopénicilla-nique présentent l'avantage (B) :1. D'être résistants en milieu acide cequi permet une absorption orale (dans lamesure où l'absorption intestinale estpossible). Tous les dérivés présentés en Bpeuvent être administrés par voie orale.La pénicilline V (phénoxyméthylpénicil-line) a les mêmes propriétés antibacté-riennes que la pénicilline G. 2. D'êtrerésistants à la pénicillinase. Les pénicil-lines isoxazolyl (oxacilline, dicloxacil-line, flucloxacilline) conviennent pour letraitement (oral) des infections par desstaphylocoques synthétisant des pénicil-linases. 3. D'avoir un spectre pluslarge. L'aminopénicilline, amoxicilline,affecte de nombreux germes Gram-, parexemple les colibacilles ou les salmo-nelles (typhus). Elle peut être protégée dela dégradation par la pénicillinase parson association avec V acide clavuliniqueun inhibiteur de cette enzyme.
L'ampicilline, de structure voisine(pas de groupe 4 OH), a le même spectred'action mais est faiblement absorbée( < 50 %) et affecte de ce fait particulière-ment la flore intestinale (effet secondairediarrhée), elle doit donc être uniquementinjectée.
Les carboxypénicillines (ticarcilline)et les acylaminopénicillines (meziocil-line, aziocilline, pipéracilline) possèdentun spectre encore plus large (par ex.contre les bactéries Pseudomonas). Cesmolécules ne sont pas résistantes en mi-lieu acide ou à la pénicillinase.
Les céphalosporines (C). Ces anti-biotiques fi-lactames proviennent égale-ment des champignons et exercent uneaction bactéricide en inhibant la trans-peptidase. La structure de base, forméepar l'acide 7-aminocéphalosporaniqueest soulignée en gris dans l'exemple de lacéfalexine. Les céphalosporines sontstables en milieu acide mais beaucoup desreprésentants de ce groupe sont mal ab-sorbés. En raison de la nécessité d'une ad-ministration parentérale, la plupart de cesmolécules, parmi lesquelles les plus ac-tives, seront presque exclusivement réser-vées à l'utilisation hospitalière. Peud'entre elles, comme par exemple la céfa-lexine, conviennent à l'administrationorale. Les céphalosporines sont résis-tantes à la pénicillinase ; mais il existe desgermes synthétisant des céphalospori-nases. Quelques dérivés sont cependantinsensibles aussi à cette p-lactamase. Lescéphalosporines ont un large spectreantibactérien. Les dérivés récents (ex.céfotaxime, cefménoxime, céfopérazone,ceftriaxone, ceftazidime, latamoxef) tou-chent également des germes résistants auxautres substances antibactériennes. Lescéphalosporines sont en général bien sup-portées par l'homme. Toutes peuvent pro-voquer une réaction allergique, certainespeuvent aussi toucher les reins, provoquerdes saignements (antagonisme de lavit. K) ou des intolérances à l'alcool.
Autres inhibiteurs de synthèse dela paroi bactérienne. Les antibiotiquesbacitracine et vancomycine perturbent letransport des éléments constitutifs de laparoi à travers la membrane cellulaire etsont actifs uniquement contre les bacté-ries Grain*. La bacitracine est un mé-lange de polypeptides très néphrotoxiqueet qui sera uniquement utilisé localement.La vancomycine est un glycopeptide.C'est l'agent de choix pour le traitement(oral) d'une inflammation intestinalepouvant intervenir comme complicationd'un traitement antibactérien (entéroco-lite pseudomembraneuse, provoquée parClostridium difficile). Elle n'est pas ab-sorbée.
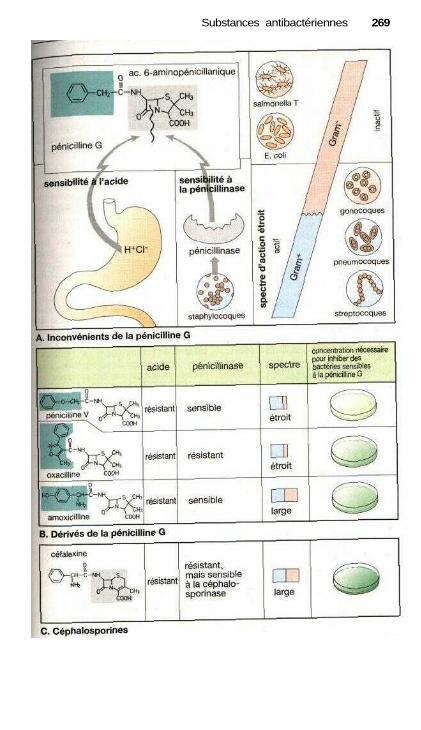
Substances antibactériennes 269

270 Substances antibactériennes
Inhibiteurs de synthèsede l'acide tétrahydrofoliqueL'acide tétrahydrofolique (THF) est unco-enzyme d'une étape de synthèse desbases puriques et pynmidiques. Celles-ci sont des éléments constitutifs de l'ADNet des ARN et sont nécessaires à la crois-sance et à la division cellulaire. En cas decarence en THF, la multiplication cellu-laire est inhibée. THF est synthétisé àpartir de l'acide dihydrofolique sous l'ac-tion de l'enzyme dihydrofolate réductase.DHF provient dans les cellules humaines,de l'acide folique, vitamine qui ne peutêtre synthétisée par l'organisme mais doitêtre captée à l'extérieur. Les bactériesn'ont pas besoin d'acide folique car ellessont capables par elles-mêmes de pro-duire de l'acide folique ou plus exacte-ment de l'acide dihydrofolique à partir deprécurseurs. Une perturbation de la voiede synthèse du THF chez les bactéries estobtenue en utilisant le sulfonamide et estpossible en présence de triméthoprime.
Sulfonamides. Leur structure res-semble à celle de l'acide para-aminoben-zoïque (PAB), un élément de base dans lasynthèse de DHF par les bactéries. Lessulfonamides, en tant que faux substrat,bloquent de façon compétitive la transfor-mation du PAB et inhibent la synthèse deDHF. Comme la plupart des bactéries nepeuvent pas capter l'acide folique du mi-lieu environnant, elles s'appauvrissent enDHF. Les sulfonamides agissent ainsicomme des bactériostatiques sur un largespectre d'agents pathogènes. Les sulfo-namides sont produits par synthèse chi-mique. La structure de base est présentéesur la formule ci-contre. Le résidu Rconditionne la pharmacocinétique du sul-fonamide considéré. La plupart des sulfo-namides sont bien absorbés par voie orale.Ils seront métabolisés en proportions va-riables et éliminés par les reins. La vitessed'élimination ainsi que la durée d'actionpeuvent varier de façon importante.Quelques représentants de cette famillesont mal absorbés au niveau intestinal etconviennent de ce fait au traitement spéci-fique des infections bactériennes de l'in-testin. Les effets secondaires sont, entreautres, des réactions allergiques en partieaccompagnées de réactions cutanées
sévères ; un déplacement de la liaisond'autres produits pharmaceutiques auxprotéines du plasma ou chez le nouveau-né à la bilirubine (risques d'ictère du nour-risson et donc contre-indication durant lesdernières semaines de grossesse ou chez lenouveau-né). En raison de l'apparition trèsfréquente de germes résistants, les sulfo-namides ne sont aujourd'hui que rarementutilisés (introduction 1935).
Triméthoprime : inhibe la DHF-réductase bactérienne, l'enzyme humaineest nettement moins sensible que l'en-zyme bactérienne (on observe rarementdes cas d'aplasie médullaire). Le trimé-thoprime, une 2,4-diaminopyrimidine estun agent chimiothérapeutique avec uneaction bactériostatique sur un largespectre de germes pathogènes. Il est prin-cipalement utilisé comme composant ducotrimoxazole.
Le cotrimoxazole est une associa-tion de triméthoprime et d'un sulfona-mide le sulfométhoxawle. En raison del'atteinte de deux étapes consécutivesdans la synthèse du THF, l'action antibac-térienne du cotrimoxazole est meilleureque celle de chacun des composants. Lesmicrobes résistants sont rares et il peut seproduire un effet bactéricide. Les effetssecondaires correspondent à ceux dechaque substance individuelle.
Sulfasalazine (salazosulfapyri-dine). Traitement de la colite ulcéreuse in-flammatoire et de la maladie de Crohn.Les bactéries intestinales décomposentcette substance en un sulfonamide (sulfa-pyridine) et en acide 5-aminosalicylique.Ce dernier est évidemment la moléculeanti-inflammatoire (inhibition de la syn-thèse de leucotriènes ?) mais doit per-sister à forte concentration au niveau de lamuqueuse intestinale. L'association à unsulfonamide empêche l'absorption pré-coce dans les segments supérieurs de l'in-testin grêle. Le sulfonamide sera absorbéaprès hydrolyse de la liaison et peut pro-voquer des effets secondaires classiques(voir ci-dessus). Les préparations demésalazine à libération lente permettentde ne pas utiliser le sulfonamide.
La sulfasalazine a été à l'origine dé-veloppée pour le traitement de la polyar-thrite rhumatoïde (p. 314).
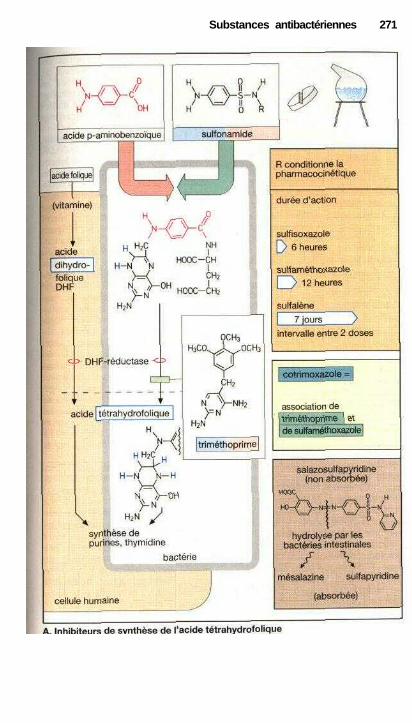
Substances antibactériennes 271

272 Substances antibactériennes
Inhibiteurs de la fonction de l'ADN
L'acide désoxyribonucléique (ADN) sertde matrice pour la synthèse des acides -ribonucléiques (ARN). Les ARN gouver-nent la synthèse de protéines et permet-tent ainsi la croissance cellulaire. Unenéo-synthèse d'ADN est la conditiond'une division cellulaire. Les substancesqui inhibent la lecture de l'informationgénomique au niveau de la matriced'ADN, perturbent le centre de contrôledu métabolisme cellulaire. Les substancesdécrites ci-dessous conviennent commesubstances antibactériennes car elles netouchent pas les cellules humaines.
Inhibiteurs de la gyrase. L'enzymegyrase (topoisomérase II) permet d'intro-duire de façon ordonnée un chromosomebactérien long d'environ 1 000 ̂ m dansune cellule bactérienne longue d'environ1 u,m. Dans le filament chromosomiquese trouvent les deux brins d'ADN en-roulés en une double hélice. Le filamentchromosomique de son côté est arrangéen spirales dont la longueur peut être di-minuée en augmentant le degré d'enroule-ment. La gyrase effectue cette torsioncomme il est montre dans le schéma enouvrant et fermant l'hélice, sans qu'il soitnécessaire de faire tourner l'ensemble dela boucle.
Les dérivés de la 4-quinolone avecune fonction acide en 3 (voir dans la for-mule la partie colorée en vert) sont des in-hibiteurs de la gyrase bactérienne. Ilssemblent empêcher la fermeture du brinouvert et ont de ce fait, une action bactéri-cide. Ces agents chimiothérapeutiquessont absorbés après prise orale. Le produitle plus ancien (acide nalidixique) agitseulement sur les bactéries Gram- et n'at-teint une concentration active que dansl'urine ; il sert au traitement des infectionsdes voies urinaires. La norfloxacine pré-sente un spectre plus large. L'ofloxacine,la ciprofloxacine, l'énoxacine atteignentpar ailleurs des concentrations activesdans l'organisme et seront utilisées égale-ment contre les infections des organesinternes.
Les effets secondaires sont endehors de troubles digestifs ou d'allergie,des altérations particulières du systèmenerveux (par ex. hallucinations, confusion
et crampes). A cause d'altérations descellules des cartilages au niveau desépiphyses et des articulations chez lesanimaux de laboratoire, les inhibiteursde la gyrase ne doivent pas être utilisésdurant la grossesse, l'allaitement et durantla croissance.
Les dérivés du nitro-imidazolepar ex. le métronidazole, altèrent l'ADNen formant des complexes avec un brin ouen le cassant. Ceci se produit chez lesanaérobies stricts, c'est-à-dire des bacté-ries se développant en l'absence d'oxy-gène. Dans ces conditions a lieu unetransformation en un métabolite réactif(par ex. hydroxylamine, voir sur la figure)qui attaque l'ADN. L'effet est bactéri-cide. C'est au même mécanisme qu'estdue l'action cytotoxique sur le proto-zoaire Trichomonas vaginalis (respon-sable d'inflammation du vagin et del'urètre) et Entamoebia histolytica (res-ponsable d'inflammation du gros intestin,dysenterie amibienne, et d'abcès hépa-tiques). Le métronidazole est bien ab-sorbé après prise orale ; il peut être admi-nistré en injection intraveineuse oulocalement (ovules vaginaux). En raisonde la crainte d'altérations génétiques,d'effets cancérigènes ou tératogènes éga-lement chez l'homme, le métronidazolene doit, si possible, pas être administrépour une période supérieure à 10 jours etpendant la grossesse ou l'allaitement. Letinidawle doit être considéré comme lemétronidazole.
La rifampicine inhibe chez les bac-téries l'enzyme qui assemble les ARN co-pies de la matrice d'ADN (transcription) :ARN polymérase - ADN dépendante. Larifampicine a une action bactéricide. Sonttouchées à côté des mycobactéries (tuber-culose, lèpre) de nombreuses bactériesGram* ou Granr. La rifampicine est bienabsorbée après administration orale. Enraison du risque de développement d'unerésistance lors d'utilisation fréquente, ellesert seulement au traitement de la tuber-culose et de la lèpre (p. 278).
La rifampicine est contre-indiquéedans le premier trimestre de la grossesseet durant l'allaitement.
La rifabutine, en principe comparableà la rifampicine, peut cependant être encoreactive en cas de résistance à celle-ci.
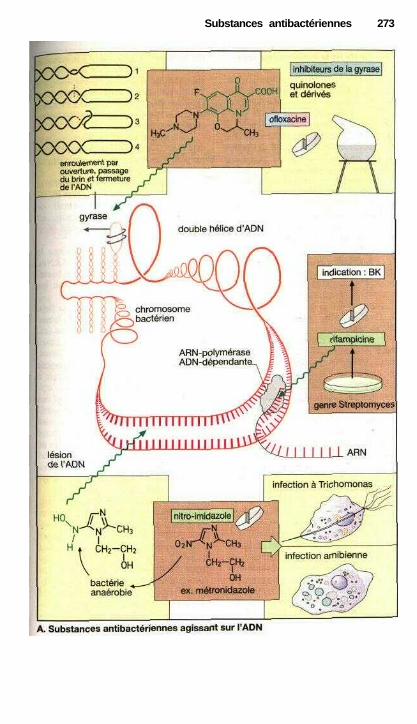
Substances antibactériennes 273

274 Substances antibactériennes
Inhibiteurs de la synthèse protéique
La synthèse protéique correspond à la tra-duction (translation) de l'information gé-nétique transmise auparavant sous formed'ARNm (p. 272), en une chaîne pepti-dique. L'assemblage de cette chaîne àpartir des acides aminés (AA) s'effectuesur un ribosome. Le transport des acidesaminés jusqu'à l'ARNm est assuré pardifférentes molécules d'ARN de transfert(ARNt) qui ont chacune lié un AA parti-culier. A un ARNt correspond une unitéde code spécifique de l'ARNm (le codon,se composant de 3 bases).
En temps normal, l'introductiond'un acide aminé comporte les étapes sui-vantes (A) :
1. Le ribosome s'associe à deux co-dons de l'ARNm. L'un (celui de gauche)est déjà lié au complexe ARNt-AA, l'AAfait déjà partie de la chaîne peptidique.L'autre (celui de droite) est prêt pourla fixation d'un nouveau complexeARNt-AA.
2. Après cette fixation, se formeune liaison entre son AA et celui du com-plexe ARNt-AA voisin (à gauche). Ceciest réalisé par l'enzyme peptide synthé-tase (peptidyl-transférase) et a pourconséquence la dissociation de l'AA et del'ARNt dans le complexe de gauche.
3. Cet ARNt se dissocie del'ARNm. Le ribosome peut se déplacer lelong de l'ARNm et s'intéresser au codonsuivant.
4. De ce fait, le complexe ARNt-AA qui était à droite, se déplace vers lagauche, et un nouveau complexe peutmaintenant s'associer à droite.
Chacune de ces différentes étapespeut être inhibée par des antibiotiquesappartenant à des groupes différents. Lesexemples décrits proviennent tous demicro-organismes de type streptomyces,quelques aminoglycosides sont égalementissus du groupe des micromonospora.
l.a) Les tétracyclines inhibent lafixation du complexe ARNt-AA au ribo-some. Elles ont une action bactériosta-tique et atteignent un large spectred'agents pathogènes.
b) Les aminoglycosides provo-quent l'association d'un complexe ARNt-
AA incorrect, ce qui conduit à la synthèsede protéines erronées. Les aminoglyco-sides ont une action bactéricide. Le pointfort de leur spectre d'action porte sur lesbactéries Gram-, Streptomycine et kana-mycine servent principalement au traite-ment de la tuberculose.
En termes de nomenclature, « ...my-cine » provient d'un organisme de typestreptomyces et « ...micine » par exemplegentamicine d'un micromonospora.
2. Chloramphénicol : il inhibe lapeptide synthétase. Il a une action bacté-riostatique sur un large éventai] degermes. La molécule, assez simple, estaujourd'hui synthétisée par voie chi-mique.
3. Érythromycine : elle bloque ledéplacement du ribosome sur l'ARNm.Elle agit essentiellement de façon bacté-riostatique et touche principalement lesgermes Gram4'.
Pour l'administration orale, la basesensible aux acides se trouve sous formede sels (par ex. stéarate) ou d'esters (ex.éthylsuccinate). L'érythromycine est biensupportée. Elle convient, entre autres,comme antibiotique de remplacementdans les cas de résistance ou d'allergie àla pénicilline. La ctarithromycine, Yay-thromycine et la roxithromycine sont desdérivés de l'érithromycine avec une sensi-bilité plus faible aux acides et unemeilleure biodisponibilité après priseorale. Les substances que nous venons deciter sont les représentants les plus impor-tants du groupe des antibiotiques macro-lides (qui renferme également la spiramy-cine).
La clindamycine a une action anti-bactérienne voisine de celle de l'érythro-mycine. Elle agit de façon bactériosta-dque principalement sur les germesGrairr1' aérobies ainsi que des germesanaérobies. La clindamycine est un ana-logue chloré, semi-synthétique, de la lin-comycine qui provient, elle, d'un strepto-myces. Après administration orale, laclindamycine, mieux absorbée que la lin-comycine, possède une action antibacté-rienne plus élevée et sera donc préférée.Les deux substances passent aisémentdans le tissu osseux.
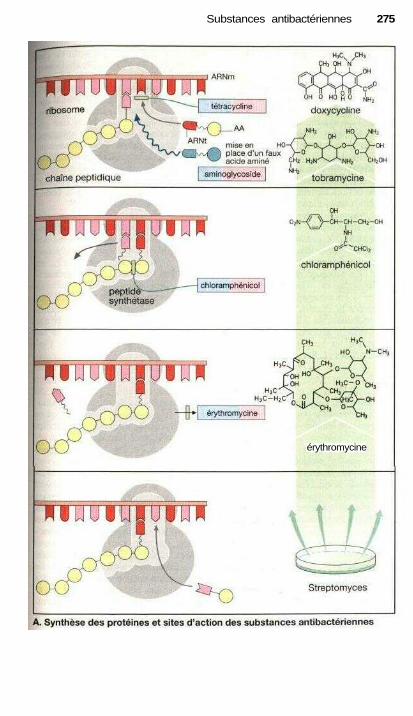
Substances antibactériennes 275
érythromycine

276 Substances antibactériennes
Les tétracyclines sont absorbéesau niveau du tractus digestif, certes enquantités variables selon la substance,mais cependant presque complètementpour la doxycycline et la minocycline.Leur administration intraveineuse estrarement nécessaire. Les effets secon-daires les plus fréquents sont destroubles gastro-intestinaux (vomisse-ments, diarrhées, nausées) dus : 1. à uneffet irritant direct de la substance sur lamuqueuse intestinale, 2. à une altéra-tion de la flore bactérienne naturelle del'intestin (antibiotique à spectre large)suivie par une colonisation par desagents pathogènes, entre autres unchampignon, le candida. La prise si-multanée d'anti-acides ou de lait pourcalmer les douleurs d'estomac est uneerreur. En présence de cations muiti-valents (par ex. Ça24-, Mg24^, A13'1',Fe^/Fe14-), les tétracyclines forment descomplexes insolubles. De ce fait, ilssont inactivés ; la capacité d'absorp-tion, l'activité antibactérienne et l'effetirritant sur les muqueuses décroissentprogressivement. Etant donné la possi-bilité de former des complexes avec leÇa2'1', la tétracycline à tendance à se dé-poser dans les dents ou les os en coursde croissance. Ceci entraîne une colo-ration irréversible des dents en brun-jaune ou une inhibition réversible de lacroissance osseuse. En raison de ceteffet secondaire, les tétracyclines nedoivent pas être administrées à partir du3e mois de grossesse et jusqu'à la8e année de la vie. D'autres effets indé-sirables sont une sensibilité accrue de lapeau à la lumière ainsi que des lésionshépatiques, essentiellement après admi-nistration i.v.
Le chloramphénicol, un antibio-tique à spectre large est complètementabsorbé après administration orale. Il sedistribue de façon uniforme dans l'or-ganisme et traverse facilement les bar-rières de diffusion comme la barrièrehémato-encéphalique. En dépit de cespropriétés favorables, l'utilisation duchloramphénicol est très rare à cause dudanger d'altérations de la moelle os-
seuse (utilisation par exemple en casd'infections du SNC). Deux formesd'aplasie médullaire sont possibles :1. Une forme toxique et réversible, dé-pendante de la dose et apparaissant du-rant le traitement. 2. Le cas échéant,une forme apparaissant après une pé-riode de latence de plusieurs semaines,indépendante de la dose et souventmortelle. Il faut également tenir comptede ce danger d'aplasie médullaire encas d'administration locale, par ex.gouttes oculaires, à cause de la bonnepénétration du produit.
Les antibiotiques aminoglyco-sides se composent de sucres aminésreliés entre eux par des liaisons glycosi-diques (voir la gentamicine C,,, uncomposant du mélange de gentami-cines). Ils contiennent de nombreuxgroupements hydroxyle et aminé quipeuvent lier les protons. Ces composéssont donc extrêmement polaires et tra-versent mal les membranes. Ils ne se-ront pas absorbés au niveau de l'in-testin. La néomycine et la paromomy-cine seront administrées par voie oralepour éliminer les bactéries intestinales(avant une opération de l'intestin oupour diminuer la production d'ammo-niaque en cas de coma hépatique). Lesaminoglycosides utilisés pour le traite-ment d'infections bactériennes plus sé-vères doivent être injectés (par ex. gen-tamicine, tobramycine, nétilmicine,amikacine). L'apport local de formes li-bérant la gentamicine est égalementpossible dans le cas d'infections des osou des viscères. Les aminoglycosidesatteignent l'intérieur de la bactérie enutilisant les systèmes de transport bac-tériens. Au niveau du rein, ils s'accu-mulent dans les cellules du tubuleproximal en empruntant un système deréabsorption destiné aux oligopeptidesbasiques. Les cellules tubulaires peu-vent être lésées (néphrotoxicité généra-lement réversible). Dans l'oreille in-terne peut se produire une lésion descellules sensorielles, de l'organed'équilibration et de l'organe auditif(ototoxicité en partie irréversible).
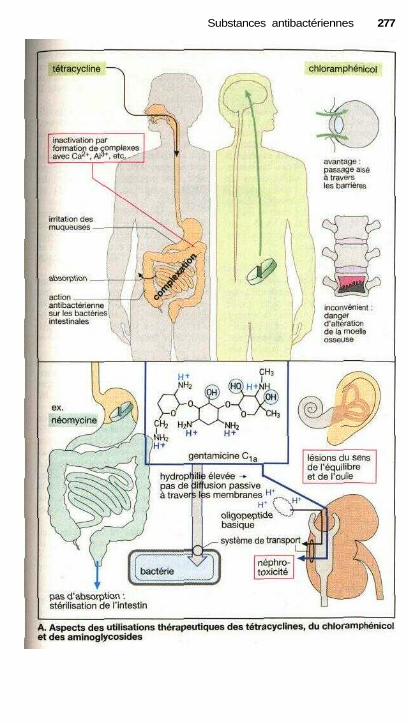
Substances antibactériennes 277

278 Substances antibactériennes
Substances contre les infectionsà mycobactéries
Les mycobactéries sont responsables dedeux maladies : la tuberculose provoquéeprincipalement par Mycobacterium tuber-culosis et la lèpre par M. leprae. Le prin-cipe général de traitement est l'adminis-tration combinée de deux substances ouplus. Le traitement combiné empêche lasélection de mycobactéries résistantes.Comme les effets antibactériens dechacun des produits s'additionnent, ilsuffit de dosages plus faibles pour chaquemolécule, ce qui fait diminuer les risquesd'effets secondaires. Les principales sub-stances ne sont dirigées que contre l'unede ces deux maladies.
Antituberculeux (1)Les médicaments de choix sont l'isonia-zide, la rifampicine, l'éthambutol et à côtéde cela la streptomycine ainsi que lepyrazinamide. Les moyens de réserve,plus mal supportés sont l'acide p-amino-salicylique, la cyclosérine, la viomycine,la kanamycine, l'amikacine, la capréomy-cine, l'éthionamide.
Isoniazide. Il a une action bactéri-cide contre les bactéries tuberculeuses encroissance. Son mécanisme d'actionn'est pas éclairci (dans la bactérie se pro-duit une transformation en acide isonico-tinique qui ne passe pas à travers lesmembranes et s'accumule dans lesgermes). L'isoniazide est très rapidementabsorbé après prise orale. L'élimination alieu dans le foie par acétylation. En fonc-tion de la vitesse d'élimination, généti-quement déterminée, on distingue deuxgroupes d'individus : les acétyleurs lentset les acétyleurs rapides. Les effets se-condaires notables sont : altération desnerfs périphériques mais également duSNC qui peut être évitée par l'adminis-tration de vit. B,, (pyridoxine) ; lésionshépatiques.
Rifampicine. Son origine, son actionantibactérienne et ses voies d'administra-tion ont été décrites page 272. Pour cettesubstance en général bien tolérée, il fautsignaler comme effets secondaires : lé-sions hépatiques ; reactions allergiquesavec entre autres, des symptômes de type
grippal ; coloration perturbante mais nondangereuse des fluides corporels enrouge/orange ; induction enzymatique(éviter les contraceptifs oraux). Pour larifabutine, voir p. 272.
Éthambutol. La raison de sa spécifi-cité d'action contre les mycobactéries estinconnue. L'éthambutol agit par voieorale. Il est en général bien supporté. Hpeut se produire une lésion particulière dunerf optique, dose dépendante et réver-sible, avec perturbation de la vision descouleurs (rouge/vert, perte de champ vi-suel).
Pyraynamide. Son mécanismed'action est inconnu. Il est administre parvoie orale, peut altérer les fonctions hépa-tiques et déclencher une hyperuricémie eninterférant avec l'élimination rénaled'acide urique.
Streptomycine. Comme tous les an-tibiotiques aminoglycosides, elle doit êtreinjectée (p. 274 et suivantes) ; elle lèsel'oreille interne, en particulier le sens del'équilibre ; en comparaison sa néphro-toxicité est faible.
Substances contre la lèpre (2)La rifampicine est souvent utilisée en as-sociation avec l'une des deux substancesdécrites ci-dessous voire même avec lesdeux.
La dapsone est un sulfone qui defaçon analogue aux sulfonamides (p. 270)inhibe la synthèse de l'acide dihydrofo-lique. Elle a une action bactéricide surles souches sensibles de M. leprae. Ladapsone est administrée par voie orale.L'effet secondaire le plus fréquent est laformation de méthémoglobine avec unedisparition accélérée des érythrocytes(hémolyse).
La clofaïimine est un colorant avecune action bactéricide contre les germesde la lèpre et par ailleurs des propriétésanti-inflammatoires. Elle est administréepar voie orale mais absorbée de façon in-complète. En raison de son hydrophobieélevée, elle se dépose dans le tissu adi-peux et les autres tissus et ne disparaît quetrès lentement de l'organisme (t 1/2 ~ 70jours). Un effet indésirable, en particulierchez les patients avec la peau la plusclaire, est une coloration rouge-brun.
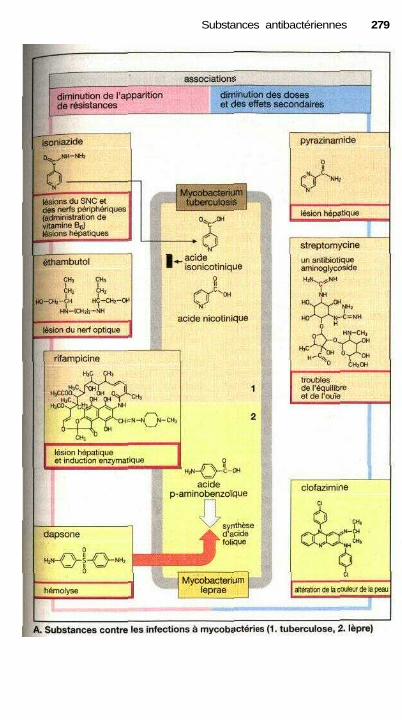
Substances antibactériennes 279

280 Antifongiques
Substances contre les infectionsprovoquées par des champignons
Les maladies infectieuses dues aux cham-pignons sont en général limitées à la peauet aux muqueuses : mycoses locales.Rarement, en cas de déficit immunitaire,on observe une atteinte des organes in-ternes : mycoses systémiques.
Les agents les plus courants des my-coses sont les dermatophytes qui aprèsune contamination de la surface externerésident dans les cheveux ou les ongles.
Candida albicans : cette levure setrouve de façon normale à la surface ex-terne de l'organisme ; une infection desmuqueuses et plus rarement de la peau oumême des organes internes peut se pro-duire en cas de diminution des défenses(par ex. altération de la flore bactériennepar des antibiotiques à large spectre, trai-tement immunosuppresseur).
Les dérivés imidazolés inhibent lasynthèse de l'ergostérol. Ce stéroïde est uncomposant essentiel de la membrane cyto-plasmique des cellules de champignon,comparable au cholestérol dans les celluleshumaines. Sous l'action d'un dérivé imida-zolé, les champignons ne se développentpas (effet fongistatique) ou même meurent(effet fongicide). Le spectre des champi-gnons touchés est très étendu. Les princi-paux dérivés imidazolés conviennent seule-ment pour une application locale, à causede leur faible absorption et de leur mau-vaise tolérance systémique (clotrimawle,éconazole, oxiconazole, et autres dérivésazolés). Très rarement, on observe une der-matite de contact. Le miconazole peut êtreutilisé localement mais aussi de façon sys-témique en courtes perfusions (bien quemal supportées).
Le kétoconawle, grâce à unemeilleure absorption, peut être donné enprises orales. Les effets secondaires sontrares (à surveiller le cas échéant un risquede lésions hépatiques mortelles).
Le fluconawle et l'itraconazolesont des dérivés triazolés nouveaux et ad-ministrables par voie orale. Le naftifineune allylaminc et Vamorolfine une mor-pholine agissent également sur la syn-thèse de l'ergostérol mais en un autresite ; tous les deux sont des antimyco-tiqueslocaux.
Les antibiotiques polyènes. Ampho-téricine B et nystatine sont d'origine bac'térienne. Ils se déposent sur la membranedu champignon (sans doute à proximitédes molécules d'ergostérol) de telle sorteque se forment des pores. L'augmentationde la perméabilité par exemple pour lesions K4' est responsable de l'effet funei-cide. L'amphotéricine B touche la maïo-rité des germes responsables des mycosessystémiques. En raison de leur mauvaiseabsorption, les antibiotiques polyènesdoivent être administrés en perfusion. Lemalade supporte assez mal le traitement(frissons, fièvres, troubles du SNC, réduc-tion de la fonction rénale, inflammationau site d'injection). Utilisés localementsur la peau ou les muqueuses, l'amphoté-ricine B sert au traitement des mycoses àCandida. Dans le cas de candidoses intes-tinales, l'administration orale permet untraitement local, à cause de la mauvaiseabsorption. La nystatine sera utilisée seu-lement localement (entre autres aussi dansla bouche) et également contre les my-coses à Candida.
La flucytosine est transformée dansles Candida en 5-fluoro-uracile sous l'ac-tion d'une cytosine désaminase spéci-fique des levures. Ce composé agitcomme un antimétabolite et perturbe lemétabolisme des ARN et de l'ADN(p. 294). L'effet est fongicide. Aprèsprise orale, la flucytosine est absorbéetrès rapidement. Sa tolérance chezl'homme est bonne. Souvent il est associéà l'amphotéricine B ce qui permet dediminuer les doses utilisées au cours dutraitement.
La griséofulvine provient de moi-sissures et n'agit que contre les dermato-phytes. Elle agit vraisemblablement dansles champignons comme un poison du fu-seau, inhibant les mitoses. Bien que di-rigée contre les mycoses locales elle doitêtre utilisée par voie systémique. Elle sedépose dans la kératine nouvellementformée. Ainsi « imprégnée » celle-ci nepeut plus servir de terrain nourricier auxchampignons. Le temps nécessaire à l'éli-mination des dermatophytes dépend de lavitesse de renouvellement de la peau, descheveux et des ongles. La gnséofulvinepeut provoquer divers effets secondairespeu caractéristiques.
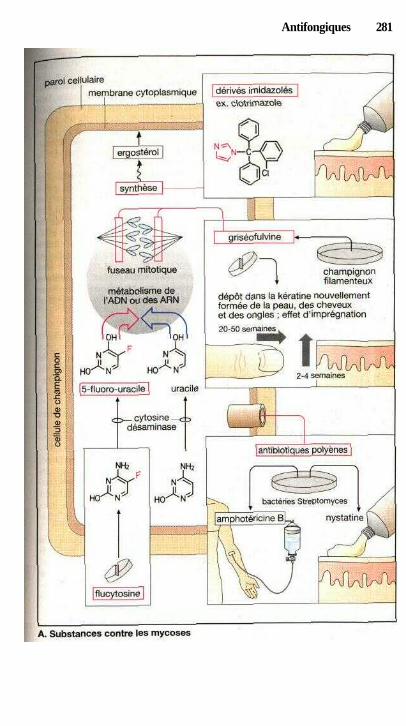
Antifongiques 281

282 Virustatiques
Médicaments antiviraux
Les virus se composent principalementde matériel héréditaire (acide nucléique,brin vert en A), et d'une capside pro-téique (hexagone bleu) ainsi que dans denombreux cas d'une enveloppe (cerclegris) formée d'une double couche phos-pholipidique dans laquelle sont inséréesdes protéines (bâtonnets bleus). Lesvirus ne possèdent aucun métabolismepropre, mais sont multipliés par les cel-lules atteintes. Pour pouvoir, à titre thé-rapeutique, bloquer la multiplication vi-rale lors d'une infection, on doit inhiberspécifiquement dans les cellules infec-tées les phénomènes métaboliques quiparticipent à la multiplication des parti-cules virales.
Multiplication virale en prenantl'exemple du virus de l'herpès simplex(A). 1. La particule virale se fixe sur lacellule cible (adsorption). 2. L'enve-loppe virale fusionne avec la membraneplasmique de la cellule cible et la nu-cléocapside (acide nucléique + capside)pénètre à l'intérieur de la cellule (péné-tration). 3. La capside s'ouvre (uncoa-ting) ; dans le cas du virus herpès, cephénomène se produit au niveau despores nucléaires, et l'ADN viral parvientau noyau cellulaire ; le matériel géné-tique du virus va maintenant pouvoirperturber le métabolisme cellulaire. 4a.Synthèse d'acides nucléiques : le maté-riel génétique du virus (ici de l'ADN) vaêtre reproduit, et des ARN produits pourpermettre une synthèse protéique. 4b.Les protéines servent « d'enzymes vi-rales » pour la multiplication du virus(ex. ADN-polymérase et thymidine ki-nase), comme éléments de la capside oude l'enveloppe ou aboutissent dans lamembrane cellulaire. 5. Les composantsindividuels sont assemblés (maturation)et il se produit 6. une libération desvirus-filles, qui peuvent ensuite se ré-pandre à l'intérieur ou à l'extérieur del'organisme. Dans le cas des virusherpès, la multiplication provoque ladestruction de la cellule cible ce qui dé-clenche les symptômes de la maladie.
Moyens de lutte antivirale propresà l'organisme (A). L'organisme peut in-
terrompre la multiplication virale grâceà des lymphocytes T cytotoxiques, quireconnaissent les cellules produisant desvirus (présence de protéines virales dansla membrane) et les détruisent, ou bien àl'aide d'anticorps qui fixent les parti-cules virales extracellulaires et les inac-tivent. L'activation des défenses immu-nitaires spécifiques est le but de lavaccination préventive.
Interférons (IFN). Ce sont desglycoprotéines qui sont libérées, entreautres, par les cellules infectées par desvirus. Dans les cellules voisines, lesinterférons déclenchent la synthèse de« protéines antivirales ». Celles-ci inhi-bent la synthèse des protéines viralesen détruisant (de façon préférentielle)les ARN viraux ou en réprimant leurlecture (traduction). Les interféronsn'agissent pas de façon spécifiquecontre un virus donné. Ils sont cepen-dant spécifiques d'une espèce et doi-vent pour une utilisation thérapeutiqueêtre d'origine humaine. Les interféronsproviennent par exemple de leucocytes(IFN-a), de fibroblastes (IFN-p) ou delymphocytes (IFN--y). Les interféronssont également utilisés pour le traite-ment de certaines tumeurs (leucémies àtricholeucocytes).
Antimétabolites virustatiques(B). Ce sont de faux constituants del'ADN. Un nucléoside (par ex. la thymi-dine) se compose d'une base (ex. la thy-mine) et d'un sucre, le désoxyribose.Dans un antimétabolite, l'un de cesconstituants est incorrect. Les nucléo-sides anormaux vont être activés dansl'organisme grâce à la fixation de troisrésidus phosphate pour donner les inhi-biteurs proprement dits (p. 284).
Idoxuridine et analogues. Ilssont intégrés dans l'ADN ce qui l'en-dommage. La synthèse de l'ADN hu-main est également atteinte si bien queces composés sont réservés à l'adminis-tration locale (ex. kératite à herpès sim-plex).
La vidarabine inhibe l'ADN poly-mérase induite par le virus plus forte-ment que celle de l'organisme. Elle nesert aujourd'hui qu'au traitement localdes infections à virus herpès.
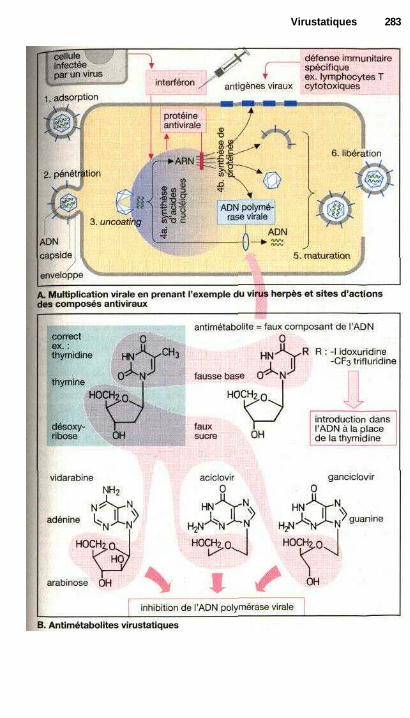
Virustatiques 283

284 Virustatiques
L'aciclovir (A) possède parmi lesantimétabolites le plus haut degré despécificité et la meilleure tolérance, carson activation n'intervient que dans lescellules infectées où il inhibe essentiel-lement la synthèse d'ADN viral : 1. Lapremière étape de phosphorylation estaccomplie par une thymidine kinase quin'est codée que par les virus herpès sim-plex et varicella zoster. 2. Compte tenude la polarité du résidu acide phospho-rique, l'aciclovir triphosphate ne passepas à travers la membrane et s'accumuledans les cellules infectées. 3. L'aciclovirtriphosphate est bien accepté commesubstrat par l'ADN polymérase virale : ilinhibe l'activité enzymatique et conduitaprès introduction dans l'ADN viral àune interruption des chaînes car il nepossède pas le groupement 3' OH dudésoxyribose, nécessaire à l'accrochaged'un nouveau nucléotide. L'intérêt thé-rapeutique important de l'aciclovir semanifeste en particulier lors de gravesinfections par les virus herpès (encépha-lite, infection généralisée) ou varicellazoster (par ex. zona). Dans ces cas, ilsera administré par perfusion i.v. L'aci-clovir peut également être utilisé parvoie orale, mais l'absorption intestinaleest incomplète (15-30 %). Il existe parailleurs des formes topiques. Comme lasynthèse d'ADN endogène n'est pas al-térée on n'a pas à craindre d'aplasiesmédullaires.
Le ganciclovir (voir formulep. 283) sert pour le traitement par perfu-sion d'infections sévères par des cyto-mégalovirus (appartenant aussi augroupe des virus herpès). Le ganciclovirest moins bien supporté, et les leucopé-nies ou les thrombopénies ne sont pasrares.
La zidovudine (azidothymidine,B), ou plus exactement son triphosphateinhibe la transcriptase inverse. Cette en-zyme est présente dans les virus HIV,responsables du SIDA, et transcritd'abord dans les cellules infectéesl'ARN viral en ADN. Cette substanceest utilisée pour freiner la maladie ; uneélimination des virus, c'est-à-dire uneguérison, n'est pas possible. La tolé-rance n'est pas bonne (entre autres leu-
copénie). La didanosine et la wlcitabineagissent de la même manière mais pré-sentent un autre spectre d'effets secon-daires.
Le saquinavir inhibe la protéasedu virus HIV. Cette enzyme découpedans les particules virales nouvellementformées un précurseur protéique degrande taille en diverses protéines fonc-tionnelles parmi lesquelles la transcrip-tase inverse. Cette molécule en associa-tion avec d'autres virustatiques (trithé-rapie) a permis de diminuer la « chargevirale » chez de nombreux patients.
i. Le foscarnet est un analogue d'un^ diphosphate.
Comme le montre la figure en A,lors de l'introduction d'un nucléotidedans le brin d'ADN un résidu diphos-phate sera éliminé. Le foscarnet inhibel'ADN-polymérase en interagissantavec le site de fixation du groupementdiphosphate sur l'enzyme. Indications :traitement systémique d'infections sé-vères à cytomégalovirus chez des ma-lades du SIDA, traitement local pour desinfections à virus herpès.
L'amantadine (C) influence spé-cifiquement la multiplication des virusA de l'influenza (virus à ARN, respon-sables de la grippe). Ces virus serontcapturés par endocytose. Il est néces-saire pour la libération de l'ARN viralque les protons contenus dans l'endo-some pénètrent à l'intérieur du virus.L'amantadine bloque vraisemblable-ment un canal protéique présent dansl'enveloppe virale et par lequel les pro-tons peuvent pénétrer dans le virus,bloquant ainsi le phénomène d'un-coating. Par ailleurs, l'amantadine in-hibe la maturation virale. Elle est uti-lisée à titre préventif, mais doit sipossible être prise avant l'apparitiondes symptômes. L'amantadine estégalement utilisée comme antiparkin-sonien (p. 186).
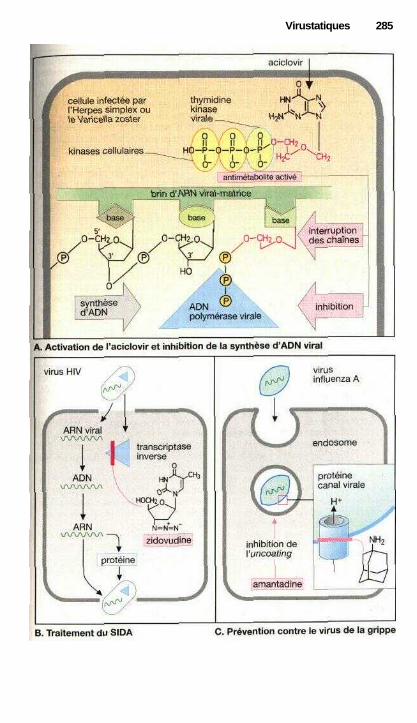
Virustatiques 285

286 Désinfectants
Désinfectants
La désinfection est une inactivation ouune élimination des germes pathogènes(protozoaires, bactéries, champignons,virus) dans l'environnement de l'hommeou sur l'individu lui-même (on parlealors d'antiseptiques). Elle peut être réa-lisée par des moyens chimiques ou pardes procédés physiques (non évoquésici). La stérilisation est l'élimination detous les germes pathogènes ou non,l'asepsie est une diminution du nombrede germes sur la peau ou les muqueuses.
Les produits pour la désinfectiondoivent si possible inactiver les germespathogènes de façon complète, rapide, etdurable et en même temps posséder unefaible toxicité (faible toxicité systé-mique, bonne tolérance tissulaire, faibleantigénicité) et ne pas altérer le matériel.Ces exigences se trouvent souvent enopposition avec les propriétés des sub-stances. Dans le choix d'une substance ilfaut donc réaliser des compromis selonles buts recherchés.
Aujourd'hui, les désinfectants uti-lisés sont les oxydants, les produitshalogènes ou libérant des halogènes,les alcools, les aldéhydes, les acides or-ganiques, les phénols et les substancestensio-actives, hier on utilisait aussi lessels de métaux lourds.
Le mécanisme d'action est unedénaturation des protéines, une inhibi-tion d'enzymes, une modification de lacharge de surface ou une déshydratation.L'effet dépend de la concentration et,dans la plupart des cas, également de ladurée d'application.
Spectre d'action. Les désinfec-tants inactivent les bactéries (bactériesGram+ > bactéries Gram- > mycobac-téries), moins bien leurs formes sporu-lées et seuls quelques-uns (fornialdé-hyde) sont virucides.
Domaines d'application. Désin-fection de la peau : une diminution dunombre de germes est souhaitée lorsd'une intervention chirurgicale ou d'uneponction de façon à diminuer le risqued'infection de la plaie. On utilisera lesalcools (propanol 1 et 2, éthanol à 60-90 %), les composés libérant de l'iode
(PVP-iode = polyvinyipyn-olidone-iodeà la place de la teinture d'iode, commeune sorte de dépôt pour le principe actifl'iode -^ rémanence), des tensio-actifscationiques ou un mélange de ces com-posants. Ils doivent agir en moins d'unquart d'heure dans les zones de la peaupauvres en glandes sébacées et en moinsde 10 minutes dans les parties riches englandes sébacées.
Désinfection des muqueuses : laquantité de germe peut être réduite,même si c'est avec une efficacitémoindre que sur la peau, avec le PVP-iode ou la chlorhexidine (durée d'action2 minutes).
Désinfection des plaies : ceci peutêtre réalisé avec le peroxyde d'hydro-gène (H^O; en solution à 0,3 - 1 %,action brève, bouillonnant au contact dusang et donc asséchant la plaie), avecdu permanganate de potassium (solutionaqueuse à 0,0015 %, légèrement astrin-gent) ainsi qu'avec le PVP-iode, lachlorhexidine et les biguanides.
Désinfection des mains par hy-giène ou avant une opération : la désin-fection des mains est nécessaire aprèsune éventuelle contamination (hygiène)ou avant une intervention chirurgicale.On se sert en premier lieu d'alcool maiségalement de mélanges d'alcools et dephénols, de tensio-actifs et d'acides. Lemélange avec d'autres substances al-longe la durée d'action (rémanence) etdiminue le caractère inflammable del'alcool.
Désinfection des instruments : lesinstruments (en particulier ceux qui nesont pas stérilisables à la vapeur ou à lachaleur) peuvent être désinfectés en uti-lisant des aldéhydes.
Désinfection des surfaces : elles'effectue avec des aldéhydes en asso-ciation à des tensio-actifs cationiques oudes oxydants, rarement avec des acidesou des bases.
Désinfection des pièces ; l'air despièces ainsi que les surfaces, dans la me-sure où les microorganismes sont aisé-ment accessibles, peuvent être désin-fectés par vaporisation ou évaporationde formaldéhyde.
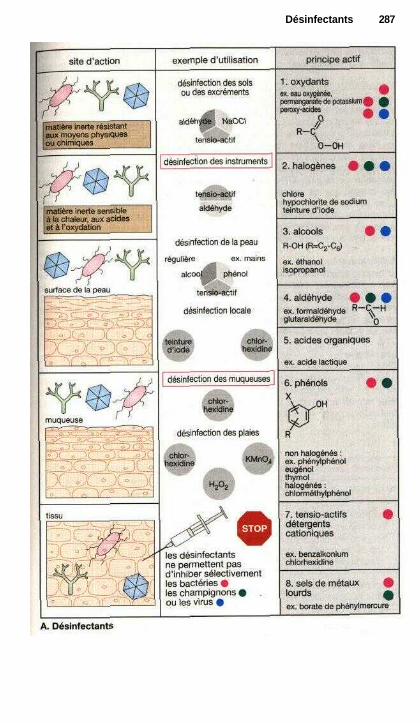
Désinfectants 287

288 Médicaments antiparasitaires
Substances antiparasitaires(endo- et ectoparasites)
En particulier dans des conditions d'hy-giène défavorables, l'homme peut êtrecontaminé par des organismes pluricel-lulaires vivant en parasites (nommés iciparasites). La peau et les cheveux sontles sites où s'installent les ectopara-sites par exemple les insectes, puces etpoux, ou les araignées (arachnides) res-ponsables de la gale. On utilise alorsdes insecticides ou des arachnicides.L'intestin ou d'autres organes internespeuvent être contaminés par des endopa-rasites. Ce sont des vers, contre lesquelssont dirigés des antihelminthiques.
AntihelminthiquesComme le montre le tableau ci-dessous,deux substances nouvelles, prazi-quantel et mébendazole, permettent letraitement de très nombreuses maladiesprovoquées par les vers. Les deux sontbien supportées par l'homme.
InsecticidesTandis que dans la lutte contre lespuces, le nettoyage des vêtements et despièces est suffisant, les poux et les aca-riens seront éliminés chez les individuscontaminés par l'utilisation d'insecti-cides.
Chlorphénothane (DDT). Il tueles insectes dès l'absorption de trèsfaibles quantités de substance, par expar contact de leurs pattes avec des sur-faces traitées (insecticides de contact).La cause de la mort est une lésion dusystème nerveux accompagnée decrampes. Chez l'homme, le DDT agitcomme un poison du système nerveuxmais seulement après la prise de quan-tités très importantes. Le DDT est chi-miquement stable et ne sera dégradédans l'organisme et dans l'environne-ment qu'extrêmement lentement. Lamolécule très lipophile est stockée dansle tissu adipeux des organismes vivants.Le DDT répandu dans l'environnementcomme pesticide peut s'accumuler defaçon dangereuse au cours de la chaînealimentaire. C'est pour cette raison queson utilisation est interdite dans denombreux pays.
Lindane. C'est l'isomère -y, actifde l'hexachlorocyclohexane. Il agitégalement chez l'insecte comme neuro-toxique (et dans certains cas chezl'homme). Après application locale, desirritations de la peau et des muqueusessont possibles. Le lindane affecte égale-ment, outre les poux et les puces, lesacariens vivant sur la peau (respon-sables de la gale). Le lindane est mieuxdégradé que le DDT.
Traitement des maladies provoquées par les vers
Vers (helminthes)
Vers plats (plathelminthes)Vers segmentés (cestodes)Vers non segmentés (trématodes)par ex. du genre schistosomes(responsables de la bilharziose)
Vers ronds (nématodes)Asticots (Enterobius vermicularisou encore oxyure)Ascaride (Ascaris lumbricoïdes)Trichines (Trichinella spiralis)
Traitement de choix
PraziquantelPraziquantel
Mébendazole
MébendazoleMébendazole
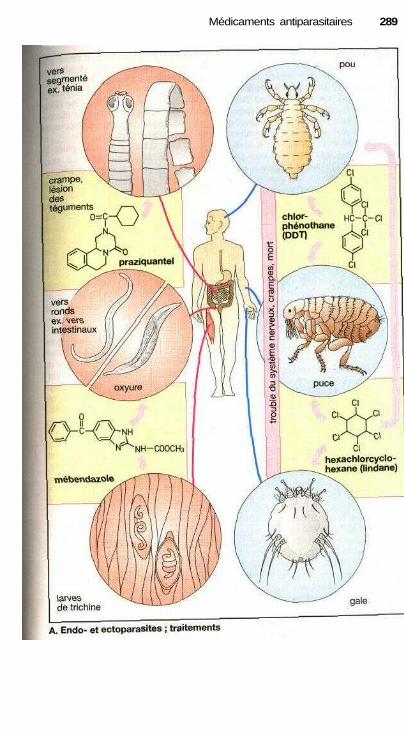
Médicaments antiparasitaires 289

290 Médicaments antiparasitaires
Antimalariens
La malaria est provoquée par lePlasmodium, un organisme unicellulaire(protozoaire). L'agent pathogène est trans-porté chez l'homme sous forme de sporo-zoïtes lors de la piqûre par un moustiqueanophèle contaminé (A). Les sporozoïtespénètrent dans les cellules du foie et se dé-veloppent en schizontes (schizontes pri-maires). Ceux-ci donnent naissance à denombreux mérozoïtes qui passent dansle sang. Ce cycle pré-érythrocytaire estasymptomatique. Dans le sang, ces para-sites envahissent les érythrocytes (cycleérythrocytaire). Les mérozoïtes formés se-ront libérés en même temps par les érythro-cytes contaminés : lyse des érythrocytesavec accès de fièvre. De nouveau les éry-throcytes seront infectés. La durée de dé-veloppement du parasite conditionne letemps avant l'apparition d'un nouvel accèsde fièvre. Dans le cas de Plasmodium (P.)vivax et de P. ovale, les sporozoïtes hépa-tiques peuvent également donner des hyp-nozoïtes qui subsisteront sous cette formependant des mois et des années avant deparvenir au stade schizonte.
Les différentes formes de développe-ment du parasite peuvent être selon les caséliminées par diverses substances. Onconnaît le mécanisme d'action de certainesd'entre elles : la chloroquine et la quinines'accumulent dans les vacuoles digestivesacides du schizonte sanguin et inhibent uneenzyme qui en temps normal polymériseles groupements hèmes libérés à partir del'hémoglobine, qui sinon sont toxiquespour le parasite. La pyriméthamine inhibela dihydrofolate réductase (p. 270) du pro-tozoaire. Le proguanil donne naissance àun composé actif appartenant à la même
1 famille que la pyriméthamine. Le sulfa-•smide, sulfadoxine inhibe la synthèse de]''acide dihydrofolique (p. 270).
Pour le choix d'une substance, ilfaut tenir compte de la tolérance et desphénomènes de résistance.
Tolérance. C'est la quinine, le pre-mier produit antimalarien utilisé, qui a lafenêtre thérapeutique la plus étroite. Lescomposés récents sont tous bien supportés.
C'est en particulier chez P. falci-parum, responsable des formes les plus
dangereuses de malaria, qu'on observe ledéveloppement de formes résistantes.La fréquence d'apparition de souches ré-sistantes augmente avec la fréquenced'utilisation d'une substance. Une résis-tance peut apparaître contre la chloro-quine et également contre l'associationpyriméthamine-sulfadoxine.
Choix d'une substance pour laprophylaxie antimalarienne. La prisecontinuelle de substances antimalariennesdurant un séjour dans les zones présentantun danger de malaria constitue lameilleure protection contre le développe-ment de la maladie mais cependant pascontre l'infection. La primaquine pourraitcertes agir contre les schizontes primairesde tous les types de Plasmodium ; ellen'est cependant pas utilisée pour une pro-phylaxie à long terme à cause d'une tolé-rance peu satisfaisante lors d'une adminis-tration de longue durée et du danger dedéveloppement d'une résistance (c'est leseul moyen contre les schizontes secon-daires de P. ovale et P. vivax). Pour la pré-vention, on utilise plutôt les substancescontre les schizontes sanguins. Le produitde choix est la chloroquine. A cause de sapersistance dans le plasma (t 1/2 plasma-tique 3 jours ou plus), une prise hebdoma-daire est suffisante. Dans les régions oùsévissent des formes résistantes, on utili-sera comme alternative la méfloquine, leproguanil ainsi que le cas échéant la doxy-cycline, une tétracycline. Les produitscontre les schizontes sanguins ne bloquentpas l'atteinte asymptomatique du foie,mais seulement l'infection des érythro-cytes responsable des symptômes de lamaladie (traitement suppressif). Contre lapersistance éventuelle de parasites dans lefoie, il est nécessaire de prendre de la pri-maquine pendant deux semaines après lafin du séjour en zone malarique.
Il est très important à titre prophy-lactique de se protéger des piqûres demoustique : moustiquaire, vêtements re-couvrant la peau...
Pour le traitement on utilise enprincipe les mêmes produits ainsi que laquinine et 1'' halofantrine contre lesschizontes sanguins, et la combinaisonpyriméthamine et sulfadoxine pour lesautomédications initiales.
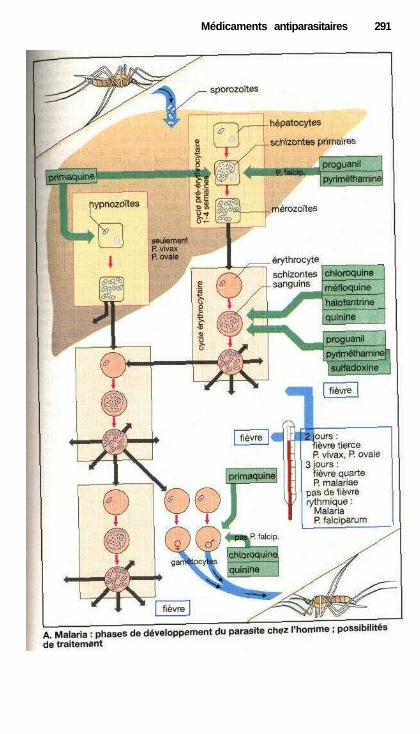
Médicaments antiparasitaires 291

•292 Cytostatiques
Substances contre les tumeursmalignes
Une tumeur (néoplasme) se compose decellules qui se multiplient sans tenircompte du « plan de développement del'organisme ». II s'agit d'une tumeur ma-ligne (cancer) lorsque le tissu tumoral pé-nètre en le détruisant dans le tissu sain en-vironnant et que les cellules tumoralesdisséminées peuvent former dans d'autresorganes des tumeurs filles (métastases).Une guérison nécessite l'élimination detoutes les cellules malignes (traitementcuratif). Si cela n'est pas possible, on peutchercher à freiner leur croissance pourprolonger la vie du malade ou améliorersa qualité de vie (traitement palliatif). Ladifficulté des traitements médicamenteuxest que les cellules malignes font partie del'organisme et ne présentent aucune pro-priété métabolique particulière.
Cytostatiques (A). Ce sont des sub-stances lésant les cellules (cytotoxiques),qui touchent en particulier les cellules envoie de division (mitose). Les cellulesmalignes se divisant rapidement serontainsi lésées de façon préférentielle.L'altération des phénomènes de divisioncellulaire peut non seulement ralentir laprolifération des cellules malignes, maiségalement déclencher un processusd'apoptose (suicide des cellules tou-chées). Les tissus avec un vitesse de divi-sion plus faible, comme les tissus sains,demeurent pratiquement intacts. Ceci estégalement valable pour les tumeurs ma-lignes formées de cellules différenciées sedivisant rarement. Quelques tissus sainsont cependant, de façon physiologique,une fréquence de division élevée. Par laforce des choses, un traitement cytosta-tique endommage également ces tissus.Ceei-a-pour conséquence les effets secon-daires typiques suivants :
La chute des cheveux se produit parsuite d'une atteinte des cellules des folli-cules pileux ; les troubles digestifs, parexemple diarrhées, proviennent d'un re-nouvellement insuffisant des cellules del'épithélium intestinal, qui ne vivent quequelques jours ; nausées et vomissementssont dus à une stimulation des chémoré-
cepteurs de l'area postrema (p. 324). Latendance à l'infection est liée à un affai-blissement du système immunitaire(p. 296). Par ailleurs, les Cytostatiquesprovoquent une dépression médullaire.La production des cellules sanguines dansla moelle est précédée par une divisiondes cellules souches et de leurs filles.L'inhibition de cette production estd'abord visible pour les granulocytes àcourte durée de vie (neutropénie), ensuitepour les plaquettes sanguines (thrombo-pénie) et finalement pour les érythrocytesdont la durée de vie est longue (anémie).Infertilité : elle peut se produire par suitede la suppression de la spermatogenèse oude la maturation des ovocytes. La plupartdes Cytostatiques affectent le métabo-lisme de l'ADN. Il existe donc un dangerd'altération du patrimoine génétique descellules saines ( e f f e t mutagène). Des leu-cémies qui se déclarent des années aprèsun traitement cytostatique peuvent en êtreune conséquence ( e f f e t carcinogène). Onpeut également craindre des malforma-tions du fœtus lorsque des agents Cytosta-tiques doivent être employés en cours degrossesse ( e f f e t tératogène).
Les Cytostatiques possèdent diffé-rents mécanismes d'action.
Altérations du fuseau achroma-tique (B). Avant que la cellule ne se di-vise, les chromosomes dédoublés vont seséparer à l'aide du fuseau achromatique.Les substances appelées poisons du fu-seau bloquent cet événement (voir entreautres colchicine, p. 310). Les microtu-bules constituent un élément fondamentalde ce fuseau achromatique. La vincristineet la vinblastine proviennent d'une planteperenne, vinca rosea, et sont donc nom-mées vinca-alcaloïdes. Elles inhibent laformation des microtubules. Un effet se-condaire particulier est une altération dusystème nerveux (phénomènes de trans-port axonal dépendants des microtu-bules).
Le paclitaxel (taxol) provient desfeuilles de l'if. Il inhibe la dépolymérisa-tion des microtubules et aboutit à la for-mation de microtubules atypiques. Le do-cetaxel est un dérivé semi-synthétique dutaxol.
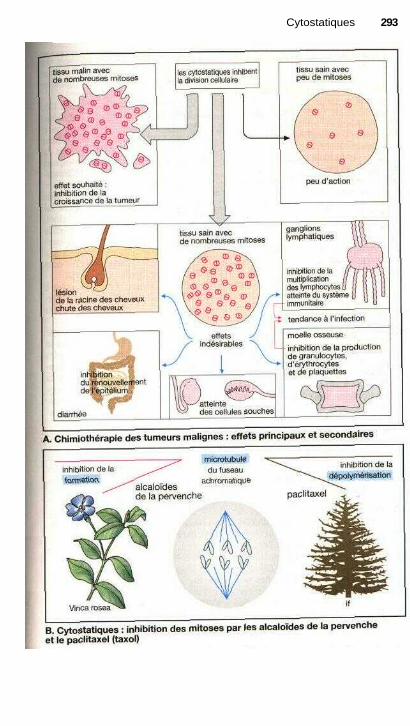
Cytostatiques 293

294 Cytostatiques
Inhibition de la synthèse d'ARNet d'ADN (A). La mitose est précédée parun doublement des chromosomes (syn-thèse d'ADN) et par une augmentation dela synthèse protéique (synthèse d'ARN).L'ADN existant (gris) sert de matricepour la nouvelle synthèse d'ADN etd'ARN (en bleu). L'inhibition de ces syn-thèses est possible par :
Une lésion de la matrice (1) : cyto-statiques alkylants. Ce sont des molé-cules réactives qui forment, via leur ré-sidu alkyle, des liaisons covalentes avecl'ADN. Par exemple, l'azote libéré parl'élimination d'un atome de chlore peutformer un pont entre les deux brinsd'ADN. La lecture correcte de l'informa-tion génétique n'est plus possible. Parmiles agents alkylants, on trouve : chloram-bucil, melphalan, thiotepa, cyclophos-phamide (p. 315), ifosfamide, busulfan,lomustine. Les effets secondaires propressont : des lésions pulmonaires (busulfan),lésion de l'épithélium de la vessie (cyclo-phosphamide) que l'on peut éviter en pre-nant du Mesna (mercapto 2 éthane suifo-nate de sodium). Le cisplatine et lecarboplatine forment également uneliaison (mais pas d'alkylation) avec lesbrins d'ADN. Les antibiotiques cytosta-tiques s'insèrent dans la double héliced'ADN. Ceci peut aboutir à une cassuredes brins (par ex. dans le cas de la bléo-mycine). Les antibiotiques de type an-thracyclines, daunorubicine et adriamy-cme (doxorubicine) peuvent provoquercomme effet secondaire particulier une lé-sion du muscle cardiaque. La bléomycinepeut entraîner une fibrose pulmonaire.
Les épipodophyllotoxines, étopo-side et téniposide interfèrent avec latopoAsomérase II, qui en temps ordinairecoupè^lesJ?rins d'ADN, les déroule et lesreferme ; en inhibant cette fermeture, cescomposés induisent des coupures dans lesbrins d'ADN.
Inhibition de la synthèse des nu-cléotides (2). L'acide téthrahydrofolique(THF) est indispensable à la synthèse desbases puriques ainsi que de la thymidine.Il provient de l'acide folique, entre autres,sous l'action de la dihydrofolate réductase(p. 270). Le méthotrexate, un analogue del'acide folique, inhibe l'activité de l'en-
zyme. Les cellules s'appauvrissent enTHF. L'effet de ces antimétabolites peutêtre inhibé en présence d'acide folinique(5 formyl THF ; leucovorine).
Insertion de faux nucléotides (3).Des bases modifiées {6-mercaptopurine,5 fluorouracile) ou des nucléosides avecdes sucres modifiés (cytarabine) agissentcomme antimétabolites. Ils inhibent lasynthèse d'ADN/ARN ou provoquentjuste après leur incorporation, la forma-tion d'acides nucléiques modifiés.
La 6-mercaptopurine se forme dansl'organisme à partir d'un précurseur in-actifYawthioprine (p. 37). L'allopurinol,un inhibiteur de la formation d'acideurique inhibe la dégradation de la 6-mer-captopurine, si bien qu'en cas d'adminis-tration simultanée il faut diminuer la dosede 6-mercaptopurine.
Il est aussi souvent possible, grâce àune association de cytostatiques d'at-teindre un meilleur effet avec des effetssecondaires plus faibles.
Après un succès initial, l'activitédes produits peut disparaître parce que sedéveloppent dans la tumeur des cellulesrésistantes. Il existe différents méca-nismes de résistance : diminution de lacapture cellulaire, par exemple diminu-tion de la synthèse des protéines de trans-port qui sont indispensables à la pénétra-tion du méthotrexate dans les cellules.
Augmentation d'un système d'ex-crétion : par ex. synthèse de la glycopro-téine P qui permet le transport hors descellules de l'anthracycline, des alcaloïdesde la pervenche, des épipodophyllo-toxines et du paclitaxel (multi-drug résis-tance, expression du gène mdr-1).
Diminution de l'activation d'une« prodrogue », par exemple de la cytara-bine, qui est toxique sous forme de cytara-bine triphosphate synthétisée dans la cel-lule.
Modification du site d'action, parex. augmentation de la synthèse de la di-hydrofolate réductase pour compenserl'effet du méthotrexate.
Réparation des lésions, par ex. aug-•mentation de l'efficacité des enzymes deréparation de l'ADN en présence de cis-platine.
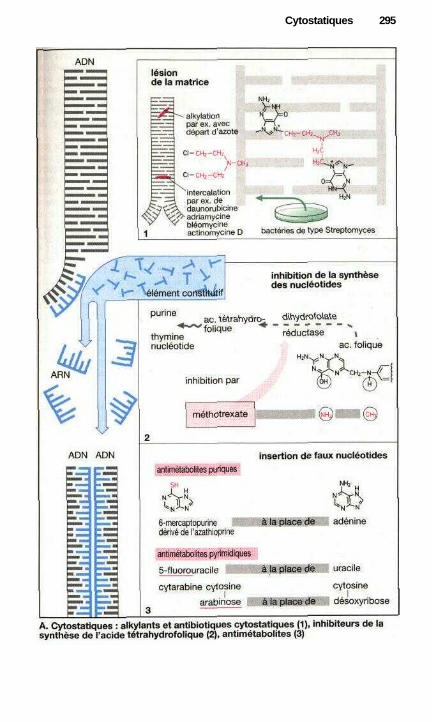
Cytostatiques 295

296 Immunomodulateurs
Inhibition des réactions immunitaires
L'inhibition des réactions immunitairesest nécessaire lors de transplantationsd'organes pour éviter le rejet ou dans lecas des maladies auto-immunes. Une im-munosuppression entraîne cependant unrisque de baisse des défenses contre lesagents infectieux et à long terme undanger de développement de cancers.
Une réaction inimunc spécifiquedébute par l'association d'un antigène surcertains lymphocytes qui comportent lesrécepteurs appropriés. Les lymphocytes Breconnaissent directement les structuressuperficielles de l'antigène au moyen derécepteurs présents sur leur membrane,qui sont voisins des anticorps synthétisésensuite. Les lymphocytes T ont besoind'une présentation des structures antigé-niques à la surface de macrophages oud'autres cellules à l'aide du MHC (com-plexe majeur d'histocompatibilité, majorhistocompatibility complex), pour pou-voir reconnaître ces antigènes grâce auxrécepteurs T. Proches de ceux-ci ontrouve les complexes CD3, ainsi que CD4(pour les cellules T auxiliaires, « helper »)ou CD8 (pour les cellules T cyto-toxiques). Les protéines CD participent àl'interaction avec le MHC. A côté de lareconnaissance de l'antigène, la stimula-tion par des médiateurs de type cytokineest indispensable à l'activation des lym-phocytes. L'interleukine 1 est formée parles macrophages et différentes interleu-kines, dont l'interleukine \2^par lescellules T auxiliaires. Les lymphocytesspécifiques d'un antigène se multiplient etla défense immunitaire se met en route.
I. Interférence avec la reconnais-sance de l'antigène. L'anticorps antiCD3 est un anticorps monoclonal dirigécontre le CD3 de souris, qui interfère avecla reconnaissance de l'antigène par leslymphocytes T (administration lors descrises de rejet) (nnironionah CD3).
IL Inhibition de la formation descytokines. Les glucocorticoïdes modu-lent l'expression de nombreux gènes.C'est ainsi que sera par ex. inhibée la syn-thèse d'IL-1 et d'IL-2, ce qui permet decomprendre la suppression des réactionsimmunitaires dépendantes des cellules T.A côté de cela, les glucocorticoïdes inter-
fèrent en de nombreux sites avec les cyto-kines et les médiateurs de l'inflammation.Les glucocorticoïdes seront utilisés lorsdes transplantations, dans les maladiesauto-immunes et allergiques. Leur admi-nistration systémique est associée audanger d'apparition d'un syndrome deCushing iatrogène (p. 246).
La cyclosporine A est produite pardes champignons et se compose de11 acides aminés, en partie atypiques.Après administration orale, l'absorptionpeut en effet être incomplète. Elle s'associedans les lymphocytes T à un récepteurcytosolique (cyclophiline). Le complexeainsi formé inhibe l'enzyme calcineurine.Cette enzyme (phosphatase) joue un rôleclef dans les événements qui conduisent àla reconnaissance des antigènes par les cel-lules T. Elle participe à l'induction de lasynthèse de diverses cytokines et en parti-culier de l'interleukine 2.
L'évolution des transplantations re-pose principalement aujourd'hui sur l'ad-ministration de cyclosporine. L'effet se-condaire majeure est une altération rénale.
Le tacrolimus est issu d'un champi-gnon de la famille des streptomycètes, il ales mêmes propriétés pharmacologiquesque la cyclosporine.
III. Perturbations du métabo-lisme cellulaire par des inhibiteurs de laprolifération cellulaire. Certains agentscytostatiques sont également utiliséscomme immunosuppresseurs, à des dosesplus faibles que celles utilisées pour letraitement des cancers. Par ex. Yawthio-prine, le méthotrexate et le cyclophospha-mide (p. 294). L'effet antiprolifératif n'estpas spécifique des lymphocytes et toucheaussi bien les cellules B que les diffé-rentes cellules T.
Le mycophénolate mofétil agit plusspécifiquement sur les lymphocytes quesur les autres cellules. Il inhibe l'inosinemonophosphate déshydrogénase qui esten particulier nécessaire à la synthèse despurines dans les cellules lymphoïdes. Ilest utilisé dans les réactions de rejet aigu.
IV. Immunsérum anti-cellules T.Il est obtenu chez l'animal après immuni-sation avec des lymphocytes T humains.Les anticorps s'associent aux cellules T etles lèsent ; la préparation sert à l'atténua-tion des réactions de rejet.
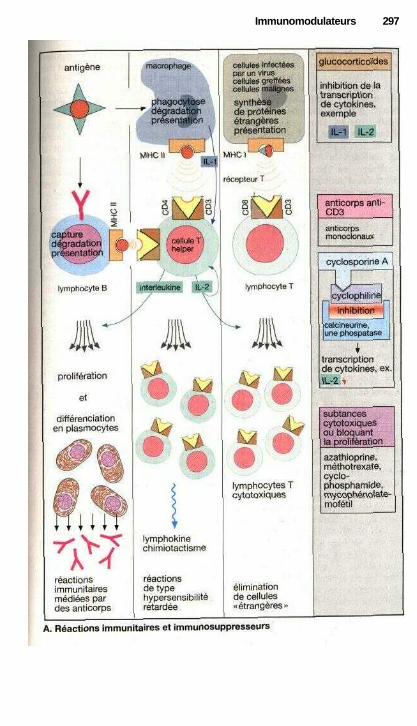
Immunomodulateurs 297

298 Antidotes
Moyens de lutte contreles empoisonnements, antidotes
Les moyens utilisés pour lutter contreles surdosages médicamenteux ont étédécrits dans les chapitres correspon-dants : par ex. la physostigmine lorsd'une intoxication par l'atropine, la na-loxone lors d'un empoisonnement parles opioldes, le flumazenil lors d'unsurdosage en benzodiazépines, desfragments d'anticorps pour une intoxi-cation par les digitaliques, la N-acétyl-cystéine pour une dose trop forte de pa-racétamol.
Les chélatants (A) servent d'anti-dotes dans les intoxications par les mé-taux lourds. Ils ont pour fonction decomplexer les ions métalliques et doncde les détoxifier. Lorsque l'on parle dechélates (du grec chele : désignant lespinces d'un crustacé) on désigne uncomplexe formé entre un ion métalliqueet des substances qui peuvent en denombreux sites établir une liaison avecl'ion métallique. Compte tenu de leuraffinité de liaison élevée, les chélatantsattirent à eux les ions métalliques pré-sents dans l'organisme. Les chélatantsne sont pas toxiques et sont principale-ment éliminés par les reins ; ils main-tiennent également l'ion métalliquesous forme liée dans l'urine concentréeet le plus souvent acide et l'entraînentainsi vers l'élimination.
Le Na, Ca-EDTA es/utilisé pourle traitement des empoisonnements auplomb. Cet antidote peut ne pas tra-verser les membranes cellulaires et doitêtre administré par voie parentérale. Acause de leur affinité de liaison élevée,les ions plomb déplacent le calcium deses sites de liaison. Le chélate conte-nant du plomb sera éliminé par lesreins. Parmi les effets secondaires leprincipal est la néphrotoxicité. Le N83Ça pentetate : est un complexe du di-éthylènetriaminopenta-acétate (DTPA)qui sert d'antidote dans les empoison-nements au plomb ou par d'autres mé-taux lourds.
Le dimercaprol a été développédurant la Seconde Guerre mondiale
comme antidote contre des composésorganiques de l'arsenic provoquant deslésions cutanées (B). Il est capable delier différents ions métalliques. Le di-mercaprol se présente sous forme d'unesubstance visqueuse, aisément décom-posable qui sera injecté en i.m. sousforme d'une solution huileuse. L'acidedimercaptopropanesulfonate dont lesel sodique permet l'administration oralea une structure et une fonction voisines.Les effets secondaires possibles sontfièvre, frissons et reactions cutanées.
La déféroxamine provient d'unchampignon, Streptomyces pilosus.Cette substance présente une forte affi-nité pour le fer, mais ne dissocie cepen-dant pas le fer associé à l'hémoglobineou aux cytochromes. La déféroxamineest mal absorbée après prise orale. Pourpouvoir éliminer le fer de l'organisme,l'antidote doit être administré par voieparentérale. La prise orale sert juste àdiminuer l'absorption de fer intestinale.
Il faut noter que la saignée, lemoyen le plus puissant qui soit pour di-minuer le fer de l'organisme, ne doit ce-pendant pas être envisagée dans lesconditions de surcharge en fer associéesà une anémie.
La D-pénicillaillinc peut stimulerl'élimination des ions cuivre (maladiede Wilson) et celle des ions plomb. Onpeut l'administrer par voie orale. Ilexiste pour ce composé deux indica-tions supplémentaires : dans le cas decystinurie avec tendances à la forma-tion de calculs de cystine dans les voiesurinaires basses, elle inhibe la forma-tion de cystine en formant avec la cys-téine un disulfide très soluble ; dans lecas de polyarthrite elle peut être utiliséecomme traitement de fond. Le fait quela D-pénicillamine réagisse avec les al-déhydes et inhibe ainsi la polymérisa-tion du collagène, peut être une des ex-plications de son action thérapeutique.Les effets secondaires sont des lésionsde la peau (entre autres diminution de larésistance mécanique avec une ten-dance à la formation de vergetures), lé-sions rénales, dépression médullaire etaltérations du goût.
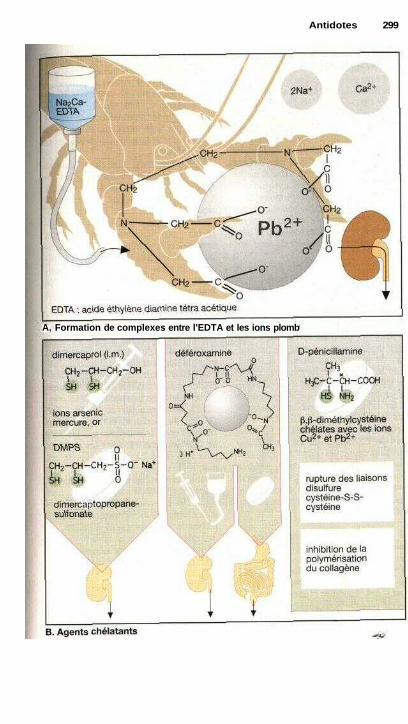
Antidotes 299
A. Formation de complexes entre l'EDTA et les ions plomb

300 Antidotes
Antidotes contre l'empoisonnementpar le cyanure (A). Les ions cyanure(CN-) parviennent essentiellement dansl'organisme sous forme d'acide ; celui-ci peut être inspire, se former dans lesuc gastrique acide à partir de sels decyanure ou être libéré dans le tractusgastro-intestinal à partir d'amandesamères. 50 mg seulement de cyanurepeuvent être mortels. Les ions CN- selient avec une très forte affinité au fertnvalent. Au niveau des cytochromesoxydases de la chaîne respiratoire, cettefixation bloque l'utilisation d'oxygènece qui provoque une asphyxie interneavec des érythrocytes chargés d'oxy-gène (couleur rouge claire du sang vei-neux).
Des petites quantités de cyanurepeuvent être transformées dans l'orga-nisme en thiocyanate relativement peutoxique sous l'action de la « rhodanidesynthétase » (thiosulfate-sulfotransfé-rase, déjà présente dans le foie). Lespossibilités de traitement sont : l'ad-ministration intraveineuse de thiosul-fate de sodium pour stimuler la forma-tion de thiocyanate. La mise en route dece traitement est lente et c'est pourquoile traitement de choix est l'injection i.v.d'un inducteur de methémoglobine lediméthylaminophénol (DMAP), quipeut convertir rapidement le fer diva-lent de l'hémoglobine en fer tri valentqui peut capter les ions CN-. L'hydro-xycohalamine est également un trèsbon antidote, car l'ion CN- s'associeavec une forte affinité sur son atome decobalt central formam la cyanocobala-mine. /
Chlorure de tolonium (bleu detoluidine). Si le fer de l'hémoglobineest sous forme trivalente, on obtient lamethémoglobine de couleur brune quine permet pas le transport d'oxygène.Dans des conditions normales, il seforme presque constamment de la me-thémoglobine qui est cependant réduitesous l'action de la glucose 6-phosphate
déshydrogénase. Les substances qui sti-mulent la formation de methémoglo-bine (B), peuvent cependant provoquerdans l'organisme une carence mortelleen oxygène. Le chlorure de toloniumest un colorant oxydoréducteur, qui estinjecté par voie intraveineuse et trans-forme le fer de la methémoglobine en saforme réduite.
L'obidoxime est un antidotecontre un empoisonnement par les in-secticides organophosphorés (p. 102).La phosphorylation de l'acétylcholi-nestérase conduit à une inhibition irré-versible et à une surcharge de l'orga-nisme en neurotransmetteur. Lesconséquences possibles sont une stimu-lation anormale des effets sympathomi-métiques ainsi qu'un blocage ganglion-naire et une altération de latransmission neuromusculaire avec unblocage respiratoire périphérique.
Les bases du traitement sont :1. protection des récepteurs muscari-niques par l'atropine à forte dose, et2. réactivation de l'acétylcholinestéraseempoisonnée par l'obidoxime qui s'as-socie à l'enzyme, capte le résidu phos-phate, se solubilise et débarrasse ainsil'enzyme de son inhibiteur.
L'hexacyanoferrate de fer(« bleu de Berlin ») est un antidotecontre l'empoisonnement par les selsde thallium (par ex. dans les poisonspour rongeurs). Les symptômes de cetempoisonnement sont d'abord destroubles intestinaux puis des dom-mages nerveux et cérébraux et unechute des cheveux. Les ions thalliumsont excrétés dans l'intestin mais réab-sorbés de nouveau. Le bleu de Berlin,colloïde insoluble non absorbable lieles ions thallium. Il sera administré parvoie orale pour bloquer l'absorptiondes ions thallium immédiatementaprès la prise du poison ou pour capterles ions thallium déversés dans l'in-testin lors d'une surcharge en thalliumdéjà établie et permettre ainsi leur éli-mination.
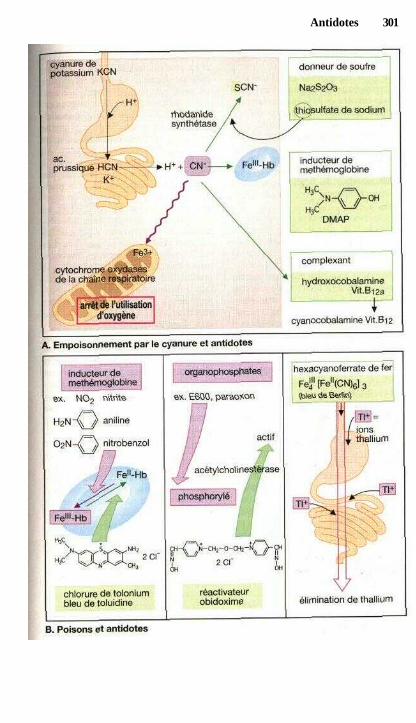
Antidotes 301

302 Traitements de maladies particulières
Angine de poitrine
L'attaque douloureuse lors d'une crised'angine de poitrine indique l'existenced'une carence en oxygène au niveau dumuscle cardiaque. Le manque d'oxy-gène est en général la conséquenced'une irrigation sanguine insuffisante(ischémie) due à un rétrécissement desartères coronaires. Celui-ci est dû :
- principalement à une altérationathéromateuse de la paroi vasculaire(coronarosclérose avec angine d'ef-fort),
- très rarement à un rétrécisse-ment de type spasme dans une artèrecoronaire morphologiquement saine(spasme coronaire avec angine surve-nant au repos),
- plus fréquemment à un spasmecoronaire dans une portion de vaisseauprésentant des lésions athéromateuses.
Le but du traitement est d'empê-cher l'état de carence en oxygène etégalement d'augmenter l'irrigation san-guine (apport en oxygène) ou de dimi-nuer le besoin en oxygène.
Paramètres gouvernant l'ap-port en oxygène. La force motrice duflux sanguin est la différence de pres-sion entre le début des artères coro-naires (pression aortique) et l'embou-chure des veines coronaires (pressiondans l'oreillette droite). Une résistances'Oppose à Y écoulement du sang. Ellese compose de trois paramètres.1. En temps normal, le diamètre desgros vaisseaux coronaires est suffisam-ment important pour qu'ils ne partici-pent pas de façon notable à la résistanceà l'écoulement. En cas d'athérome oude spasme.coronarien, c'est à ce niveauque réside l'obstacle pathologique àl'écoulement. L'artériosclérose corona-rienne, fréquente, ne peut pas être in-fluencée par des moyens pharmacolo-giques, le spasme coronaire, plus rare,peut être éliminé par des vasodilata-teurs convenables (nitrate, nifédipine).2. Le diamètre des vaisseaux résistifsartériolaires régule l'irrigation san-guine dans le lit vasculaire coronarien.Le diamètre des artérioles est fixé en
fonction du contenu du myocarde en 0,et en produits métaboliques et s'ajusteautomatiquement au débit nécessaire(B, sujet bien portant). Cette autorégu-lation métabolique du débit sanguin ex-plique pourquoi, dans le cas d'une athé-rosclérose coronaire, la crise d'anginede poitrine se produit d'abord au coursd'un effort (B, chez un malade). Aurepos, la résistance pathologique àl'écoulement sera compensée par unediminution correspondante de la résis-tance artériolaire : l'irrigation du myo-carde est suffisante. En cas d'effort, unélargissement supplémentaire des arté-rioles n'est plus possible, le débit est in-suffisant et la douleur se manifeste. Lesmédicaments qui dilatent les artériolesne présentent pas d'intérêt : au repos, seproduit dans la zone du territoire vascu-laire sain une stase sanguine (steal ef-fect) liée à une dilatation artériolairesuperflue, ce qui peut déclencher unecrise d'angine de poitrine.3. La pression interne des tissus, la ten-sion des parois, dépend des capillaires.Pendant la contraction systolique desmuscles, on aboutit à un arrêt du fluxsanguin ; celui-ci se produit principale-ment pendant la diastole. La tension desparois pendant la diastole (pré-charge)dépend de la pression et du volumeavec lequel le ventricule sera rempli.Les nitrates abaissent cette composantede la résistance à l'écoulement en dimi-nuant l'apport de sang au cœur.
Paramètres gouvernant les be-soins en oxygène. Le muscle cardiaqueutilise la majeure partie de son énergiepour la contraction. Le besoin en oxy-gène augmente en même temps que :1. ^fréquence cardiaque, 1. la vitessede contraction, 3. la tension de la paroidéveloppée pendant la systole (post-charge) ; celle-ci dépend du volume deremplissage du ventricule et de la pres-sion qui doit être atteinte durant la sys-tole. Avec une augmentation de la résis-tance périphérique, la pression aortiqueaugmente et par la même la résistance àl'éjection. Les (3-bloquants, les antago-nistes calciques ainsi que les nitrates(p. 304) diminuent les besoins en oxygène.
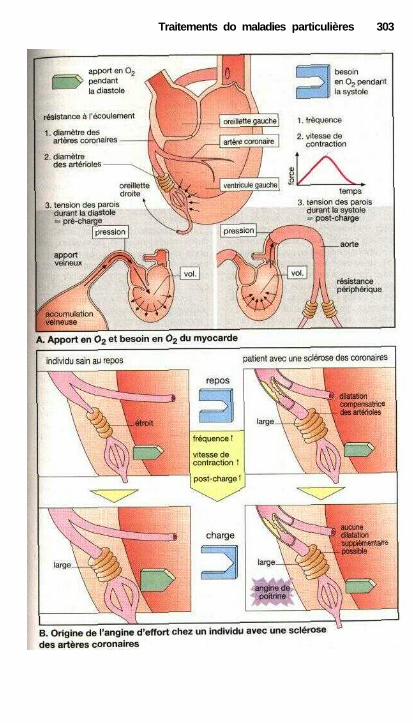
Traitements do maladies particulières 303

304 Traitements de maladies particulières
Anti-angineux
Les substances appartenant aux troisgroupes dont les propriétés pharma-cologiques ont déjà été présentées en dé-tail dans d'autres pages peuvent être utili-sées comme anti-angineux : ce sont lesnitrates organiques (p. 120), les antago-nistes calciques (p. 122), et lesP-bloquants (p. 92 et suivantes).
Les nitrates organiques (A) aug-mentent le débit sanguin ou l'apporten oxygène. Grâce à la diminution del'apport de sang veineux au cœur, la ten-sion de la paroi pendant la diastole (pré-charge) diminue. Ainsi réussit-on, en uti-lisant les nitrates, à diminuer la résistanceà l'écoulement même en cas d'une anginede poitrine due à une athérosclérose coro-naire. En cas d'angine de poitrine avecspasme coronaire, l'action vasodilatatricesur les artères entraîne une disparition duspasme et une normalisation du débit. Lebesoin en oxygène décroît à cause de ladiminution des deux paramètres qui gou-vernent la tension systolique (post-charge) : le volume de remplissage duventricule et la pression dans l'aorte.
Antagonistes calciques (B). Ils ré-duisent le besoin en oxygène en dimi-nuant la pression aortique, qui est l'un descomposants de la post-charge.
La nifédipine, une dihydropyridine,n'a pratiquement aucun effet cardiodé-presseur : elle peut provoquer une tachy-cardie réflexe avec une augmentation dubesoin en oxygène. Les, susbtances am-phiphiles cationiques, vérapamil et dil-tiazem sont cardiodépressives. La diminu-tion de la fréquence cardiaque et de laforce de contraction entraîne d'un côté laréduction du besoin en oxygène maispeut, d'un autre côté, altérer de façon dan-gereuse la fonction cardiaque par une bra-dycardie, un bloc AV ou une insuffisancede contraction. Dans les angines coro-naires spastiques, les antagonistes cal-ciques peuvent abolir le spasme et amé-liorer le débit sanguin.
P-Bloquants (C). Ils protègent lecœur contre une stimulation sympathiqueconsommant de l'oxygène en bloquantune augmentation de fréquence ou de vi-tesse de contraction médiée par les récep-teurs.
Utilisation des anti-angineux (D)Les substances qui ne sont pas cardiodé^pressives et qui peuvent être prises rapi-dement servent au traitement des crisesLe moyen de choix est la nitroglycérine(NTG, 0,8-2,4 mg en sublingual ; débutde l'action en 1 à 2 minutes, durée en-viron 30 min.). Le dinitrate d'isosorbide(DNI) peut être également utilisé (5 à10 mg, sublingal) ; en comparaison de laNTG, son action est un peu retardée maisdure plus longtemps. Finalement, la nifé-dipine peut également convenir (5 à20 mg, en cassant la capsule et en avalantson contenu).
Les nitrates conviennent, sous cer-taines conditions, à la prévention desaccès tout au long de la journée ; ainsipour éviter le développement d'une ac-coutumance aux nitrates, il paraît judi-cieux d'instituer une pause d'environ12 heures dans l'administration. Pourpouvoir prévenir durant toute la duréel'apparition d'une crise, on peut donner lematin et à midi par exemple du DNI (parex. 60 mg sous forme retard) ou son méta-bolite le mononitrate d'isosorbide. Àcause de son élimination présystémiquedans le foie, la NTG convient peu pourune administration orale. L'apportcontinu de NTG au moyen d'un emplâtrecutané n'apparaît pas, non plus, réelle-ment recommandable à cause du dévelop-pement d'une accoutumance. Dans le casde la nwlsidomine, le risque d'une accou-tumance est nettement plus faible, maiselle présente des limitations d'emploi.
Lors du choix d'un antagoniste cal-cique, il faut faire attention aux effets dif-férents de la nifédipine ou du vérapamil etdu diltiazem sur les performances car-diaques (voir ci-dessus).
Lorsque l'on donne un ^-bloquant,il faut également penser à la limitation desperformances cardiaques qui découle del'inhibition du sympathique. À cause dublocage des récepteurs (i;, vasodilata-teurs, on ne peut pas exclure la possibilitéqu'un vasospasme puisse se produire plusfacilement. Une monothérapie par les (3-bloquants ne sera recommandée que dansles cas de sclérose coronaire mais pasdans les angines spastiques.
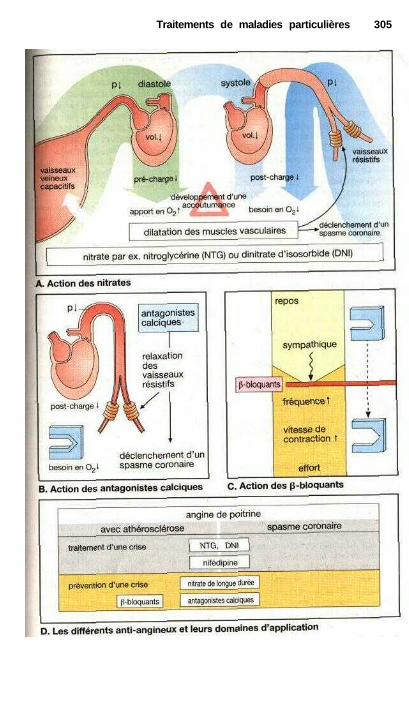
Traitements de maladies particulières 305

306 Traitements de maladies particulières
Hypertension et antihypertenseurs
Une hypertension artérielle (pression san-guine élevée) ne modifie en général pasl'état de santé des patients atteints maisprovoque cependant à long terme des lé-sions vasculaires et des maladies asso-ciées (A). Le traitement antihypertenseura pour but d'éviter le développement deces lésions et donc de normaliser l'espé-rance de vie.
L'hypertension est rarement laconséquence d'une autre maladie(exemple : tumeur sécrétant des catécho-lamines, phéochromocytomes) ; dans laplupart des cas son origine n'est pas dé-tectable : hypertension essentielle. Si onne parvient pas à l'aide d'une réductiondu poids et d'une alimentation pauvreen sel à une réduction suffisante de la ten-sion, il faut alors utiliser des anti-hyper-tenseurs. En principe, une diminution dudébit cardiaque ou des résistances péri-phériques peut conduire à une diminutionde la pression sanguine (p. 308, para-mètres gouvernant la pression sanguine).Différentes substances agissent sur l'unde ces paramètres ou sur les deux. Pourarriver à un schéma thérapeutique conve-nable, on tiendra compte de l'efficacité etde la tolérance des produits. Le choixd'une substance donnée est pris sur labase d'une réflexion concernant le rapportrisque/efficacité des différentes substancesutilisables et en tenant compte du cas dechaque patient.
Pour les substances .utilisées en mo-nothérapie, il faut par exemple envisagerles P-bloquants (p. 92), us sont tout à faitrecommandés en cas d'hypertension chezun adulte jeune avec une tachycardie et undébit cardiaque élevé ; en cas de tendanceau bronchospasme, les P-bloquants mêmecardiosélectifs (pi) sont contre-indiqués.Les diurétiques thiazidiques (p. 160)conviendront bien dans le cas d'une hy-pertension associée à une insuffisancecardiaque mais ne seront pas adaptés aucas d'une tendance à l'hypokaliémie. S'ilexiste à côté de l'hypertension une anginede poitrine, le choix tombera plutôt sur undiurétique que sur un p-bloquant ou unantagoniste calcique (p. 122). Dans le casdes antagonistes calciques, il faut souli-
gner que le vérapamil, au contraire de lanifédipine présente des propriétés cardio-dépressives. Il faudra penser à un ai-blo-quant en paticulier chez des patients ayantune hyperplasie bénigne de la prostate etdes difficultés de miction. Il faut noterque jusqu'à présent, c'est seulement dansle cas des P-bloquants et des diurétiquesqu'ont été entreprises des études degrande ampleur qui ont montré une rela-tion entre la baisse de la pression artérielleet une diminution de la morbidité et de lamortalité.
En traitement combiné, il faut sur-tout définir quels sont les produits qui secomplètent de façon judicieuse. En asso-ciation avec un p-bloquant (bradycardie,cardiodépression par blocage sympa-thique) la nifédipine convient bien (tachy-cardie réflexe), mais un autre antagonistecalcique, le vérapamil (bradycardie, car-diodépression) n'est pas adapté. Une mo-nothérapie avec les inhibiteurs de l'en-zyme de conversion (p. 124) conduit chezenviron 50 % des patients à une diminu-tion suffisante de la pression artérielle ; encombinaison avec un diurétique (thiazi-dique, p. 156) ce pourcentage atteint90 %. Lors de l'administration d'un vaso-dilatateur, la dihydralazine ou le mi-noxidil (p. 118), les P-bloquants servent àempêcher une tachycardie réflexe, lesdiurétiques inhibent la rétention de fluide.
L'arrêt brutal d'un traitementcontinu peut entraîner une élévation de lapression sanguine à un niveau supérieur àcelui de la pression avant traitement.
Médicaments pour le traitementd'une crise hypertensive. Ce sont la ni-fédipine (capsules cassées entre les dentsou avalées) la nitroglycérine (sublin-guale), la clonidine (per os ou i.v., p. 96)la dihydralazine (i.v.) le diazoxid (i.v.,p. 118), le nitroprussiate de sodium (per-fusion, p. 120). C'est seulement encas de phéochromocytome qu'est indiquél'a-bloquant non sélectif phentolamine(P. 90).
Antihypertenseurs utilisés durantla grossesse. Ce sont les p-bloquants car-diosélectifs (Pi), l'a-méthyl-DOPA (p. 96),en cas d'éclampsie (élévation massive de lapression artérielle avec symptômes cen-traux) la dihydralazine (en perfusion i.v.).
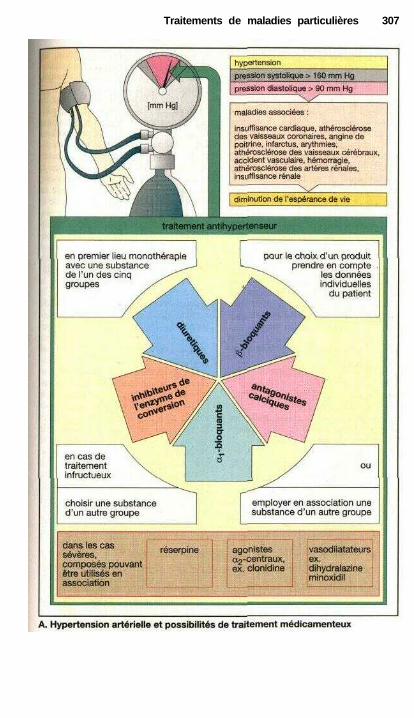
Traitements de maladies particulières 307

308 Traitements de maladies particulières
Différentes formes d'hypotensionet leur traitement médicamenteux
85 % du volume sanguin sont localisésdans le système vasculaire veineux ; àcause de la faible pression qui y règne(pression moyenne environ 15 mmHg),on parle de système basse pression. Les15 % restants remplissent le lit artérielque l'on nomme système haute pression àcause de la pression élevée (environ100 mmHg). La pression sanguine dans lesystème artériel est la force motrice pourl'irrigation des organes et des tissus. Lesang déversé par ce système s'accumuledans le système basse pression et est re-pompé par le cœur dans le système hautepression.
La pression artérielle (en abrégéPA) dépend : 1. de la quantité de sang« injectée » par le cœur dans le systèmehaute pression, par unité de temps. Ledébit cardiaque est fonction du volumed'éjection c'est-à-dire du volume sanguinpropulsé à chaque battement cardiaque, etde la fréquence cardiaque ; le volumed'éjection est entre autres conditionné parle retour veineux,2. de la résistance contre laquelle l'écou-lement du sang doit lutter, c'est-à-dire dela résistance périphérique ou de l'étroi-tesse des artérioles.
Baisse prolongée de la pression ar-térielle (PA syst. demeurant < 105 mmHg).L'hypotonie essentielle primaire n'adans la plupart des cas aucun caractèremaladif. Si des symptômes tels que fa-tigue et étourdissements surviennent, ondoit recommander un entraînement dusystème circulatoire plutôt-que des médi-caments. (
L' hypotention secondaire est laconséquence d'une maladie sous-jacenteet c'est elle qu'il convient de traiter. Si levolume d'éjection est faible par suited'une insuffisance cardiaque, un glyco-side cardiaque pourra augmenter la forcede contraction et le volume d'éjection. Sila diminution du volume d'éjection est laconséquence d'un volume sanguin insuf-fisant, on pourra y remédier en cas deperte de sang par une solution remplaçantle plasma, en cas de carence en aldosté-rone par l'administration d'un minéralo-
corticoïde. En cas de bradycardie, unagent parasympatholytique (ou un stimu-lateur cardiaque) pourra stimuler la fré-quence cardiaque.
Accès d'hypotension. Troubles dela régulation orthostatique. Lors du pas-sage de la position couchée à la positiondebout (orthostase), le sang présent dansle système basse pression s'écoule en di-rection des pieds, parce que sous le poidsde la colonne de sang les veines de lamoitié inférieure du corps s'élargissent.La chute du volume d'éjection est enpartie compensée par une élévation de lafréquence cardiaque. La diminution res-tante du débit cardiaque peut être équili-brée par une élévation des résistances pé-riphériques, de sorte que la pressionartérielle et l'irrigation sanguine soientmaintenues. Une altération de la régula-tion orthostatique se produit lorsque lacontre-régulation n'est pas suffisante : lapression sanguine chute, l'irrigation ducerveau décroît et apparaissent en consé-quence des malaises tels des étourdisse-ments, « tout devient noir devant lesyeux », ou même des pertes deconscience. Dans la forme sympathoto-nique, les réflexes sympathiques agissentde façon accrue (augmentation plus im-portante de la fréquence cardiaque et de larésistance périphérique, c'est-à-dire de laPA diast.) et ne peuvent donc compenserla réduction de l'apport veineux. Entermes de prévention, l'utilisation desympathomimétiques ne présente doncque peu d'intérêt. L'important seraitd'abord un entraînement du systèmecardio-vasculaire. Par voie médicamen-teuse, l'augmentation de l'apport veineuxest possible de deux façons. Une augmen-tation de l'apport de sel de cuisine accroîtles réserves d'eau et de sel et par la mêmele volume sanguin (contre-indications :par exemple hypertension et insuffisancecardiaque). Une constriction des vais-seaux veineux capacitifs peut être déclen-chée par la dihydroergotamine. Il reste àdéterminer dans quelle mesure cet effet nepeut pas également être atteint sur le planthérapeutique par un ci-sympathomimé-tique. Dans la forme asympathotonique,très rare, les sympathomimétiques sontpar contre certainement recommandés.
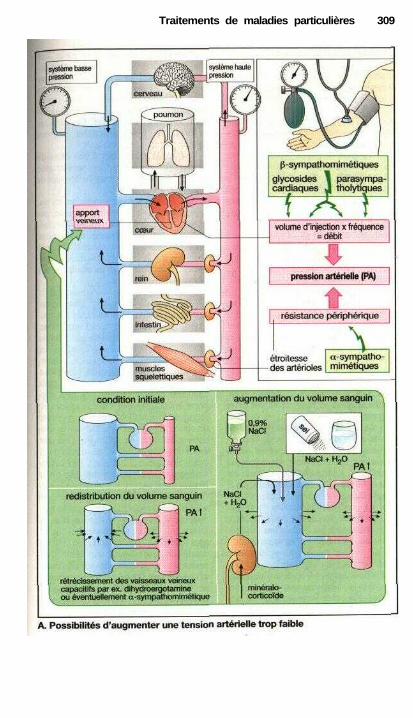
Traitements de maladies particulières 309

310 Traitements de maladies particulières
La goutte et son traitement
L'origine de la goutte, une maladie méta-bolique, est une élévation de la concentra-tion sanguine en acide urique, le produitde dégradation des purines (hyperuri-cémie). Par accès, se produit une précipi-tation de cristaux d'urate de sodium dansles tissus.
La crise de goutte typique consisteen une inflammation très douloureusedu gros orteil et des articulations de lacheville. L'inflammation se développed'abord à cause des efforts de l'organismepour se débarraser des cristaux par phago-cytose (1-4). Les granulocytes neutro-philes enveloppent les cristaux grâce àleurs mouvements amiboïdes et les cap-turent (2). La vacuole de phagocytosefusionne avec un lysosome (3). Les en-zymes lysosomiales ne peuvent cepen-dant pas détruire l'urate de sodium. Si lescristaux se déplacent au cours de mouve-ments amiboïdes ultérieurs, la membranedes phagolysosomes se rompt. Les en-zymes se répandent dans le granulocyte,le détruisent et lèsent le tissu environnant.Des médiateurs inflammatoires commepar exemple les prostaglandines sont li-bérés (4). Des granulocytes attires s'accu-mulent et périssent de la même façon.L'inflammation se renforce et une crisede goutte se déclenche.
Le but du traitement de la crise degoutte est d'interrompre la réaction in-flammatoire. Le remède de choix est lacolchicine, un alcaloïde de la colchique(Colchicum automnale). Ce composé estconnu comme un poison du fuseau, car ilbloque les mitoses en métaphase en inhi-bant les protéines contractiles du fuseauachromatique. Son action dans les crisesde goutte repose sur l'inhibition des pro-téines contractiles dans les neutrophiles,ce qui bloque leurs mouvements ami-boïdes et donc la phagocytose. Les effetssecondaires les plus fréquents d'un traitê7ment par la colchicine sont des douleursabdominales, des vomissements et desdiarrhées, correspondant tout à fait à l'in-hibition des mitoses dans l'épithélium del'estomac et de l'intestin, qui se divisetrès rapidement en temps normal. La col-chicine est principalement administrée
par voie orale (0,5 mg/h par ex. jusqu'à ceque les douleurs cèdent ou qu'apparais-sent des troubles gastro-intestinaux ; dosemaximale 10 mg). Une crise de gouttepeut également être traitée avec des anti-inflammatoires tels que ex. l'indométa-ciné ou la phénylbutazone. Dans les cassévères, les glucocorticoïdes peuventégalement être prescrits.
Pour la prévention d'une crise degoutte, il faut ramener la concentrationd'acide urique dans le sang en dessous de6 mg/100 ml.
Régime : les aliments riches en pu-rine (noyaux cellulaires) sont à éviter, parexemple les abats. Le lait, les produits lai-tiers et les œufs sont pauvres en purine etsont recommandés. Le café et le thé sontautorisés car la caféine, une méthylxan-thine, ne participe pas au métabohsme despurines.
Uricostatiques : ils diminuent laproduction d'acide urique. L'allopurinolet son métabolite, l'alloxanthine (oxypu-rinol), qui s'accumule dans l'organisme,inhibent la xanthine oxydase qui catalysela transformation de l'hypoxanthine enxanthine puis en acide urique. Ces précur-seurs sont facilement éliminés par le rein.L'allopurinol est administré par voie orale(300-800 mg/jour). Il est très bien sup-porté à l'exception de rares réactions al-lergiques et constitue le moyen préventifde choix. Au début du traitement se pro-duisent des crises de goutte que l'on peutéviter en donnant en même temps de lacolchicine (0,5-1,5 mg/jour). Les urico-suriques comme le probénécide ou lahenzbromarone (100 mg/jour) ou la sul-finpyrazone stimulent l'élimination ré-nale d'acide urique. Ils occupent le sys-tème de réabsorption des acides dans letubule proximal de sorte que celui-ci n'estplus disponible pour le transport d'acideurique. En cas de dosage trop faible, c'estseulement le système de sécrétion desacides qui sera inhibé car il a une activitéde transport plus faible ; l'éliminationd'acide urique est alors interrompue etune crise de goutte est possible. Chez lespatients avec des calculs dans les voiesurinaires, les uricosuriques sont contre-indiqués.
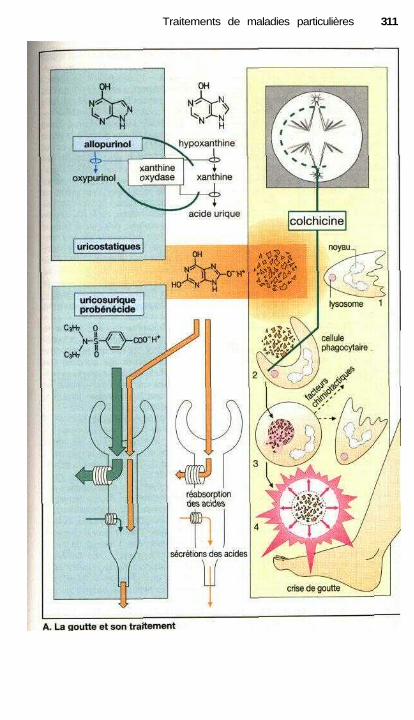
Traitements de maladies particulières 311

312 Traitements de maladies particulières
Ostéoporose
L'ostéoporose correspond à une diminu-tion de la niasse osseuse (« fonteosseuse »), qui touche de la même ma-nière la trame de l'os et les substances mi-nérales. On aboutit à un tassement desvertèbres avec des douleurs osseuses, undos rond et un raccourcissement du tronc.Le col du fémur et le radius distal sont fré-quemment atteints par des fractures. Labase de cette fonte osseuse est un déplace-ment de l'équilibre entre synthèse osseusepar les ostéoblastes et dégradation os-seuse par les ostéoclates, en direction dela dégradation. Classification : ostéopo-rose idiopathique : type 1 : chez lesfemmes atteignant la ménopause ; type 2 :chez les hommes et les femmes vers70 ans. Ostéoporose secondaire commeconséquence de maladies sous-jacentes(maladie de Cushing par ex.) ou provo-quées par des médicaments (par ex gluco-corticoïdes ou traitement chronique àl'héparine). Dans ce cas, l'origine peut enêtre éliminée.
Ostéoporose post-ménopause.Après la ménopause se déclenche unepoussée de dégradations. Plus la masseosseuse de départ est faible et plus tôt onatteindra une proportion de perte osseusequi déclenchera les douleurs.
Les facteurs de risques sont : uneménopause précoce, une activité physiqueinsuffisante, le tabagisme ou l'abusd'alcool, un poids insuffisant ou unenourriture pauvre en calcium.
Prévention. La poussée de dégrada-tions osseuses après la ménopause peutêtre empêchée par l'administration d'oes-trogènes. On utilisera souvent des œstro-gènes conjugués (p. 252)^CoîBrne le trai-tement par les œstrogènes seuls augmentele risque d'un cancer de\l'endomètre, ondoit administrer en même temps des pro-gestatifs comme par ex. dans le cas d'unecontraception orale combinée (exception,après une hystérectomie). Durant ce trai-tement, les règles sont maintenues. A ladifférence de ce que l'on observe pour lacontraception orale, le risque de troublesthromboemboliques n'est pas augmentémais plutôt diminué. L'apport hormonalpeut se poursuivre pendant 10 ans et plus.
L'apport quotidien de calcium doit repré-senter 1 g/jour avant la ménopause (cor-respondant à ~ 1 1 de lait) et 1,5 g après.
Traitement. La néosynthèse de l'ossera induite par des fluorures administréspar exemple sous forme de fluorure de so-dium. Il stimule les ostéoblastes. Dansl'hydroxyapatite, il sera inséré à la placedu groupement hydroxyle (p. 263), ce quirend plus difficile la dégradation par lesostéoclastes. Pour garantir la minéralisa-tion de l'ostéoïde nouvellement synthé-tisé il faut se préoccuper d'un apport encalcium suffisant mais cela ne doit pasêtre fait en même temps, car le fluorure decalcium non absorbé, précipite déjà dansl'intestin. Cette difficulté n'est pas ren-contrée lorsque le fluorure est administrésous forme de monofluorophosphate desodium. Comme on ne sait pas encoreavec certitude dans quelles conditions latendance aux fractures décroît, l'adminis-tration de fluorure n'est pas encore untraitement de routine.
La calcitonine (p. 262) inhibe lefonctionnement des ostéoclastes et la dé-gradation de l'os. En tant que peptide elledoit être administrée par injection (ouégalement via la muqueuse nasale en pul-vérisation). La calcitonine de saumon estplus active que la calcitonine humaine,car elle est éliminée plus lentement.
Les biphosphonates ont une struc-ture voisine de celle d'un composant del'organisme, le pyrophosphate, qui inhibela dissolution et la perte de la substanceminérale des os. Ils ralentissent la dégra-dation des os par les ostéoclastes, maiségalement en partie la minéralisation os-seuse. Les indications de ces composéssont : la dégradation osseuse provoquéepar une tumeur, l'hypercalcémie, la ma-ladie de Paget. Lors d'études cliniquesavec Yétidronate dans l'optique du traite-ment de l'ostéoporose, cette moléculeétait administrée par phases alternant avecdes plages d'arrêt. Pour les molécules in-troduites ultérieurement comme le clo-dronate, le pamidronate et égalementl'alendronate l'inhibition des ostéoclastesest l'action majeure, ce qui permet dans letraitement de l'ostéoporose une adminis-tration continue.
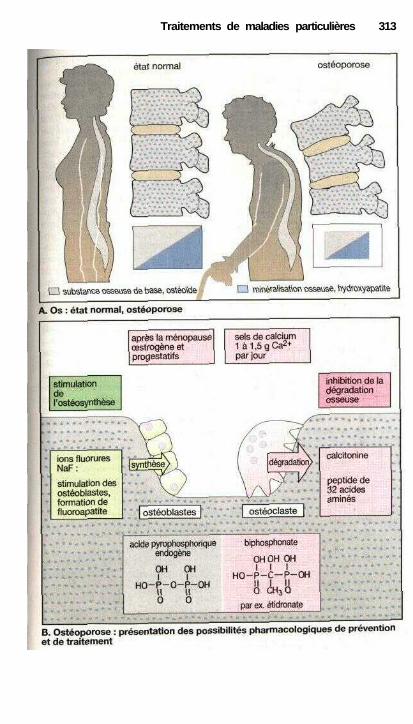
Traitements de maladies particulières 313

314 Traitements de maladies particulières
Polyarthrite rhumatoïdeet son traitement
L'arthrite rhumatoïde ou polyar-thrite chronique est une maladie in-flammatoire évolutive des articulationsqui atteint par poussées successives lesarticulations, principalement les petitesarticulations des doigts et des pieds. Lapolyarthrite rhumatoïde a vraisembla-blement pour base une réaction anor-male du système immunitaire. La reac-tion erronée peut être favorisée etdéclenchée par différentes conditions(par exemple dispositions génétiques,usure due à l'âge, refroidissement, in-fection). L'élément nuisible conduit àune inflammation de la membrane sy-noviale (membrane tapissant les articu-lations), dont la conséquence est la libé-ration d'un antigène qui entretient lephénomène inflammatoire. L'inflam-mation de la membrane synoviale s'ac-compagne de la libération de média-teurs de l'inflammation, qui stimulentpar chimiotactisme un afflux (diapé-dèse) de cellules sanguines phagocy-taires (granulocytes, macrophages)dans le tissu synovial. Ces cellules pha-gocytaires libèrent des enzymes des-tructrices qui participent à la destruc-tion des tissus. L'inflammation s'étend,entre autres, à cause de la formation deprostaglandines et de leucotriènes(p. 194) à l'ensemble de l'articulation.On aboutit à une lésion du cartilage etfinalement à une destruction et un rai-dissement de l'articulation.
Traitement pharmacologique :les symptômes de l'inflammation peu-vent être soulagés de façon aiguë pardes inhibiteurs de synthèse des pros-taglandinesf (p. 198 ; anti-inflamma-toires non stéroidiens, AINS, commepar exemple le diclofénac, l'indométa-cine, le piroxicam) et par les glucocor-ticoïdes (p. 246 et suivantes). Lorsd'une administration nécessairementchronique peuvent se manifester lese f f e t s secondaires des AINS (p. 198 et246). La progression de la destruction
des articulations n'est arrêtée ni par lesAINS ni par les glucocorticoïdes.
L'utilisation de traitement defond peut conduire à une diminutiondes besoins en AINS. Le terme de trai-tement de fond ne signifie pas qu'il estpossible d'atteindre les mécanismes pa-thogènes de base. Bien plus, sur la based'un traitement avec ces composés,l'utilisation de molécules agissant rapi-dement est non seulement possible maispeut se révéler nécessaire. Les traite-ments de fond ont en commun une ins-tallation lente de leur action après plu-sieurs semaines de traitement. Parmiles modes d'action envisagés, on a pro-posé une inhibition de l'activité des ma-crophages et de la libération d'enzymeslysosomiales. Parmi les traitements defond on trouve : la sulfasalazine (inhi-biteur de lipooxygénases ?, p. 270) ; lamésalazine est en générale insuffisantepour cette indication ; la chloroquine(accumulation lysosomiale) et les selsd'or (accumulation lysosomiale ; i.m. :aurothioglucose, aurothiomalate ; moinsactif p.o. : auranofine) ainsi que la D-pénicillainine (complexation de ca-tions métalliques indispensables à l'ac-tivité d'enzymes, p. 298). Les effetssecondaires fréquents sont : altérationde la peau et des muqueuses, atteintesdes fonctions rénales, modifications dela formule sanguine. Certains agentscytostatiques et immunosuppresseurs,azatliioprinc, cyclophosphamide eten particulier le méthotrexate (à dosefaible une fois par semaine) seront uti-lisés comme traitement de fond. A côtéde son action immunosuppressive leméthotrexate a un effet anti-phlogis-tique et vient après la sulfasalazine pourson rapport risque/efficacité.
L'élimination chirurgicale de lasynovie enflammée (synovectomie)procure souvent aux patients des phasesplus longues sans souffrir. Lorsqu'elleest réalisable, elle est entreprise, cartous les moyens pharmacologiques sontassociés à des effets secondaires impor-tants.
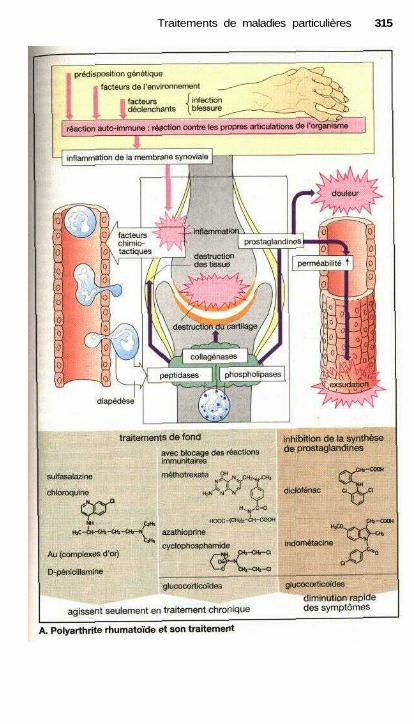
Traitements de maladies particulières 315

316 Traitements de maladies particulières
La migraine et son traitement
Le terme de migraine désigne un tableaudouloureux qui est associé en premierlieu avec de violents maux de tête et desnausées et qui survient par accès de fré-quence irrégulière et d'une durée de plu-sieurs heures. Chez une partie des pa-tients, il existe une « aura » typique quiannonce une crise et peut être décrite parune chute du champ visuel, prenant sou-vent la forme d'une image avec descontours extérieurs crénelés (spectre defortifications) et en dehors de cela parl'incapacité des yeux à se fixer sur desobjets précis, par une hypersensibilitédes organes des sens, par une photo-phobie et une fringale de certains ali-ments. L'origine exacte de ces maux estinconnue. Vraisemblablement la crisemigraineuse a pour origine une libéra-tion de médiateurs inflammatoires àl'extrémité des fibres nociceptives affé-rentes (inflammation neurogène) ou unealtération de la circulation cérébrale. Acôté d'une prédisposition individuelle,un facteur déclenchant de la crise est né-cessaire, par exemple, une forte tensionpsychique, un manque de sommeil. Letraitement pharmacologique a deuxbuts : interruption de la crise et préven-tion d'accès futurs.
Traitement des crises. De façonsymptomatique, les maux de tête seronttraités par des analgésiques (paracé-tamol, acide acétylsalicylique), les nau-sées par le métoclopramide ou la dom-péndone (pp. 144 et 324). Compte tenude l'inhibition de la vidange gastriqueliée à la crise de migraine, l'absorptiondes médicaments peut être ralentie defaçon telle qu'aucune concentrationplasmatique efficace ne puisse être at-teinte. Le métoclopramide qui stimule lavidange^gastrique, augmente l'absorp-tion des substances analgésiques et favo-rise ainsi^Taction des antalgiques. Sil'acide acétylsalicylique est administrépar voie i.v., sa disponibilité est assurée,c'est pourquoi l'administration i.v. estrecommandée en cas de crise migrai-neuse.
Si les antalgiques s'avèrent n'êtrepas suffisamment efficaces, on peut
alors dans la plupart des cas interrompreune crise ou empêcher le déclenchementd'une crise qui s'annonce par l'ergota-mine ou le sumatriptan. Ces deux sub-stances n'agissent qu'en cas de mi-graine et n'ont aucun effet sur d'autresmaux de tête. L'action particulière deces deux substances est vraisemblable-ment liée à leur propriété commune destimuler les récepteurs 5HTio, un sous-type de récepteurs de la sérotonine.L'ergotamine présente également uneaffinité pour les récepteurs de la dopa-mine (-» nausée et vomissements) ainsique pour les récepteurs a-adrénergiqueset 5HT; (-> altérations vasculaires, aug-mentation de l'agrégation plaquettaire).Les effets secondaires vasculaires peu-vent entraîner en cas d'utilisations fré-quentes des altérations circulatoiressévères (ergotisme). De plus, en cas deprise fréquente (> 1 fois par semaine),l'ergotamine peut de façon paradoxaledéclencher elle-même des maux de têtequi, bien que leurs caractéristiquessoient différentes (douleurs perfo-rantes), peuvent conduire le patient àreprendre de l'ergotamine. Il s'installeainsi un cercle vicieux qui risque, aprèsune utilisation chronique et inappro-priée d'analgésiques et d'ergotamine,d'aboutir à des lésions rénales et destroubles circulatoires irréversibles.
L'ergotamine et le sumatriptann'ont qu'une biodisponibilité réduite parvoie orale. La dihydroergotamine peutêtre administrée en injection intramus-culaire ou par injection intraveineuselente, le sumatriptan par voie sous-cutanée.
Prévention des crises. La prise ré-gulière de molécules aussi différentesque le propranolol ou le métroprolol (P-bloquants), la flunarizine (action commeantagoniste de l'histamine et de la dopa-mine et comme anti-calcique), le pizoti-fène (un antagoniste de la sérotoninedont la structure est proche de celle d'unantidépresseur tricyclique) et le méthy-sergide (antagoniste sérotoninergiquepartiel) peut réduire la fréquence descrises de migraine. Le traitement de pre-mière intention est l'un des (î-bloquantscités plus haut.
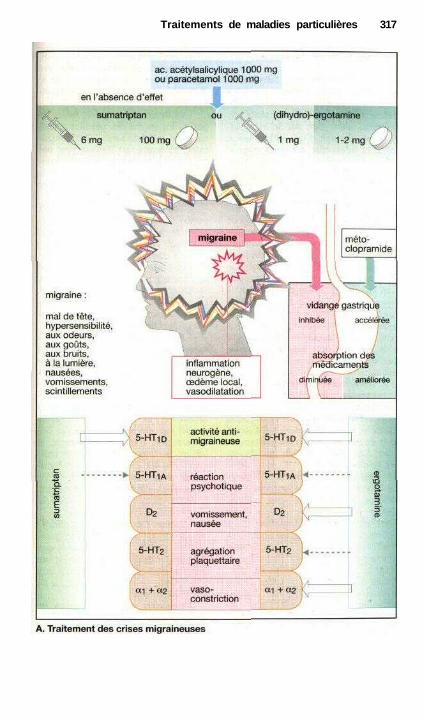
Traitements de maladies particulières 317

318 Traitements de maladies particulières
Traitement des refroidissements
Lorsque l'on parle de refroidisse-ments, en langage courant « coupsde froid », « grippe », « infection grip-pale » (la grippe est au sens strict uneinfection assez rare par le virus in-fluenza), il s'agit d'une inflammationaiguë et infectieuse des voies respira-toires supérieures. Les symptômes,étemuements, coryza (à cause d'unerhinite), enrouement (laryngite), dou-leurs de gorge et difficultés à déglutir(pharyngite, amygdalite), toux avec ca-tarrhe d'abord séreux puis muqueux(trachéite, bronchite), douleurs muscu-laires, fièvre et dégradation de l'état gé-néral peuvent apparaître isolément ouavec diverses combinaisons, simultané-ment ou successivement. La dénomina-tion provient de l'idée répandue autre-fois qu'un refroidissement était àl'origine de ces maux. En général, cettemaladie est provoquée par des virus(rhino, adeno, parainfluenza virus) quisont transportés par des « projections »provenant de la toux ou d'étemue-ments.
Moyens thérapeutiques. Un trai-tement causal avec des antiviraux n'estpas possible à l'heure actuelle. Lessymptômes d'un refroidissement /es-tompent spontanément. L'administra-tion de médicaments n'est pas obliga-toire. Les moyens utilisés adoucissentles symptômes.
Rhume. La production de sécré-tions peut être interrompue par des pa-rasympatholytiques. Il faut s'acco-moder des autres actions de typeatropinique (p. 104 et suivantes). C'estpourquoi les parasympatholytiques sontà peine utilisés ; il est vraisemblable ce-pendant que lors de l'utilisation d'anti-histaminique H, (composant de nom-breux traitements) ce soit leur actionparasympatholytique qui soit utilisée.Administres localement (gouttes na-sales), les a.-sympathomimétiques pro-voquent une vasoconstriction et un dé-gonflement de la muqueuse nasale (ilest à nouveau possible de respirer par lenez) et, de façon secondaire, une dimi-
nution des sécrétions nasales (p. 90).Lors d'une administration régulièrependant une longue période existe ledanger d'une lésion de la muqueuse na-sale (p. 90).
Difficultés à avaler et maux degorge. En suçant des pastilles conte-nant des anesthésiques locaux (benzo-caïne, tétracaïne, p. 206) on peut ob-tenir, mais seulement pendant quelquesinstants, une disparition de ladouleur/gêne. Il faut cependant penserau risque de sensibilisation.
Toux. Étant donné que la touxpermet d'expectorer les sécrétions for-mées et accumulées dans le tractusbronchial au cours d'un refroidisse-ment, l'interruption de ce processusphysiologique n'a de sens que lorsquese manifeste une toux d'irritation (touxsèche, sans production de sécrétions).La codéine et la noscapine (p. 210) blo-quent la toux, en inhibant au niveaucentral le réflexe de toux.
Accumulation de mucosités. Lesexpectorants stimulent l'expectorationdu mucus bronchique en rendant lemucus plus fluide : soit en dégradant lessubstances contenues dans le mucus(mucolytiques comme par ex. la N-acétylcystéine) ou bien en favorisant laproduction de mucus moins épais(bouillottes chaudes). On peut se de-mander, lors d'un refroidissement, siles mucolytiques sont vraiment indi-qués et si les expectorants tels l'am-broxol et la bromhexine changent defaçon efficace la consistance du mucus.L'acétylcystéine est indiquée dans lamucoviscidose.
Fièvre. Les analgésiques antipy-rétiques (acide acétylsalicylique, para-cétamol, p. 196) ne sont indiqués quedans des fortes fièvres. La fièvre est uneréaction naturelle de l'organisme auxinfections et un indicateur commode deleur déroulement.
Douleurs articulaires, maux detête. On peut utiliser les analgésiquesantipyrétiques contre les douleurs arti-culaires ou les maux de tête accompa-gnant un refroidissement.
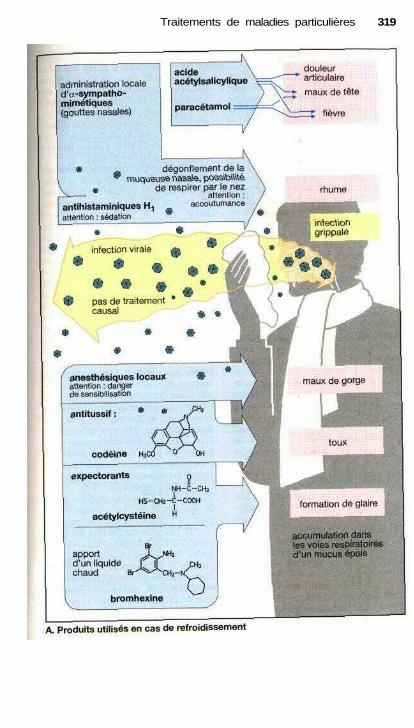
Traitements de maladies particulières 319

320 Traitements de maladies particulières
Traitement anti-allergique
La réaction allergique médiée par les IgE(p. 72) s'accompagne de la libérationd'histamine (p. 114) et de la formationd'autres médiateurs (entre autres les leu-cotriènes p. 194) par les mastocytes. Lesconséquences sont : relaxation desmuscles vasculaires ; la dilatation desvaisseaux entraîne localement une rou-geur, comme par exemple au niveau dutissu conjonctifde l'œil, et de façon systé-mique une chute de la pression artérielle(en cas de choc anaphylactique). Éléva-tion de la perméabilité vasculaire avecpassage de fluide dans les tissus : gonfle-ment du tissu conjonctif au niveau du nez(« rhume des foins ») ou de la muqueusebronchiale ; urticaires cutanées. Contrac-tion de la musculature des bronches avecasthme bronchique. Stimulation de lamusculature de l'intestin avec des diar-rhées.
1. Stabilisation des mastocytes.Le cromoglycate bloque la libération desmédiateurs pas les mastocytes mais sim-plement après une administration chro-nique. Il semble également exercer uneaction inhibitrice dans les réactions in-flammatoires d'origine allergique en in-terférant avec l'effet des médiateurs surles cellules impliquées. Il est administrépar voie locale : œil, muqueuse nasale,arbre bronchique (inhalation)i muqueuseintestinale (voie orale, pratiquement au-cune absorption). Indications : préventiondu rhume des foins, de Y asthme aller-gique et également des allergies alimen-taires. Le nédocromil a la même action.
2. Blocage du récepteur de l'his-tamine. Ce sont principalement les récep-teurs H] qui participent aux réactionsallergiques. Les antihistaminiques H,(p. 114) sont en général administrés parvoie orale. Leur effet thérapeutique estcependant souvent décevant. Indication :rhume des foins.
3. Antagonistes fonctionnels desmédiateurs de l'allergie
a) Les a-sympathomimétiques telsla naphazoline, l'oxymétazoline, la tétry-zoline sont utilisés localement sur les mu-queuses nasales et le tissu conjonctif, ilsagissent en rétrécissant les vaisseaux et, àcause de la diminution du flux sanguin, en
diminuant l'œdème et les sécrétions(p. 90), par ex. dans le rhume des foins.Compte tenu du risque de lésion des mu-queuses ils doivent dans tous les cas êtreadministrés pendant de courtes périodes.
b) Adrénaline : administrée en i.v.elle constitue le traitement le plus efficaceen cas de choc anaphylactique : ellecontracte les vaisseaux, diminue leur per-méabilité et dilate les bronches.
c) Les P;-sympathomimétiques,tels la terbutaline, le fénotérol, le salbu-tamol, sont utilisés dans l'asthme bron-chique ', en général localement, par inha-lation, en cas d'urgence par voieparentérale. Même par inhalation, desquantités non négligeables de produitactif peuvent parvenir dans la circulation(effets secondaires, battements de cœur,tremblement, agitation, hypokaliémie).
d) Théophylline : elle appartientaux méthylxanthines. Tandis que la ca-féine (1,3,7 triméthylxanthine, théine) aprincipalement une action stimulante surle SNC et contracte les vaisseaux céré-braux, la théophylline présente simultané-ment une action notable, bronchodilata-trice et diurétique. Les effets sont dus àl'inhibition d'une phosphodiestérase(augmentation d'AMPc, p. 66) ainsi qu'àune action antagoniste au niveau des ré-cepteurs de l'adénosine. En cas d'asthmebronchique, la théophylline peut êtredonnée par voie orale pour prévenir unecrise, par voie parentérale pour inter-rompre une crise. En cas de surdosage,peuvent se produire des crampes et desarythmies cardiaques.
e) Ipratropium (p. 104) : il peutêtre inhalé pour dilater les bronches en casde bronchoconstriction d'origine aller-gique ; il n'est souvent pas assez puissant.
f) Les glucocorticoïdes (p. 246)agissent de façon très efficace dans le trai-tement des allergies, vraisemblablementparce qu'ils interviennent à différents en-droits dans le processus. Indications :rhume des foins, asthme bronchique (sipossible administration locale de produitsayant une forte élimination présysté-mique, par ex. béclométhasone, budéso-nide, ainsi que choc anaphylactique (i.v. àdose élevée) ; il est probable que se pro-duisent également des effets non géno-miques rapides.
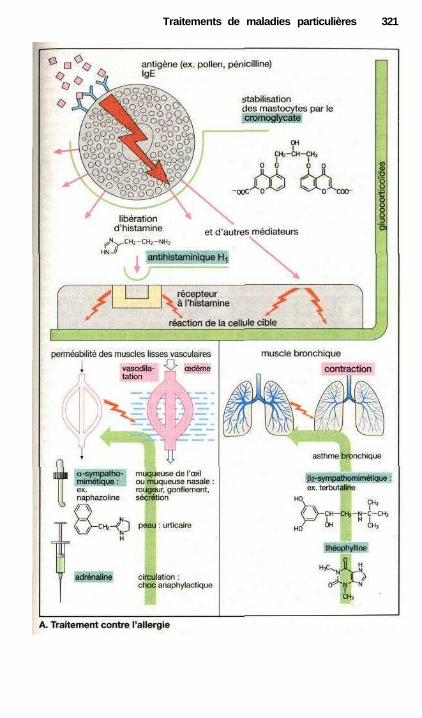
Traitements de maladies particulières 321

322 Traitements de maladies particulières
Asthme
Définition. Blocage respiratoire surve-nant par crises à la suite d'un rétrécisse-ment des bronches lié à une hypersensi-bilité bronchique.
Il n'est pas rare que le patient asth-matique sous-estime le degré de gravitéréelle de sa maladie. Dans ces condi-tions, la mesure de la vitesse maximaled'expiration forcée (peak-flow) par lespatients est un moyen important associéau traitement. Après un apprentissageadapté, le patient peut reagir de lui-même à une modification de l'intensitéde la crise par un changement de médi-cation (dans le cadre d'un plan de trai-tement préalablement établi par le mé-decin).
Pathophysiologie. La maladie estessentiellement due à une inflammationd'origine allergique de la muqueusebronchique. C'est ainsi par exempleque les leucotriènes qui sont synthéti-sées au cours d'une reaction immune àIgE (p. 320) ont un effet chimiotactiquesur les cellules inflammatoires. A l'in-flammation est associée une hypersen-sibilité des bronches envers des stimulispasmogènes. Si bien qu'à côté des an-tigènes, d'autres stimuli peuvent dé-clencher des crises d'asthme (A). Parexemple dans l'asthme d'effort, l'inspi-ration profonde de l'air froid environ-nant est un agent déclenchahtjrnpor-tant. Un exemple de déclenchementprovoqué par un médicament est celuides inhibiteurs de cyclooxygénase(p.198).
Bases de traitement. L'élimi-nation des déclencheurs des crisesd'asthme est un moyen important maispas toujours réalisable. Les médica-ments qui diminuent l'inflammation al-lergique ou atténuent l'hypersensibilitébronchique touchent au centre des évé-nements pathophysiologiques : gluco-corticoïdes et les agents stabilisant lesmastocytes. Les bronchodilatateurs (?,-sympathomimétiques, théophylline etipratropium) agissent de façon sympto-matique.
Le schéma par degré (B) fournitun axe concernant les possibilités d'in-tensification des traitements médicamen-teux en cas d'aggravation de la maladie.
Les médicaments de choix pour letraitement d'une crise d'asthme sont les^-mimétiques à courte durée d'action.Utilisés par inhalation comme le salbu-tamol et le fénotérol. Leur action com-mence quelques minutes après l'inhala-tion et dure de 4 à 6 heures.
S'il est nécessaire d'utiliser les ?;-mimétiques plus de trois fois par se-maine, cela indique une empreinte plusforte de la maladie. On ajoutera alors autraitement un produit anti-inflamma-toire, chez les enfants et les adolescentséventuellement un stabilisateur de mas-tocytes, plus tard un glucocorticoïde.L'utilisation par inhalation doit être ef-fectuée de façon régulière, l'améliora-tion apparaît en l'espace de quelquessemaines. La « crainte de la cortisone »n'est pas fondée dans le cas d'une utili-sation correcte par inhalation de gluco-corticoïdes ayant une élimination pré-systémique élevée (effets secondaireslocaux possibles : muguet buccal, en-rouement). L'apparition d'un muguetpeut être évitée par l'utilisation avant lepetit déjeuner ou le repas du soir. Plusl'utilisation de p^-mimétiques inhalésà la demande est faible et meilleur estle traitement bloquant les réactions in-flammatoires.
Dans les cas sévères il est cepen-dant nécessaire de renforcer le traite-ment bronchodilatateur : ^-mimétiquepar voie systémique ou le cas échéantthéophylline (utilisable uniquement parvoie systémique ; fenêtre thérapeutiquefaible ; contrôle du taux plasmatique).le salmétérol est un P^-mimétique in-halé avec une longue durée d'action(~ 12 heures), qui contrairement auxdeux traitements que nous venons deciter présente l'avantage d'une faiblecharge systémique ; il peut par exempleêtre utilisé la nuit pour prévenir lescrises nocturnes. L'ipratropium inhalés'avère donner de bons résultats chezde nombreux patients.
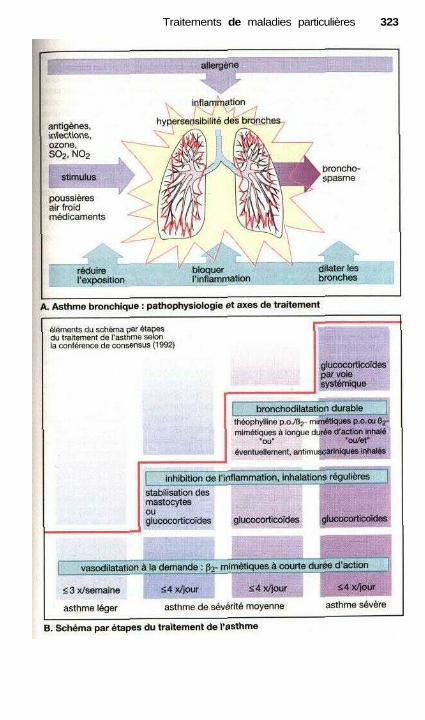
Traitements de maladies particulières 323

324 Traitements de maladies particulières
Vomissements et anti-émétiques diphénylméthane (ex. diphenhydra-nùne, méclozine). Tous les parasympa-tholytiques ou tous les antihistami-niques H| ne conviennent cependantpas de façon systématique. L'efficacitédes substances baptisées anti-émétiquesdépend de l'état présent de l'individu(remplissage de l'estomac, excès d'al-cool), des circonstances extérieures(exemple du comportement des autresvoyageurs) et du type de mouvement.Les médicaments seront avalés 30 minavant le début du voyage et la prise serarepétée toutes les 4-6 heures. La scopo-lamine peut également assurer une pro-tection de 1 à 3 jours en utilisant un em-plâtre placé sur la peau 6-8 heures avantle début du voyage.
Vomissements durant la gros-sesse. Ils se produisent principalementpendant le premier trimestre de la ges-tation ; en conséquence, le traitementpharmacologique tombe pendant la pé-riode de sensibilité maximale du fœtusà une atteinte chimique. C'est pourquoiles anti-émétiques (antihistaminiques etéventuellement neuroleptiques, p. 234)doivent être utilisés en premier lieulorsque survient, par suite des vomisse-ments, une altération sérieuse de l'eauet des électrolytes maternels qui peutmettre en danger l'embryon.
Vomissements associés à l'utili-sation de médicaments. Pour empê-cher les vomissements après administra-tion de cytostatiques (en particulier lecisplatine), on peut utiliser les antago-nistes 5HT,, ondansétron, granisétron ettropisétron. On peut également envi-sager les antagonistes dopaminergiques(lévopromazine, halopéridol) ou les an-tagonistes ayant un effet sur les récep-teurs dopaminergiques et 5HT (méto-clopramide), éventuellement associésaux glucocorticoïdes (dexaméthasone).
Les vomissements survenantaprès une opération, pendant un traite-ment par des radiations ionisantes,une crise d'urémie ou des maladies ac-compagnées d'une augmentation de lapression intracérébrale, pourront éga-lement être traités par des neurolep-tiques ou le métoclopramide.
Le vomissement est une vidange del'estomac dirigée en sens inverse. Lepylore est fermé, tandis que le cardia etl'œsophage se détendent, de telle sorteque sous la pression produite par lacontraction des muscles de la paroi ab-dominale et du diaphragme, le contenude l'estomac est refoulé vers la bouche.L'accès aux voies aériennes est fermépar l'épiglotte. En général un vomisse-ment est précédé par une phase de sé-crétion de salive et de bâillement. Lacoordination de ces phénomènes a lieudans le centre médullaire du vomisse-ment, qui peut être stimulé par diffé-rents effecteurs. Ils sont médiés parl'organe de l'équilibre, les yeux, lenez, la langue et des terminaisons sen-sitives dans la muqueuse du tractus di-gestif. A côté de cela, des événementspsychiques peuvent également sti-muler le centre du vomissement. Lesmécanismes à la base des cinétoses(mal de mer ou mal des transports) oudes vomissements durant la grossessene sont pas connus.
Le centre du vomissement ne peutpas être atteint directement par dessubstances polaires car il est situé sousla barrière hémato-encéphalique. Defaçon indirecte, des substances qui nepénètrent pas dans le cerveau peuventcependant activer le centre du vomisse-ment en stimulant les ehémorécep-teurs de l'area postrema.\ )
Traitement anti-émètique. Levomissement peut être une réactionnormale de l'organisme, par exemplelors de l'absorption orale d'un poison.Les anti-émétiques seront indiqués dansle mal des transports, dans les vomisse-ments de la grossesse, pour éviter lesvomissements post-opératoires ou asso-ciés à la prise de médicaments, et ceuxaccompagnant un traitement par les ra-diations ionisantes.
Cinétoses. Il est possible à titrepréventif d'empêcher les symptômesd'une cinétose avec la scopolamine (unparasympatholytique, p. 106), avec desantihistaminiques H] (p. 114) de type
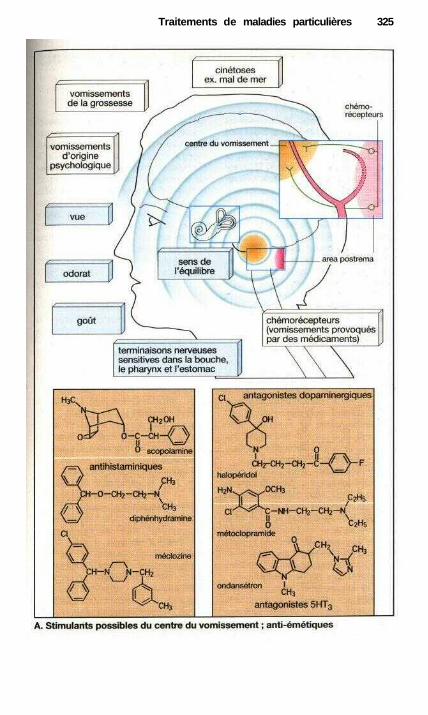
Traitements de maladies particulières 325

326327
Lectures complémentaires 1 Liste des médicaments
Bowman W.C., Rand M.J., Textbookof Pharmacology, 2e édition. BlackwellScientific Publication, Oxford, 1980.
Eighozi J.L., Duval D., Aide Mémoirede Pharmacologie, 2e édition. Flam-marion Médecine-Sciences, Paris,
Hardman J.G., Linbird L.E., MolinoffP.B., Ruddon R.W., Goodman-GilmanA., Thé pharmacological Basis ofTherapeutics, 9e édition, New YorkMcGraw-Hill 1996.
Schorderet M., Pharmacologie, desconcepts fondamentaux aux applica-tions thérapeutiques. Frison-Roche,Slatkine, Paris, Genève, 1989 2e édi-tion 1992.
Guide National de Prescription 1997Edition du Vidai, Paris.
Nomenclature : le concept de sub-stance active ou de produit pharmaco-logique désigne une substance capabled'influencer un phénomène biologiquesans que l'on se préoccupe de savoir sile produit exerce sur l'organisme uneffet bénéfique ou néfaste. Un poisonest également un produit pharmacolo-gique. Au sens strict du terme, un agentpharmaceutique désigne une substanceutilisée à des fins thérapeutiques.L'appellation de médicament de façonclaire indique une substance de ce type.Une substance médicamenteuse peutêtre désignée sous plusieurs noms :- une dénomination chimique,- une dénomination INN (internationalnon-proprietary name) ou dénominationcommune internationale (DCI),- une dénomination commerciale.L'exemple du diazépam permet d'ex-pliquer ce phénomène de façon plus dé-taillée.
La dénomination chimique ducorps est : 7-chloro-l,3-dihydro-l-mé-thyl-5-phényl-2H-1,4-benzodiazépine-2-on, ce qui n'est pas très pratiqued'emploi. Un nom plus simple est dia-zépam. Ce n'est pas un nom brevetémais un nom générique, une dénomina-tion INN, qui s'instaure lorsqu'unecommission internationale s'est mised'accord sur cette appellation.
Les formes galéniques contenantdu diazépam ont d'abord été commer-cialisées par la firme Hoffmann laRoche sous le nom de Valium®. Cenom est une marque déposée protégéepar un brevet, ce qu'indique le signe ®.Lorsque la protection du brevet portantsur la fabrication de médicamentscontenant du diazépam est tombée,d'autres firmes pharmaceutiques ont étéautorisées à produire des médicamentscontenant cette substance. Chacune adonné à sa préparation un nom qui luiest propre. C'est ainsi qu'existent, en
1996, plus de 15 noms commerciaux depréparations contenant du diazépam.Certains de ces noms révèlent aisémentla nature du composant principal puis-que seul le nom de la société a été ra-jouté au nom INN, par ex. diazépamRatiopharm® ; on parle dans ce casd'un médicament générique. D'autressont des créations nouvelles tel parexemple le Neurolytryl®. Pour des mé-dicaments dont les ventes sont bonnes,il existe plus de 20 préparations com-merciales. Le nombre des noms com-merciaux est ainsi nettement supérieurà celui des substances actives. Pour desraisons de clarté, seul le nom génériquea été utilisé dans cet atlas pour désignerles substances actives, dans le cas décritci-dessus le nom de diazépam.
Utilisation des listes
Les buts des listes sont les suivants :1. Le lecteur désire connaître
pour une substance active une prépara-tion commerciale dans laquelle figurecette substance. Le renseignement luiest fourni par la liste nom des sub-stances —> dénomination commerciale.
2. Le lecteur veut savoir quellessont les propriétés pharmacologiquesd'une substance contenue dans une pré-paration commerciale donnée. Pourconnaître le nom générique de cettesubstance, on utilise d'abord la dénomi-nation commerciale —> nom des sub-stances. A l'aide de l'index, on pourraensuite trouver la partie du texte corres-pondant à la substance en question.
Compte tenu de leur nombre, laliste des noms commerciaux ne peutêtre complète. Pour les médicamentsqui sont vendus sous plusieurs nomscommerciaux on a cité le plus vendu etdans quelques cas le nom de prépara-tions dont la vente est moins impor-tante, mais qui sont souvent prescrites.

328 Nom de la substance ->• dénomination commerciale
Les noms commerciaux qui per-mettent de reconnaître sans ambiguïté lenom de la substance active n'ont pas étérépertoriés. A l'exception de quelquescas, les préparations composées n'ontpas été envisagées. Pour les substancesactives qui ne sont présentes que dansdes préparations composées ou cellespour lesquelles il n'existe pas de prépa-rations commerciales, mais qui peuventêtre préparées dans les pharmacies, on adonné l'appellation de la pharmacopée.Le lecteur ne retrouvera donc pas dansl'index de nombreux noms commer-ciaux. C'est là qu'interviennent les in-
formations inscrites sur les boîtes demédicament ou dans les notices d'ac-compagnement, dans lesquelles le nomgénérique ou la désignation INN (DCI)est fournie. Il est alors possible en utili-sant l'index d'aboutir à la bonne « en-trée » dans le texte.
Note du traducteur : de nombreusessubstances citées dans cet ouvrage sontcommercialisées en Allemagne, enSuisse ou en Belgique mais pas enFrance ; elles n'ont donc pas été in-cluses dans ces listes.
Nom de la substance Dénomination commerciale
Aabciximabacarboseacébutololacénocoumarolacétazolamideacétylcystéineacétyldigoxineacétylsalicylique (acide)
acitrétineACTHactinomycine D
aciclovirADHadrénalineadriamycinealcuronium-^aldostérone '1
alfuzosineallopurinolalprénololalprostadilaluminium (chlorure)amantadineambroxolamikacineamiloridee-amino caproïque (acide)amiodaroneamitriptyline
Réopro®Glucor®Sectral®Sintrom®Diamox®Exomuc®, Fluimucil®, Mucolator®, Tixair®Acylanide®aspirine, Aspégic®, Catalgine®, Juvépirine®,Détoxalgine®, Rhonal®Soriatane®synacthènedistribuée en Suisse ou en Belgique(Cosmegen®)Zovirax®Minirin®, Diapid®Anahelp®, Anakit®, Adrénaline Aguettant®Adriblastine®Alloférine®non utilisée sur le plan thérapeutique,voir fludrocortisoneUrion®, Xatral®Xanturic®, Zyloric®Aptine®Prostine VR®, Caverject®, Edex®Etiaxil®Mantadix®Muxol®, Surbronc®Amikiin®Modamide®Capramol®, Hémocaprol®Corbionax®, Cordarone®, Rythmarone®Elavil®, Laroxyl®

Nom de la substance ->• dénomination commerciale 329
amodiaquineamorolfineamoxicillineamoxicilline +
acide clavuliniqueamphotéricine Bampicillineamrinoneaprotininearticaïneastémizoleaténololatracuriumatropineauranofineaurothiopropanol sulfonateazapropazoneazathioprineazidothymidine / zidovudineazithromycineaziocilline
Bbacitracinebacitracine + néomycinebaclofènebéclométasonebénazéprilbensérazide
+ L-DOPAbenzathine pénicillinebenzbromarone
bétaxoloibézafibratebifonazolebipéridènebisacodylbisoprololbléomycinebromazépambromhexinebromocriptinebudésonidebumétanidebuprénorphinebusérélinebuspironebusulfan
Flavoquine®Locéryl®Agram®, Amodex®, Clamoxyl®, Bristamox®Augmentin®
Fungizone®Totapen®, Ampicilline GNRInocor®Iniprol®, Zymofren ®Alphacaïne®Hismanal®Ténormine®Tracrium®Atropine Aguettant®, chibro-atropineRidauron®Allochrysine®Tolyprine®Imurel®Retrovir®Zithromax®Securopen®
bacitracine diamantbacitracine néomycine monotLiorésal®Bécotide®Briem®, Cibacène®Modopar®
Extencilline®Désuric®, avec Allopurinol Anrate®,Désatura®Kerlone®Béfizai®Amycor®Akineton®Contalax®, Dulcolax®Détensiel®, Soprol®bléomycine Roger Bellonlexomil RocheBisolvon®Parlodel®Pulmicort®Burinex®Temgésic®Suprefact®Buspar®Misulban®
calcifédiol / 25-OH-D3calcitonine
Dédrogyl®Calcitar®, Calsyn®, Cibacalcine®, Staporos®

330 Nom de la substance -+ dénomination commerciale
calcitriol/l,25-OH2-D3captoprilcarbacholcarbamazépinecarbénicillinecarbénoxolonecarbidopa + lévodopacarbimazolcarboplatinecartéoloicéfalexinecefménoximecéfopérazonecéfotaximeceftazidimeceftriaxoneceliprololChénodésoxycholique
(acide)chlorambucilchloramphénicol
chlorhexidinechlormadinonechloroquinechlopromazinechlortalidonecholécalciférolcholestyramineciclosporinecilazaprilcimétidineciprofloxacinecisapridecisplatineclindamycineclodronique (acide)clofazimineclofibrateclométiazoleclomifèneclonazépamclonidineclotiazépamclotrimazolecodéine
colchicinecortisol / hydrocortisone^^cortisone ( "cotrimoxazole (triméthoprime +
sulfaméthoxazole)
Rocaltrol®Captolane®, Lopril®Isopto-carbachol®Tégrétol®Pyopen®retiré du commerceSinemet®Neo-mercazol®Paraplatine®Mikelan®Keforal®, Céporexine®Cemix®Cefobis®Claforan®Fortum®Rocéphine®Celectol®Chenodex®
Chloraminophène®Tifomycine®, Solnicol®, Cébénicol®,Ophtaphenicol®Hibidil®, Plurexid®, Urgo-spray®Lutéran®Nivaquine®Largactil®Hygroton®vitamine D^Questran®Neoral® Sandimmun®Justor®Tagamet®, Édalène®Ciflox®Prepulsid®cisplatine, Cisplatyl®Dalacine®Clastoban®, Lytos®Lamprène®Lipavlon®, Clofirem®, Athérolip 500®Hémineurine®Clomid®, Pergotime®Rivotril®Catapressan®Vératran®Trimysten®Bromocodyl®, Bexol®, Camphopneumine®,Édulcor®, Euphon®, Eucalyptine Le Brun®,Néo-codion®colchicine Houdé, Colchimax®hydrocortisone Rousselcortisone Roussel
Bactrim®, Eusaprim®

Nom de ta substance -»• dénomination commerciale
'cromoglycate(acide cromoglicique)
cyanocobalaminecyclophosphamidecyprotérone (acétate)cytarabine
Cromoptic®, Opticron®, Lomudal®Docémine®, vit Bi2Endoxan®Androcur®Aracytine®
Ddaltéparine sodiquedantrolènedapsonedaunorubicinedéféroxaminedésipraminedesfiuranedesmopressinedésogestreldexaméthasonedextrans 40, 60diazépamdiazoxidediclofénacdicloxacillinedidanosinediéthylstilbestroldigitoxinedigoxinedihydralazinedihydroergotaminedihydroergotoxinediltiazemdimenhydrinatedimercaproldiméticonedinoprostonediphénhydraminediphénoxylatedisopyramidedobutaminedompéridonedorzolamidedoxorubicinedoxyçycline
doxylaminedropéridol
Eéconazoleénalaprilenfluraneéphédrine
Fragmine®Dantrium®Disulone®Cérubidine®Desféral®Pertofran®Suprane®Minirin®Vamoline®Décadron®Plasmacair®, Hémodex®Valium®, Novazam®Hyperstat®Voltarène®, Voldal®Diclocil®Videx®Distilbène®digitaline Nativelle, Acylanide®digoxine Nativelle, Coragoxine®Népressol®Dergotamine®, Ikaran®, Séglor®Capergyl®, Ergodose®Tildiem®Dramamine®, Cloranautine®, Nausicalm®B.A.L.Pepsane®, gel de polysilane UPSAProstine E,®Allerga®Diarsed®Rythmodan®Dobutrex®Motilium®, Péridys®Trusopt®Adriblastine®Vibramicyne®, Tolexine®, Spanor®,Doxygram®Méréprine®, Donormyl®Droleptan®
Pévaryl®Renitec®Ethrane®Chilral®, Kiadone®, Tedralan®,Asthmalgine®

332 Nom de la substance dénomination commerciale
ergotamineérythromycine
érythropoïétineestradiolestradiol (benzoate)estradiol (percutané)estradiol (valérianate)estriolétacrynique (acide)éthambutoléthinylestradiol
éthionamideéthosuximideétidronique (acide)étiléfrineétomidateétoposideétrétinate
Ffamotidinefelbamatefélodipinefenfluraminefénotérolfentanyïfinastérideflécaïnidefluconazoleflucytosinefludrocortisoneflumazénilflunarizineflunisolideflunitrazépamfluoxétineflupentixolfluphénazineflutamidefluvastatinefluvoxaminefluorouracilefolique (acide)foscametfosinoprilfurosémide
Ggabapentineganciclovirgélatines
Gynergène®Propiocine®, Estemid®, Érythrocine®,érythrogramErypo®Estrofem®Benzo-gynoestryl®Estraderm®, Oestrogel®Progynova®Ovestin®, Estrofem®Édécrin®Myambutol®, Dexambutol®éthinylestradiol Roussel, Gynoviane®,Millianoviar®, Stédiril®, Ovariostat®Trécator®Zarontin®Didronel®Effortil®Hypnomidate®Celltop®, Étoposide Pierre PâbreTigason®
Pepdine®Taloxa®Hodil®Pondéral®Bérotec®fentanyïJanssenChibro-Proscar®Flécaïne®Triflucan®Ancotil®fludrocortisoneAnexate®Sibélium®Nasalide®Noriel®, Rohypnol®Prozac®
, Fluanxol®- ) Moditen®, Modécate®
Eulexine®Fractal®, Lescol®Floxyfral®fluorouracile RocheSpeciafoldine®Foscavir®Fozitec®Lasilix®
Neurontin®Cymevan®Plasmion®, Haemaccel®, Plasmagel®

Nom de la substance ->• dénomination commerciale 333
gemfibrozilgentamicinegestodèneglibenclamideglycéryl trinitrate
(nitroglycérine) = trinitrinegonadorélinegosérélinegranisétrongriséofulvineguanéthidine
HhalofantrinehalopéridolhalothaneHCGhéparinehéparine de bas poids
moléculairehexachlorocyclohexane
(lindane)hydrochlorothiazidehydrocortisone
hydroxycobalaminehydroxyprogestérone
Lipur®Gentalline®, Gentogram®Harmonet®, Méliane®, Moneva®Daonil®, Euglucan®
Lénitral®, Elbétrine®, Natirose®Lutrelef®Zoladex®Kytril®Griséfuline®Isméline®
Halfan®Haldol®Fluothane®Gonadotrophine chorionique Endo®Liquémine®, Calciparine®, Cuthéparine®
Fragmine®, Hémoclar®, Fraxiparine®
Aphtiria®, Élentol®Esidrex®Hydrocortisone Roussel, HydrocortisoneUpjohnDodécavit®, Novobédouze®Tocogestan®, progestérone retard Pharlon
1ibuprofèneidoxuridineifosfamideiloprostimipramineindométacineinterféron a2ainterféron a2binterféron Pipratropiumisoconazoleisofluraneisoniazideisoprénalineisosorbide (dinitrate)isotrétinoïneisradipineitraconazole
Brufen®, Advil®Iduviran®Holoxan®Ilomédine®Tofranil®Indocid®, Dolcidium®Roféron A®Introna®BêtaféronAtrovent®Fazol®Forane®Rimifon®Isuprel®Risordan®, Langoran®Roaccutane®Icaz LP®Sporanox®
josamycine
Kkanamycine
Josacine®
Kamycine®

334 Nom de la substance ->• dénomination commerciale
kétaminekétoconazole
Llactuloselamotriginelansoprazoleleuprorélinelévodopalévodopa + bensérazidelévodopa + carbidopalévomépromazinelévothyroxinelidocaïne
lincomycinelindanelisinoprillisuridelithiumlomustinelopéramide •loratadinelorazépamlosartanlynestrénollypressine
Mmannitolmaprotilineméclozinemédroxyprogestérone
(acétate)méfloquinemelphalanménadione6-mercaptopurinemésalazinemesnamestérolonemétamizoleméténolonemetformineméthimazol = thiamazolméthohexitalméthotrexateméthyl-DOPAméthysergidemétoclopramidemétoprololmétronidazolemexilétine
Kétalar®, Kétamine PanphannaKétoderm®, Nizoral®
Duphalac®Lamictal®Lanzor®, Ogast®Lucrin®Larodopa®Modopar®Sinemet®Nozinan®Levothyrox®, L-thyroxine RocheLidocaïne Aguettant, Xylocaïne®Xylocard®, Xylocaïne®Lincocine®Aphtiria®Prinvil®, Zestril®Arolac®, Dopergine®Neurolithium®, Téralithe®Bélustine®Imodium®Clarityne®Temesta®Cozaar®Ovariostat®, Ovamezzo®Diapid®
Manicol®, mannitol. Ludiomil®
Agyrax®
Prodasone®, Dépo-provera®Lariam®Aïkéran®Bilkaby®, Cépévit®Purinéthol®Pentasa lavement®Mucofluid®Proviron®Novalgine®, Viscéralgine®Primobolan®Glucinan®, Glucophage®, Stagid®Néo-Mercazol®Brietal®Ledertrexate®Aldomet®, Equibar®désemil SandozPrimpéran®, Anausin métoclopramideLopressor®, Seloken®Flagyl®Mexitil®

Nom de la substance ->• dénomination commerciale 335
meziocillinemiansérinerniconazolernidazolammifépristoneminocyclineminoxidilmisoprostolmoclobémidemolsidominemorphine
NN-acétyleystéinenadololnalbuphinenalidixique (acide)naloxonenaltrexonenandrolonenaproxèneN-butylscopolaminenédocromilnéomycinenéostigminenétilmicinenicardipinenifédipinenimodipinenitrazépamnitrendipinenitroglycérine / trinitrinenitroprussiate de sodiumnoradrénalinenordiazépam/nordazépamnoréthistéronenorfloxacinenortriptylinenoscapinenystatine
Baypen®Athymil®, Miansérine MerckDaktarin®Hypnovel®Mifégyne (RU 486)Mynocine®Lonoten®, Alopexil®Cytotec®Moclamine®Corvasal®Moscontin®
Exomuc®, Fluimucil®Corgard®Nubain®Negram®Narcan®Nalorex®Durabolin®, Anadoi®Apranax®Buscopan®Tilade®néomycine DiamantProstigmine®Nétromicine®Loxen®Adalate®Nimotop®Mogadon®Baypress®, Nidrel®Trinitrine®Nipride®Levophed®Praxadium®Norfor®, Ovariostat®Noroxine®Psychostyl®, Altilev®Tussisédal®Mycostatine®, Nysporil®
0octréotideofloxacineoméprazoleondansétronorciprénalineoxacillineoxazépamoxiconazoleoxprénololoxymétazolineoxytocine
Sandostatine®Oflocet®Antra®Zophren®Alupent®Bristopen®Seresta®Fonx®Trasicor®Iliadine®Syntocinon®

336 Nom de la substance ->• dénomination commerciale
Ppaclitaxelpamidronique (acide)pancuroniumpantoprazolepapavérineparacétamolparomomycineparoxétinepenbutololD-pénicillaminepénicilline Gpénicilline Vpentazocinepentoxifyllinepérindoprilpéthidinephénobarbitalphénolphtaléinephénoxyméthyl-pénicillinephentolaminephénylbutazonephénytoïnephysostigmine / ésérinephytoménadione / vit. Kpilocarpinepindololpipéracillinepirenzépinepirétanidepiroxicampizotifèneplicamycinepolymixine Bpravastatineprazépampraziquantelprazosineprednisolone ,prednisone y
prilocaïneprimidoneprobénécideprobénécide + ampicillineprocaïneprogestéroneproguanilprométhazinepropafénonepropofolpropranololpropylthiouracileprotamine
Taxol®Arédia®Pavulon®Eupantol®Albatran®, Dicertan®, Oxadilène®Akindoi®, Doliprane®, Efferalgan®, Tylenol®Humagel®Deroxat®Betapressine®Trolovol®Spécilline G®, Biclinocilline®, Extencilline®Oracilline®, Ospen®Portai®Torental®Coversyl®Dolosal®Gardénal®Mucinum®, Purganol®Oracilline®, Ospen®Régitine®Butazolidine®, Camdol®Di-hydan®génésérinevitamine K|Vitacarpine®, pilo 1Visken®Pipérilline®Gastrozépine®Eurelix®Feldène®, Olcam®Sanmigran®Mithracine®Colimycine®Elisor®Lysanxia®Biltricide®Minipress®Solupred®, Hydrocortancyl®Cortancyl®Citanest®Mysoline®Bénémide®Prototapen®Novocai'ne®Progestogel®, Utrogestan®Paludrine®Phénergan®, Dolsom®Rythmol®Diprivan®Aviocardyl®, Beprane®propylthiouracile PCHprotamine Choay

Nom de la substance ->• dénomination commerciale 337
pyrazinamidepyridostigminepyridoxinepyriméthamine + sulfadoxine
Qquinalapnlquinidinequininequinoléine (dérivés)quinolone
Rramiprilranitidineréserpinerifabutinerifampicineroxithromycine
Ssalbutamolsalicylique (acide)salmétérolscopolaminesélégilinesimvastatinesitostérolsomatostatinesorbitolsotalolspiramycinespironolactonestreptokinasestreptomycinesuccinylcholine/suxaméthoniumsucralfatesulbactamsulfadoxine + pyriméthaminesulfaméthoxazole
+ triméthoprimesulfasalazine /salazosulfapyridinesulfinpyrazonesulprostonesumatriptan
Ttacrinetacrolimustazobactamtémazépamténiposideterbutaline
Pirilène®Mestinon®Bécilan®, Aspardoxine®, vitamine B(,Fansidar®
Acuitel®, Korec®Cardioquine®, Longacor®Arsiquinoforme®, Quinimax®Direxiode®, Intétrix®voir acide nalidixique et dérivés
Triatec®Azantac®, Raniplex®Serpasil®Ansatipine®Rifadine®, Rimactan®Claramid®, Ruiid®
Salbumol®, Ventoline®acide salicyliqueSerevent®Scopos®Déprényï®Lodales®, Zocor®p-sitostérolModustatine®, somatostatine UCBsorbitol Delalande, nombreuses préparadonsSotalex®Rovamycine®Aldactone®, Spiroctan®Kabikinase®, Streptase®streptomycine DiamantCélocurine®Kéal®, Ulcar®Unacim®Pansidar®
Bactrim®, Eusaprim®, Bactékod®Salazopyrine®Anturan®Nalador®hnigrane®
Cognex®Prograf®Tazocilline®Nonnison®véhem SandozBricanyï®

338 Nom de la substance -»• dénomination commerciale
terfénadinetestostéronetétracaïne
tétryzolinethalidomidethéophyllinethiopentalthiotépathyroxineticarcillineticlopidinetimololtinidazoletobramycinetolbutamidet-PAtrandolapriltranexamique (acide)tranylcyprominetriamcinolonetriamtérènetriazolamtrifluopérazinetrifluridinetriiodothyroninetriméthoprime + sulfamidetriptorélinetropicamidetropisetrontyrothricine
Teldane®Pantestone®, Androtardyl®, Lontanyï®Drill®, Oromédine collutoire, Hexomédinecollutoire, Otyloi®Constrilia®thalidomide PCHInophylline®, théophylline BruneauPentothal®thiotépa Roger BellonLevothyrox®, L-thyroxine RocheTicarpen®Ticlid®Timacor®Fasigyne®Nebcine®Dolipol®Actilyse®Gopten®, Odrik®Exacyl®, Frénolyse®Tylciprine®Tédarol®, Kenacort®, Tibicorten®Tériam®Halcion®Terfluzine®Virophta®Cynomel®Bactrim®, Eusaprim®Décapeptyl LPMydriaticum®Navoban®Maxi-tyro®, Pharyngine®
Uurokinaseursodésoxycholique (acide)
Actosolv®, urokinase ChoayArsacol®, Ursolvan®
Vvalproïque (acide)
vancomycinevécuroniumvérapamilvidarabinevigabatrinevinblastine
vincristinevitamine B,,vitamine Buvitamine D
Dépakine®, valproate de sodium, Roland-MarieVancocine®Norcuron®Arpamyl®, isoptine®Vira-A®Sabril®Velbé®Oxovinca®, Pervincamine®, Pariéval®,Vincafor®Oncovin®Bécilan®, Aspardoxine®Dodécavit®, Novobedouze®Stérogyl®, Vitadone®

Nom de la substance -> dénomination commerciale 339
Coumadine®Wwarfarine
Xxylométhazoline Otrivine®
Hivid®Retrovir®Ivadal®, Srimox®Imovane®

340 Dénomination commerciale -»• nom de la substance
Dénomination commerciale
AActilyse®Actosolv®Acuitel®Acylanide®Adalate®Adrénaline Aguettant®Adriblastine®Advil®Agram®Agyrax®Akindol®Akineton®Albatran®Aldactone®Aldomet®Aïkéran®Allerga®Allochrysine®Alloférine®Alopexy®Alphacaïne®Altilev®Alupent®Amikiin®Amodex®Amycor®Anador®Anahelp®Anakit®Anausin®Ancotil®Androcur®Androtardyl®Anexate®Anrate®Ansatipine®Antra®Anturan®Aphtiria®Apranax®Aptine® "'Aracytine®Aredia®Arolac®Arpamyl®Arsacol®Arsiquinofonne®Ascofer®Aspardoxine®aspirine
Nom de la substance
t-PAurokinasequinalaprilacétyl-digoxinenifédipineadrénalinedoxorubicineibuprofèneamoxicillineméclozineparacétamolbipéridènepapavérinespironolactoneméthyl-DOPAmelphalandiphénhydramineaurothiopropanol sulfonatealcuroniumminoxidilarticaïnenortriptylineorciprénalineamikacineamoxicillinebifonazolenandroloneadrénalineadrénalinemétoclopranrideflucytosinecyprotérone (acétate)testostéroneflumazénilbenzbromarone + allopurinolrifabutineoméprazolesulfinpyrazonehexachlorocyclohexane/lindanenaproxènealprénololcytarabineacide pamidroniquelisuridevérapamilacide ursodésoxycholiquequinineferpyridoxine/vitamine B(acide acétylsalicylique

Dénomination commerciale ->• nom de la substance 341
Asthmalgine®Athérolip 500®Athymil®Atropine Aguettant®Atrovent®Augmentin®Aviocardyl®Azantac®
éphédrineclofibratemiansérineatropineipratropiumclavulanique (acide) + amoxicillinepropranololranitidine
bacitracine DiamantBactekod®Bactrim®Baypen®Baypress®Bécilan®Bécotide®Béfizai®Bélustine®Bénémide®Benzo-gynoestryl®Beprane®Berotec®Bétaferon®Bétanol®Betapressine®Bexol®Biclinocil®Bilkaby®Biltricide®Bisolvon®bléomycine Roger BellonBricanyï®Briem®Brietal®bristamoxBristopen®Bromocodyl®Brufen®Burinex®Buscopan®Butazolidine®
bacitracinesulfaméthoxazole + triméthoprimesulfaméthoxazole + triméthoprimemeziocillinenitrendipinepyridoxine/vitamine B^béclométasonebézafibratelomustineprobénécideestradiol (benzoate)propranololfénotérolinterféron (îmétipranololpenbutololcodéinepénicilline GménadionepraziquantelbromhéxinebléomycineterbutalinebénazeprilméthohexitalamoxicillineoxacillinecodéineibuprofènebumétanideN-butyl scopolaminephénylbutazone
CCalciparine®Calcitar®Calsyn®Camphopneumine®Capergyl®Capramol®Captolane®
héparinecalcitoninecalcitoninecodéinedihydroergotoxineacide e-aminocaproïquecaptopril

342 Dénomination commerciale ->• nom de la substance
quinidinephénylbutazoneacide acétylsalicyliqueclonidinealprostadilchloramphénicolcéfopérazonecéliprololétoposidesuccinylcholine/suxaméthoniumcefménoximeménadionecéfalexinedaunorubicineacide chénodésoxycholiquefinastérideatropineéphédrinechlorambucilmagnésium (hydroxyde)calcitoninebénazéprilciprofloxacineprilocaïnecisplatinecisplatinecéfotaximeamoxicillineroxithromycine
. loratadineacide clodroniqueclofibrateclomifènedimenhydrinatetacrinecolchicinecolchicine
'polymixine Btétryzolinebisacodyldigoxineamiodaroneamiodaronenadololprednisonecortisonemolsidominewarfarinepérindoprillosartancromoglycate = acide cromogliciquehéparineganciclovir
Cardioquine®Carudol®Catalgine®Catapressan®Caverject®Cébénicol®Cefobis®Celectol®Celltop®Célocurine®Cemix®Cépévit®Céporexine®Cérubidine®Chenodex®Chibro-proscar®chibro-atropineChilral®Chloraminophène®Chlorumagène®Cibacalcine®Cibacène®Ciflox®Cinatest®cisplatineCisplatyl®Claforan®Clamoxyl®Claramid®Clarityne®Clastoban®Clofirem®Clomid®Cloranautine®Cognex®colchicine HoudéColchimax®Colimycine®Constrilia®Contalax®Coragoxine®Corbionax®Cordarone®Corgard®Cortancyl®cortisone RousselCorvasal®Coumadine®Coversyl®Cozaar®Cromoptic®Cuthéparine®Cymevan®

Dénomination commerciale —>• nom de la substance 343
Cynomel®Cytotec®
triiodothyroninemisoprostol
DDaktarin®Dalacine®Dantrium®Daonil®Décadron®Décapeptyl®Dédrogyl®Dépakine®Dépo-provera®Déprényï®Dergotamine®DeroxatDésatura®désemil SandozDesférol®Désuric®Detensiel®Détoxalgine®Dexambutol®Di-hydan®Diamox®Diapid®Diarsed®Dicertan®Diclocil®Didronel®digitaline NativelleDiprivan®Direxiode®Distilbène®Disulone®Dobutrex®Docémine®Dodécavit®Dolcidium®Dolipol®Doliprane®Dolosal®Dolsom®Donormyl®Dopergine®Doxygram®Dram aminé®Drill®Droleptan®dulcolaxDuphalac®Durabolin®
miconazoleclindamycinedantrolèneglibenclamidedexaméthasonetriptorélinecalcifédiol/25-OH-D3valproïque (acide)médroxyprogestérone (acétate)sélégilinedihydroergotamineparoxétinebenzbromaroneméthysergidedéféroxaminebenzbromarone + allopurinolbisoprololacide acétylsalicyliqueéthambutolphénytoïneacétazolamidevasopressine (lysine)diphénoxylatepapavérinedicloxacillineacide étidroniquedigitoxinepropofolquinoléine (dérivés)diéthylstilbestroldapsonedobutaminecyanocobalaminehydroxocobalamine/vitamine Bnindométacinetolbutamideparacétamolpéthidineprométhazinedoxylamidelisuridedoxycyclinedimenhydrinatetétracaïnedropéridolbisacodyllactulosenandrolone

344 Dénomination commerciale -»• nom de la substance
EEdalène®Édécrine®Edex®Édulcor®Efferalgan®Effortil®Elavil®Elbétrine®Élentol®Elisor®Endoxan®Equibar®Ergodose®Erypo®Érythrocine®érythrogramEsidrex®Estemid®Estraderm®Estrofem®éthinylestradiol RousselEthrane®Etiaxil®Eucalyptine Le Brun®Euglucan®Eulexine®Eupantol®Euphon®EurelixEusaprim®Exacyl®Exomuc®Extencilline®
cimétidineacide étacryniquealprostadilcodéineparacétamolétiléfrineamitriptylineglycéryltrinitrate (nitroglycérine)hexachlorocyclohexane/lindanepravastatinecyclophosphamideméthyl-DOPAdihydroergotoxineérythropoïétineérythromycineérythromycinehydrochlorothiazideérythromycineestradiol (percutané)estradiol + estrioléthinylestradiolenfluranealuminium (chlorure)codéineglibenclamideflutainidepantoprazolecodéinepirétanidecotrimoxazoleacide tranexamiqueacétylcystéinebenzathine pénicilline/pénicilline G
FFansidar®Fasigyne®Fazol®Feldène®fentanyïJanssenFero-grad®Ferrostrane®Fiboran®Flagyl®Flavoquine®Flécaïne®Flodil®Floxyfral®Pluanxol®fludrocortisoneFluimucil®
pyriméthanune + sulfadoxinetinidazoleisoconazolepiroxicamfentanyïferferaprindinemétronidazoleamodiaquineflécaïnidefélodipinefluvoxamineflupentixolfludrocortisoneacétylcystéine

Dénomination commerciale ->• nom de la substance 345
fluorouracile RocheFluothane®FonxForane®Fortal®Fortum®Foscavir®Fozitec®Fractal®Fragmine®Fraxiparine®Frénolyse®Fumafer®Fungizone®
fluorouracilehalothaneoxiconazoleisofluranepentazocineceftazidimefoscarnetfosinoprilfluvastatinehéparine de bas poids moléculairehéparine de bas poids moléculaireacide tranexamiqueferamphotéricine B
GGardénal®Gastrozépine®Gel de polysilaneGénésérine®Gentalline®Gentogram®Glucinan®Glucophage®Glucor®Gonadrotrophine
chorionique Endo®Gopten®Griséfuline®Gynergène®Gynoviane®
phénobarbitalpirenzépinediméticonephysostigmine/ésérinegentamicinegentamicinemetforminemetformineacarbose
HCGtrandolaprilgriséofulvineergotamineéthinyiestradiol
HHaemaccel®Halcion®Haldol®Halfan®Harmonet®Hémineurine®Hémocaprol®Hémoclar®Hémodex®Hexomédine®Hibidil®Hismanal®Hivid®Holoxan®Humagel®Hydrocortancyl®hydrocortisone RousselHygroton®
gélatinestriazolamhalopéridolhalofantrinegestodèneclométiazoleacide e-aminocaproïquehéparine de bas poids moléculairedextrantétracaïnechlorhexidineastémizolezaïcitabineifosfamideparomomycineprednisolonecortisol/hydrocortisonechlortalidone

346 Dénomination commerciale ->-nom de la substance
Hyperstat®Hypnomidate®Hypnovel®
1Icaz®Iduviran®Ikaran®Ilomédine®Imigrane®Imodium®Imovane®Imurel®Indocid®Iniprol®Inocor®Inophylline®Intétrix®Introna®Isméline®Isoptine®Isopto-carbachol®Isuprel®Ivadal®
JJosacine®JustorJuvépirine®
KKabikinase®Kamycine®Kaologeais®Kéal®Kéforal®Kenacort®Kerlone®Kétalar®Kétoderm®Kiadone®Korec®Kytril®
LLaccoderme Dalibour®Lamictal®Lamprène®Langoran®
diazoxideétomidatemidazolam
isradipineidoxuridinedihydroergotamineiloprostsumatriptanlopéramidezopicloneazathioprineindométacineaprotinineamrinonethéophyllinequinoléine (dérivés)interféron a2bguanéthidinevérapamilcarbacholisoprénalinezolpidem
josamycinecilazaprilacide acétylsalicylique
streptokinasekanamycinekaolinsucralfatecéfalexinetriamcinolone
. bétaxoloikétaminekétoconazoleéphédrinequinalaprilgranisétron
zinc (oxyde)lamotrigineclofazimine.isosorbide (dinitrate)

Dénomination commerciale ->• nom de la substance 347
1————————————Lanzor®Largactil®Lariam®Larodopa®Laroxyl®Lasilix®Ledertrexate®Lénitral®Lescol®Levophed®Levothyrox®lexomil RocheLincocine®Liorésal®Lipavlon®Lipur®
; Liquemine®Locéryl®Lomudal®Longacor®Lonoten®Lontanyï®Lopressor®Lopril®Loxen®L-thyroxine RocheLucrin®Ludiomil®Lutéran®Lutrelef®Lysanxia®Lytos®
MMag2®Manicol®mannitol
; Mantadix®• Maxi-tyro®
Médrocyl®Médrol®Méliane®Méréprine®Mestinon®Méthergin®métoclopramideMexitil®Mifégyne®Mikelan®millianoviarMinipress®Minirin®
lansoprazolechlopromazineméfloquinelévodopaamitriptylinefurosémideméthotrexateglycéryltrinitrate (nitroglycérine)fluvastatinenoradrenalinethyroxinebromazépanlincomycinebaclofèneclofibrategemfibrozilhéparineamorolfinecromoglycate = acide cromogliciquequinidineminoxidiltestostéronemétoprololcaptoprilnicardipinethyroxineleuprorélinemaprotilinechlormadinonegonadorélineprazépamacide clodronique
magnésium (hydroxyde)mannitolmannitolamantadinetyrothricineméthylprednisoloneméthylprednisolonegestodènedoxylamidepyridostigmineméthylergométrinemétoclopramidemexilétinemifépristonecartéoloiéthinylestradiolprazosinedesmopressine/ADH

348 Dénomination commerciale ->• nom de la substance
Misulban®Mitracine®Moclamine®Modamide®Modécate®Moditen®Modopar®Modustatine®Mogadon®Moneva®Moscontin®Motilium®Mucinum®Mucofluid®Mucolator®Muxol®Myambutol®Mycostatine®Mydriaticum®Mynocine®Mysoline®
NNalador®Nalorex®Narcan®Nasalide®Natirose®Nausicalm®Navoban®Nebcine®Négram®Néo-codion®Néo-mercazole®néomycine DiamantNéoral®Népressol®nétromicineNeurolithium®Neurontin®Nidrel®Nimotop®Nipride®Nivaquine®Nizoral®Norcuron®Norfor®Noriel®Normison®Noroxine®Novalgine®Novazam®
busulfanmithramycine / plicamycinemoclobémideamiloridefluphénazinefluphénazinelévodopa + bensérazidesomatostatinenitrazépamgestodènemorphinedompéridonephénolphtaléineme&naacétylcystéineambroxoléthambutolnystatinetropicamideminocyclineprimidone
sulprostonenaltrexolonenaloxoneflunisolideglycéryltrinitrate (nitroglycérine)dimenhydrinatetropisétrontobramycineacide nalidixiquecodéinecarbimazolenéomycineciclosporinedihydralazinenétilmicinelithiumgabapentinenitrendipinenimodipinenitroprussiate de sodiumchloroquinekétoconazolevécuroniumnoréthistéroneflunitrazépamtémazépamnorfloxacinemétamizolediazépam

Dénomination commerciale ->• nom de la substance 349
Novobédouze®Novocaïne®Nozinan®Nubain®Nysporil®
0Odrik®Œstrogel®Oflocet®Ogast®Olcam®Oncovin®Ophtaphénicol®Opticron®Oracilline®Oromédine®Ospen®Otrivine®Otyloi®Ovamezzo®Ovariostat®Ovestin®Oxadilène®Oxovinca®
PPaludrine®Pantestone®Paraplatine®Pariéval®Parlodel®Pavulon®Pentasa lavement®Pentothal®Pepdine®Pepsane®Péridys®Pergotime®Pertofran®Pervincamine®Pévaryl®Phanurane®Pharyngine®Phénergan®phospholine iodidepilo 1Pipérilline®Pirilène®Plasmacair®Plasmagel®
hydroxycobalamine/vitanune B,,procaïnelévomépromazinenalbuphinenystatine
trandolaprilestradiol (percutané)ofloxacinelansoprazolepiroxicamvincristinechloramphénicolcromoglycate = acide cromogliciquepénicilline Vtétracaïnepénicilline Vxylométhazolinetétracaïnelynestrénoléthinylestradiol + lynestrénolestriolpapavérinevincamine
proguaniltestostéronecarboplatinevincaminebromocriptinepancuroniummésalazinethiopentalfamotidinediméticonedompéridoneclomifènedésipraminevincamineéconazolecanrénonethyrothricineprométhazineecothiopatepilocarpinepipéracillinepyrazinamidedextransgélatines

350 Dénomination commerciale ->• nom de la substance
Plasmion®Plurexid®Pondéral®Praxadium®Prepulsid®Primobolan®Primpéran®Prinvil®Prodasone®progestérone retard PharionProgestogel®Prograf®Progynova®Propiocine®Prostigmine®Prostine E;®Prototapen®Proviron®Prozac®Psychostyl®Pulmicort®Purganol®Purinéthol®Pyopen®
gélatineschlorhexidinefenfluraminenordazépamcisaprideméténolonemétoclopramidelisinoprilmédroxyprogestérone (acétate)hydroxyprogestéroneprogestéronetacrolimusestradiol (valérate)érythromycinenéostigminedinoprostoneprobénécide + ampicillinemestérolonefluoxétinenortriptylinebudésonidephénolphtaléine6-mercaptopurinecarbénicilline
QQuestran®Quinimax®
colestyraminequinine
RRaniplex®Regitine®Renitec®Réopro®Rétro vir®Rhonal®Ridauran®Rifadine®Rimactan®Rimifon®Risordan®Rivotril®Roaccutane®Rocaltrol®Rocéphine®Roféron A®Rohypnol®Rovamycine®Ruiid®Rythmarone®Rythmodan®Rythmol®
ranitidinephentolamineénalaprilabciximabazidothymidine/zidovudineacide acétylsalicyliqueauranofinerifampicinerifampicineisoniazideisosorbide (dinitrate)clonazépamisotrétinoïnecalcitriol/l,25-OH2-D3ceftri axoneinterféron a2aflunitrazépamspiramycineroxithromycineamiodaronedisopyramidepropafénone

Dénomination commerciale -» nom de la substance 351
Ife S• Sabril®• Salazopyrine®1; Salbumol®BF Sandimmun®| Sandostatine®| Sanmigran®| Scopos®t Sectral®•L Securopen®• Séglor®• Seloken®• Séresta®H Sérévent®f Serpasil®| Sibélium®f Sinemet®^B Sinex LachartreB Sintrom®K Sisolline®R (3-SitostérolJ Solnicol®B Soludactone®H| Solupred®1 Soprol®• , Soriatane®• Sotalex®H| Spanor®K Spasmag®• Speciafoldine®1 SpécillineG®• Spiroctan®• Sporanox®•I Stagid®• Staporos®• Stédiril®• Stérogyl®• Stiinox®• Streptase®f ' streptomycine Diamant| Suprane®| Suprefact®| Surbronc®| synactènesf Syntocinon®\
;. T| Tagamet®( Taloxa®î Taxol®; Tazocilline®
Tédarol®
vigabatrinesulfasalazine/salazosulfapyridinesalbutamolcyclosporine / ciclosporineoctréotidepizotifènescopolamineacébutololaziocillinedihydroergotaminemétoprololoxazépamsalmétérolréserpineflunarizinecarbidopa + lévodopaoxyrnétazolineacénocoumarolsisomicinesitostérolchloramphénicolcanrénoneprednisolonebisoprololalcitrétinesotaloldoxycyclinemagnésium (sulfate)acide foliquepénicilline Gspironolactoneitraconazolemetiorminecalcitonineéthinylestradiolvitamine DzolpidemstreptokinasestreptomycinedesfluranebusérélineambroxolACTHoxytocine
cimétidinefelbamatepaclitaxeltazobactamtriamcinolone

352 Dénomination commerciale -»• nom de la substance
Tédralan®Tégretol®Teldane®Témesta®Temgésic®Ténormine®Téralithe®Terfluzine®Tériam®Thalidomide®théophylline Bruneauthiotépa Roger BellonTibicorten®Ticarpen®Ticlid®Tifomycine®Tigason®Tilade®Tildiem®Timacor®Tixair®Tocogestan®Tofranil®Tolexine®Tolyprine®Torental®Totapen®Tracrium®Transcycline®Trasicor®Trécator®Triatec®Triflucan®Trimysten®trinitrineTrolovol®Trusopt®Tussisédal®Tylciprine®Tylenol®
UUlcar®Unacim®Urgo-spray®Urion®urokinase Choay
éphédrinecarbamazépineterfénadinelorazépambuprénorphineaténolollithiumtrifluopérazinetriamtérènethalidomidethéophyllinethiotépatriamcinoloneticarcillineticlopidinechloramphénicolétrétinatenédocromildiltiazemtimololacétylcystéinehydroxyprogestéroneimipraminedoxycyclineazapropazonepentoxifyllineampicillineatracuriumrolitétracyclineoxprénololéthionamideramiprilfluconazoleclotrimazolenitroglycérine/trinitrineD-pénicillaminedorzolamidenoscapinetranylcypromineparacétamol
sucralfatesulbactamchlorhexidinealfuzosineurokinase

Dénomination commerciale ->• nom de la substance 353
Ursolvan®Utrogestan®
VValium®valproate de sodium
Roland-MarieVancocine®Vamoline®Véhem®Velbé®Ventoline®Vératran®Vibramicyne®Videx®Vincafor®Vira-A®Virophta®Viscéralgine®Visken®Vitacarpine®Vitadone®vitamine B(vitamine B 12vitamine T)yvitamine KiVoldal®Voltarène®
XXanturic®Xatral®Xylocaïne®Xylocard®
ZZarontin®Zestril®Zithromax®Zocor®Zoladex®Zophren®Zovirax®Zyloric®Zymofren®
acide ursodésoxycholiqueprogestérone
diazépam
acide valproïquevancomycinedésogestrelténiposidevinblastinesalbutamolclotiazépamdoxycyclinedidanosinevincaminevidarabinetrifluridinemétamizolepindololpilocarpinevitamine Dpyridoxinecyanocobalaminecholécalciférolphytoménadionediclofénacdiclofénac
allopurinolalfuzosinelidocaïnelidocaïne
éthosuximidelisinoprilazithromycinesimvastatinegosérélineondansétronaciclovirallopurinolaprotinine

355
Index
Abciximab 148Acarbose 168Accommodation 98Accouchement 241- inhibition/stimulation 126- et prostaglandines 194ACE, Angiotensin Converting
Enzyme 35, 124Acébutolol 94Acénocoumarol 145Acétazolamide 160,161Acétique (acide) 35Acétylateurs rapides ou lents 278Acétylcholine, ACh 34,82,98, 180
et antidépresseurs 228dégradation 100formation du suc gastrique 164
• libération 100et maladie de Parkinson 186et neuroleptiques 234structure 35, 101, 103,185
• synthèse 100Acétylcholine (récepteurs)- muscariniques 98— nicotiniques 98Acétylcholinestérase 100,102, 184- inhibiteurs, 184Acétylcoenzyme A
intermédiaire de synthèse d'ACh100réaction de conjugaison 38
N-Acétylcystéine- comme expectorant 318- et empoisonnement
par le paracétamol 196Acétyldigoxine 132N-AcétylgIucosamine 266N-Acétylmuramique (acide) 266Aciclovir 284Acétylsalicylique (acide) 34,142,148,
196,198,200• et agrégation plaquettaire 148• métabolisme 34• migraine 316• régulation thermique 200
Acidesanti-inflammatoires 196
- biliaires 152- gras 20, 153- inhibiteurs de la production 164- neutralisation 164- organiques et désinfection 286- et pénicillines 266- sécrétion 164- et urée 310Acidose lactique 260Acipimox 154Acitrétine 74Acroléine 113,294Acromégalie 240ACTH 240,248Activité intrinsèque 60,62Acylaminopénicilline 268Acyltransférase 38Adaptine 26Adénine 295Adénohypophyse 240Adénome thyroïdien 244Adénosine (récepteurs) 320Adénosine monophosphate 3' 5'
cyclique (AMPc) 66Adénylate cyclase 66ADH 158, 162, 240- et sels de lithium 232ADN 272, 282- polymérase 282Adrénaline 82, 134,204- et anesthésie locale 204- et choc anaphylactique 320- libération 108- structure 87Adriamycine 294Adsorbants 176Aérosols 12, 14Affinité 56- et énantiosélectivité 62Agoniste 60- inverse 60- partiel 94, 212- et transduction des signaux 66AIDS voir SIDAAjmaline 136Albumine- liaison du calcium 262- liaison des médicaments 30

356 Index
Alcaloïdes 4Alcool- benzylique 284- comme désinfectant 286- et régulation thermique 200Alcool déshydrogénase 44Alcuronium 182Aldéhyde (désinfection) 286Aldostérone 158, 246- antagonistes 162Alendronate 312Alfuzosine 90Alkylants 294Allergie 320- et leucotriènes 95- traitement 320- types de réactions 72Allopathie 76Allopurinol 284, 310Alloxanthine 310Aloès 172Alprénolol 95Alprostadil 118Aluminium- hydroxyde 164- sels astringents 176- silicate 176Amanita muscaria 238Amantadine 186, 284- effet antiparkinsonien 186- effet virustatique 284Ambroxol318Amide (liaison) 34Amidon- comme excipient 8- propriétés colloïdales 150Amikacine 276, 278Amiloride 162,163Aminé biogène 114p-Amino-benzoïque (acide) 2707-Amino-céphalosporinique (acide) 248Aminoglycosides 265, 274, 276- passage à travers les membranes 26p-Aminométhyl-benzoïque (acide) 1466-Amino-pénicillanique (acide) 266,269Aminopénicilline 268Aminoquinuride 2565-Amino-salicylique (acide) 270p-Amino-salicylique (acide) 278Amiodarone 136Anutriptyline 228Ammoniaque 168Amorolfine 280
Amoxicilline 268AMPc 66Amphétamine 88- et dépression 228Amphiphilie 206Amphotéricine B 280Ampicilline 268Ampoule 12Amrinone 118Anabolisants 250Analgésiques 194- antipyrétiques 194- et asthme 196Androgène 250Androstérone 250Anémie- par carence en fer 138- et cytostatiques 292- hémolytique 70, 72- mégaloblastique 138- pernicieuse 138Anesthésie locale 128, 202,206,318- spinale 202, 216Angine de poitrine 302- prévention d'une crise 304- traitement 304Angiotensin Converting Enzyme, ACE
35,124Angiotensinase A 35Angiotensine 1 34, 124Angiotensine II 35, 124Angiotensinogène 124Anhydrase carbonique (inhibiteurs de)
160Aniline 301Anions 58Anophèles 290Anorexigènes 88Anse (diurétiques de) 20,156,160Antagonistes 60- allostériques 62- compétitifs 60- fonctionnels 60Antagonistes calciques 20,118,122,128- et angine de poitrine 302- et hypertension 306Anthracyclines 294Anthranol 175Anthrone 175Anti-acides 164- et tétracyclines 276Anti-androgène 250Anti-anémique 138

Index 357
Anti-angineux 304Anti-arythmiques 134- actions 134- bloquant les canaux sodiques 134Antibiotiques 264- cytostatiques 292- à spectre large 264Anticholinergiques 186Anticoagulants 144- et grossesse 74- oraux 144Anticonvulsivants 188Anticorps 72Antidépresseurs 106- comme co-analgésiques 192- tricycliques 228,230Antidiabétiques- oraux 260- sulfonylurés 260Anti-diarrhéiques 176Anti-émétiques 324Anti-épileptiques 181, 188- pendant la grossesse 74Anti-helminthiques 288Antihistaminiques 14, 106, lt6Antihistaminiques H[ 116- et allergie 320- et rhume 318- utilisation comme somnifères 220- et vomissements 324Antihistaminiques H, 116, 164Anti-hypertenseurs 306Anti-inflammatoires non stéroïdiens
198,314Anti-malariens 290Anti-métabolites- cytostatiques 292,294- virustatiques 282, 284Anti-mycotiques 280Anti-parasitaires 288Antiparkinsoniens 106, 181,186Antipyrétiques 196, 318Antiseptiques 286Anti-sympathotoniques 96,128Antithrombine III 146, 148Anti-thrombotiques 146Anxiolyse 224- avec des neuroleptiques 234Apofemtine 140Apolipoprotéines 152Apoptose 292Appareil juxtaglomérulaire 124Aprindine 136
Aprotinine 146Arabinose 283Arachidonique (acide) 194Area postrema 324Area Under Curve, AUC 46Arécoline 103Arihalonium lewinii 238ARN, voir Ribonucléique (acide)Artériosclérose et nicotine 112Arthrite- et allergie aux médicaments 72- rhumatoïde 314Arthus (réactions d') 72Articaïne 206Arythmies (traitement des) 134Ascaris lumbricoides 288Ascorbique (acide) 140Astémizole 114Asthme bronchique 322- et p-bloquants 92Aténolol 94Atracurium 182Atropa belladona 105Atropine 70, 98, 104,300- empoisonnement 106Auranofîne 314Aurothioglucose 314Aurothiomalate 314Autorégulation métabolique 302Axolemme 204Axoplasme 204Azapropazone 199Azathioprine 294, 296- métabolisme 37Azidothymidine 284Azithromycine 274Aziocilline 268Azoles 280
BB»,x 56Bacitracine 265,268Baclofène 180Bactéricidie 264Bamipine 114Barbituriques 220- et régulation thermique 200Barrière(s)- externes 22- hémato-encéphalique 24,26- placentaire 74- vasculaires 24

358 Index
Bases puriques 292Bateman (fonction de) 47Bathmotrope 84Béclométhasone 248- et allergie 320- en inhalation 14Béfunolol 95Bénazépri] 124Bensérazide 186Benzaikonium 287Benzathine-pénicilline G 266Benzatropine 106, 186Benzbromarone 310Benzène 36Benzocaïne206,318Benzodiazépines 224- comme anti-épileptiques 188- demi-vie d'élimination 227- dépendance 226- effets 224- pharmacocinétique 226- récepteurs 188,224- comme somnifères 220Benzopyrène 113- métabolisme 36Benzylpénicilline 266Bétaxoloi 95Bézafibrate 154Bicouche membranaire 20Bifonazol 280Biguanides 260Bile 32Bilharziose 288Biodisponibilité 42,46Bipéridène 186,236Biphosphonate et hypercalcémie 312
' Bisacodyl 172- sulfatation 38Bisoprolol 94Blastocyste 75Bléomycine 294Blocage muqueux 140ci-Bloquants 90, 118- dans l'hypertension 307P-Bloquants 92, 128,135- et angine de poitrine 302- cardiosélectifs 94- différentes molécules 94- effets indésirables 92- effets thérapeutiques 92- et hypertension 306- propriétés pharmacocinétiques 94- comme racémiques 62
- et stabilisation de membrane 94- structure de base 94Bopindolol 95Bordure en brosse (cellules à) 22Bowman (capsule de) 158Bradycardie sinusale 135Bradykinine 124- et douleur 202Bromazépam 227Bromhexine318,319Bromocriptine 114, 126,240- et maladie de Parkinson 186- structure 187Bronches 14- effet des prostaglandines 194- structure de l'épithélium 22Bronchodilatateurs 126Brotizolam 227- comme somnifère 222Buchheim, Rudolf 3Budésonide 248- et allergie 320- en inhalation 14Bufoténine 238Bumétanide 160Bunitrolol 95Buprénorphine 208Buséréline 240, 241Buspirone 116Busulfan 292Butizide 160N-Butylscopolamine 104,107,126Butyrophénone 234Butyrylcholinestérase 100
CÇa ATPase 128Calcium- carbonate de 164- et coagulation sanguine 142- complexants du 142- - et hypercalcémie 262- etostéoporose312Caféine 320- et troubles du sommeil 222Calcifédiol 262, 263Calcinose 262Calcitonine 262Calcitriol 262, 263Calmoduline 84CAM (concentration alvéolaire
moyenne) 227

Index 359
Camazépam 227Campylobacter pylori 166Canal- calcique 20, 128,136- potassique 136- sodique 202Canaux ioniques activés
par un ligand 64Cancérogénicité 6Candida 276, 280Cannabis indica 238Canrénone 162Capréomycine 278Capillaires (structure) 24Capsule de Bowman 158Captopril34, 118Carazolol 95Carbachol 102-104Carbamate 102Carbamazépine 188,189Carbénicilline 268Carbénoxolone 166Carbidopa 186, 187Carbimazol 244, 245Carboxypénicilline 268Carcinogène 292Cardiosélectivité 94Cardiostéroïde 130Cartéoloi 95Carvedilol 95Cascara sagrada 172 .Catécholamines 84, 86Catéchol-0-méthyl transférase 82Céfalexine 268, 269Cefménoxime 268Cefopérazone 268Céfotaxime 268Ceftazidime 268Ceftriaxone 268Celiprolol 95Cellule pariétale 165Cellulose 168- acétate de 10Centre d'asymétrie 62Céphalosporinase 268Céphalosporines 265,268Certoparine 146Cérulétide 178Cestodes 28817-Céto-stéroïdes250Champignons (lutte contre les) 280Charbon médicinal 176Charges partielles 58
Chélatants 298Chénodésoxycholique (acide) 178Chimiotactisme 70Chimiothérapie 264Chlorambucil 292Chloramphénicol 265, 274-276Chlorhexidine 286Chlore (perméabilité au) 224Chlormadinone (acétate) 252Chlorméthylphénol 287Chloroforme 201Chloroquine- comme anti-malarien 290- comme anti-rhumatismal 314- structure 291Chlorphénoxamine 114Chlorpromazine 206, 228- métabolisme 36- structure 235Chlorprothixène 236Chlortalidone 160Cholécalciférol 262, 263Cholécystokinine 168, 172Cholestérol 152Choline 20- comme précurseur de
l'acétylcholine 100Choline acétyltransférase 100Cholinestérase 100- non spécifique 184- sérique 184 . .Chronotropie 82Chylomicrons 152Cilazapril 124 -Cimétidinell5,164,i65Cinétique plasmatique d'un
médicament 48- durant une administration régulière
48- surface sous la courbe/AUC 46Ciprofloxacine 272Cire 10Cisapride 116Cisplatine 294Citrate 142Claviceps purpurea 127Clavulanique (acide) 268Clearance 18, 22, 28, 44,48 .Clemastil 114Clemizol-penicilline G 266Clindamycine 265, 274Clinique (investigations) 6, 74Clodronate et hypercalcémie 262

360 Index
Clofazimine 278, 279Clofibrate 154Clométhiazol 190Clomifène 254Clonazépam 272Clonidine 96, 97- et hypertension 306Clostébol 250Clostridium botulinum 180Clostridium difficile 268Clotiazépam 220Clotrimazole 280, 281Clozapine 236Coagulation sanguine 138- facteurs de 138,140Co-analgésiques 192Cocaïne 88, 89Codéine 4, 210, 318Colchicine310- et goutte 310Colchicum automnale 310Colestipol 154Colestyramine 130,154Collagène 150Colloïde 150 •- et thyroïde 244Complément 70Complexes immuns- et vasculitis 70Compliance 50Comprimés effervescents 8COMT 80, 84Conjugaison 38.Constantes- de dissociation à l'équilibre
56- de vitesse 44Constipation et laxatifs 168Contraceptifs oraux 254Contracture 131Cortex surrénalien- atrophie 248- hormones 246- insuffisance 248Cortisol 246, 247Cortisone (métabolisme) 36Cotrimoxazole 270- et diarrhée 176Coumarine 142, 144Couplage électromécanique 262- cœur 128- muscle 180Crampes 188
Crèmes- hydrophiles 16- lipophiles 16CRF 240, 248Cromoglycate 320, 322- et inhalation 14Cross-over (études) 74Curare 182Cushing (syndrome de) 246Cyanocobalamine 138Cyanure, empoisonnement
et antidote 300Cycle entéro-hépatiquë 38Cyclofénil 254Cyclooxygénase 148,194-198- et inhibiteurs 198Cyclophosphamide 294, 296Cyclopropane 217Cyclopyrrolone 220Cyclosérine 278Cyclosporine 296Cyclothymie 228Cyproheptadine 114Cyprotérone (acétate) 252Cytarabine 294Cytochrome 32Cytomégalovirus 284Cytosine désaminase 280Cytostatiques 292-294- alkylants 292- effets secondaires 296- mécanismes d'action 292- et vomissements 324
DDaltéparine 146Dantrolène 180Dapsone 278, 279Daunorubicine 294DDT 102, 288Découplage psychosomatique 228, 2327-Déhydrocholestérol 262,263Delirium alcoolique 190Demi-vie 44Dents (maux de) 197Dépendance- et benzodiazépines 226- et opioïdes 208- et sympathomimédques indirects
88Dépolarisation d'une membrane 136- et influx de calcium 137

Index 361
Dépression endogène 228Dermatophytes 280Désalkylation 36Désamination 36Desflurane216Désinfection 286Désintégration 10Désipramine 230Desmopressine 162Desmosomes 22Désogestrel 252Désoxyribose 283Désulfuration 36Détergents 286Dexaméthasone 246Dexétimide- énantiosélectivité 62- structure 63Dextran 150, 151Diabète sucré de l'adulte (type II)
256Diabètes insipidus 162Diabètes mellitus 256
' - et P-bloquants 92- et glucocorticoïdes 246- insulinodépendant 256Diacylgiycérol 662-4 Diaminopyridine 270Diaphragme et pores endothéliaux 24Diarrhée 176Diastéréo-isomères 62Diazépam 128, 226Diazoxidell8Diclofénac 198,314 'Dicloxacilline 268Didanosine 284Diète- et diabète 256- et goutte 310Diéthyléther214,215Diéthylstilbestrol et grossesse 72Diffusion- barrière de 20- et passage membranaire 26Digitalis purpurea 130, 131Digitoxine 132- accumulation 50- cycle entéro-hépatiquë 38Digoxine 132Dihydralazine 118- et hypertension 306Dihydroergotamine 126- et hypotension 308
Dihydrofolate 270, 290Dihydrofolate réductase 270, 290-294Dihydropyridine 122Dihydrotestostérone 250, 2511,25 Dihydroxycholécalciférol 262Dilatation bronchique et médicaments
122Diltiazem 122- et angine de poitrine 304Dimenhydrinate 114Dimercaprol 298- structure 299Dimercapto propane sulfonate,
structure 299Diméthylaminophénol 300Diméthylopolysiloxane 178Diméticone 178Dimétindène 114Dinoprost 126Dinoprostone 126Diphenhydramine 324- comme somnifère 220- et vomissements 324Diphénolméthane 172Diphénoxylate 176 -Dipôle 58Disopyramide 136Dispersion 12Disse (espace de) 24, 32Dissociation (constante de) 206Distribution 18, 22,28,46- volume de 44Diurétiques 156, 160, 306- effets secondaires 156- indications 156- osmotiques 158- sulfonamides 160- thiazidiques 160Dobutamine 114- racémique 62Docetaxel 292DOPA 97DOPA-décarboxylase 96,186- inhibiteurs 186L-DOPA 24, 186, 187- passage à travers les membranes 26Dopage 88Dopamine 82, 88, 186- agonistes/antagonistes 114- antagonistes et vomissements 324- et maladie de Parkinson 186- récepteurs 236- — et neuroleptiques 234

362 Index
Dopamine [3-hydroxylase 82Dorzolamide 158Dose d'un médicament 2, 68- et durée d'action 66- d'entretien 50- relation avec l'effet 52Douleurs 192- et prostaglandines 194- viscérales 192Double aveugle (étude) 74Doxazosine 90Doxorubicine 294Doxycycline 276Doxylamine 220Dragée 8Dromotrope 84Dropéridol214,234Dynorphine 208Dyskinésie tardive et neuroleptiques 236
EE 600 36, 102, 103E 605 36, 102ECso 54Éconazole 280Écothiopate 102Ectoparasites (traitements des) 288Eczéma de contact 70EDRF116, 120EDTA 60, 142, 298EEG- et crise d'épilepsie 188- et stades du sommeil 220Eicosatétraénoïque (acide) 194Eicosanoïdes 194Élimination d'un médicament 46- et accumulation 50- fonction exponentielle 44- hépatique 32, 38- et molécules hydrophiles/lipophiles
42- et protéines plasmatiques 30-; rénale 40Élimination présystémique 15, 18,42,
46- et p-bloquants 94- et estradiol 252- et opioïdes 208- et testostérone 250Embryon 73Emplâtre transdennique 12,16,18Émulsion 8, 16
Énalapril 34, 124Énantiomères 62Encaïnide 136Endocytose 26Endoparasite (traitements) 288Endoperoxydes cycliques 194P-Endorphine 208Endothélium- barrière entre sang et tissus 24- formation d'EDRP/NO 120- formation de prostacyclines 148- du glomérule 40- types 24Enflurane 216Enképhalines 34, 192,208Enoxacine 272Énoxaparine 146Entamoebia histolytica 272Enterobius vemùcularis 288Éphédrine 86, 87Epilepsie 188Épipodophyllotoxine 294Épithélium- bronchique et tabac 112-; stratifié 22Epoxy (formation de composés) 36Ergocomine 126Ergocristine 126Ergocryptine 126Ergométrine 126Ergostérol 280Ergot de seigle 126Ergotamine et migraine 316Ergotisme 126Ergotoxines 126Erythroblastes 138Erythrocytes (agrégation) 148Erythromycine 34, 265, 274Érythropoïèse 138Érythropoïétine 138Esmolol 95Espace- endoneuronal 204- extracellulaire 28- interstitiel 28- plasmatique 28Estérase 34Esters et hydrolyse 34Estomac (action des prostaglandines)
194Estradiol 252- benzoate 253- valérianate 253

Index 363
Estriol 252, 253Estrone 252, 253Étacrynique (acide) 160Éthambutol 278Éthanol- désinfection 286- élimination 44- libération d'ADH 162- régulation thermique 200Éther(éthylique)216,217Éthinylestradiol 252-254Éthinyltestostérone 252, 253Éthionamide 278Éthistérone 252Ethoforme 206Éthosuximide 190Ethyldésoxyuridine 286Étidronique (acide) 312Étiléfrine 86, 87Étofibrate 154Étomidate218Etoposide 294Étrétinate et grossesse 74Étude(s)- croisées 74- préclinique 6- randomisée 74Eugénol287Euthyréose 242Expectorants 318Expériences de liaison 56
FFab (fragments) 130Facteur- intrinsèque 138- plaquettaire 142Famotidine 166Felbamate 188Félodipine 122Felypressine 162Fenêtre thérapeutique 70Fenfluramine 88Fenotérol 84- et allergie 320,322Fentanyï 210Fer- carence en 138,140- sulfate 140Femtine 140Fibres nerveuses et anesthésie locale
203
Fibrine 34, 143Fibrinogène 143Fibrinolyse 146Fibrinolytiques 142, 146- traitements 146Fick (loi de) 44Fièvre 200- et prostaglandines 194- et régulation thermique 200Filtration glomérulaire 40,158Finastéride 250Flécaïnide 136Flucloxacilline 268Fluconazole 280Flucytosine280,281Fludrocortisone 246Plumazénil 224Flunarizine et migraines 316Flunisolide et inhalation 14,248Flunitrazépam 2275-Fluorouracile 280, 294Fluorures 312Fluoxétine 116,230Flupentixol 234, 236Fluphénazine 234, 236Flutamide 250Fluticason (propionate) 248Flutter auriculaire 134Fluvastatine 154Fluvoxamine 230Flux ioniques et myocarde 136Foie- abats et vitamine D 262- cycle entéro-hépatique 38- et élimination des médicaments 32- etiactulose 168- réactions de conjugaison 38- sinus hépatiques 32- structure des capillaires 24Folia sennae 172Folinique (acide) 294Folique (acide) 138,294Fonction de Bateman 47Fonction exponentielle 44Fongicide/fongistatique 280Formaldéhyde 286Formes- galéniques 8, 9- racémiques 625-Formyl-tétrahydrofolate 294Foscamet 282Fosinopril 124Fructose 151

364 Index
FSH 240, 250-254Fumeur (toux du) 112Furosémide 160- et hypercalcémie 262Fuseau (poison du) 292- etcolchicine310- et griséofulvine 280
GGABA 180, 188- antiépileptiques 188- benzodiazépine 224Gabapentine 188 -Galien Claude 2Gallamine 182Gallopamil 122Gamma-aminobutyrique (acide),
GABA 180, 188, 224Ganciclovir 284Ganglions 80, 108- blocage et d-tubocurarine 182Ganglioplégiques 108,128Gastrine 164Gaz hilarant 216Gélatine 16, 150Gemfibrozil 154Gentamicine 274, 276 ,Gestodène 252GH220Glandes- endocrines 24- surrénales 108Glaucome 104, 158, 160- à angle fermé 104Glibenclamide 260 yGlobulines et liaison du calcium 262B-Globulines et liaison
des médicaments 30Glomérule 40, 158Glomus carotidien 110Glucocorticoïdes 246- et allergie 320- asthme bronchique 322- crise de goutte 310- hypercalcémie 262- poly arthrite rhumatoïde 314Glucose, formule 169Glucose-6-phosphate-déshydrogénase
70Glucuronidase 38Glucuronidation 38Glucuronique (acide) 39
Glucuronyltransférase 32,38Glutamate 64, 188Glutamine et reactions de conjugaison
38Glutaraldéhyde 286Glutathion 121,196Glycérol 20Glycine 180- et réactions de conjugaison 38Glycogénolyse 82Glycosides cardiaques 20,130- et hypotension 308Glycyrrhizique (acide) 166GMPc 120GnRH 240, 250Goitre et carence en iode 242Gonadotropines 250,252, 254Goséréline 240Goudrons 113Goutte (traitement) 310- et diurétiques 160Gouttes- nasales 90, 318- oculaires 8Gradient 44Grand mal (crise de) 188Granisétron 116,324Granulocytes 70Granulocytopénie 70GRH 240GRIH 240Grippe 196, 318Griséofulvine 280Grossesse- et prise de médicaments 72- et vomissements 324Growth Hormone voir GHGTP 121Guanéthidine 96Guanylate cyclase 120Gynécomastie- et cimétidine 166- et spironolactone 162Gyrase (inhibiteurs de) 265, 272Gyrus post-centralis 192
HH^ et sensation douloureuse 192HVK^ATPase^Hahnemann, Samuel 76Hallucinogènes 238Halofantrine 290

Index 365
Halogène (désinfectant) 286Halopéridol 237, 325- et vomissements 324Halothane216- et hépatite 216- métabolisme 36Haptène 70Haschich 4, 238HCG250HDL 152Helicobacter pylori 166Helleborus niger 131Helminthes 288Hème 138Hémodilution 148,150Hémoglobine (synthèse) 138Hémosidérose 140Henle (anse) 160
. Héparine 142, 144Hépatocytes 32Héroïne 210Herpès simplex 282Herpès zoster 282Hexachlorocyclohexane 288Hexaméthonium 108Hexobarbital 220- comme somnifère 220Hibernation artificielle 236His (faisceau) 135Histamine 114, 320- et allergie aux médicaments 70- et antihistaminiques 114 '•"•- et douleur 192- libération et d-tubocurarine 182- métabolisme 36- sous-types de récepteurs 114HIV 284HMG CoA réductase 152- inhibiteurs 154Hohenheim Theophrastus von 2Homéopathie 74Homogénéisation 56Honnone(s)- antidiurétiques 162- hypophysaires 240- hypothalamiques 240- lutéinisante 240- sexuelles 250- somatotropes 240- thyréotrope 240,242- thyroïdienne 2425-HT/sérotonine 114Hydrochloromiazide 160,161
Hydrocortisone 246Hydrogel 16Hydrolyse 34Hydromorphone 208Hydrophilie 16,42- et élimination 42Hydrophobie et liaison aux protéines
30Hydroxyapatite 31225-Hydroxycholécalciférol 263Hydroxycobalamine 138- et empoisonnement par le cyanure
300Hydroxycoumarine 144Hydroxyéthylamidon 150,151Hydroxylases 32 .Hydroxyméthyl glutaryl coenzyme
A réductase 152Hydroxyprogestérone 252Hydroxytryptamme/5 HT 114Hypercalcémie 262Hyperglycémie 238Hyperkaliémie (après succinylcholine)
184Hyperlipoprotéinémie 152Hypersensibilité 68Hypertension 306Hyperthermie- et empoisonnement atropinique 106- maligne 180Hyperthyréose 242Hyperuricémie 310Hypnotiques 220Hypoglycémie 258Hypokaliémie- et diurétiques 160- et laxatifs 62- et syndrome de Cushing 246Hypolipidémiants 152Hypotension 308Hypothyréose 242Hypoxanthine310
1Ibuprofène 198Idoxuridine 282Ifosfamide 294Iloprostll8Imidazole (dérivés) 280Imidazopyridine 220 'Imipramine 206, 228-229,230-231Immunogène 70

366 Index
Immunosuppresseurs 292- et polyarthrite rhumatoïde 314Indométacine 198Induction enzymatique 32, 50,60- et antiépileptiques 188- et rifampicine 278Influenza virus 282Inhalation 14, 214- et anesthésiques 14, 200Inhibiteurs de l'enzyme de conversion
118, 132- dans l'hypertension 306Inositol triphosphate 64Inotrope 84Insecticide 102, 266, 288Insuffisance rénale 156Insuline (origine, récepteurs) 256Interactions- dipôle-dipôle 58- électrostatiques 58- hydrophobes 58- ion-dipôle 58Interféron (IFN) a, (i, -y 282Intemeurone 180Inuline 28Iode- carence 242- prophylaxie par les sels 242- teinture 288131 Iode 242lodure de potassium 242Ipratropium 104, 126,320, 322- et allergie 320 *Isoconazole 280Isofurane 216Isoniazide 278Isonicotinique (acide) 278,279Isopropanol 286Isoprotérénol 86- et inhalation 14- structure 95Isosorbide (dinitrate) 120- et angine de poitrine 304Isotrétinoïne et grossesse 74Isoxazolyl-pénicilline 268Isradipine 122Itraconazole 280
KKd56Kanamycine 274Kaolin 176
Kératine 280Kétamine218Kétoconazole 280Kétotifène 114Kininase II 124
LP-Lactamase 268P-Lactame (cycle) 266Lactique (acide) 287Lactose 168Lactulose 160Lait- formation et sécrétion 241- passage des médicaments dans le 72Lamotrigine 188Langendorff (préparation de) 128Langerhans (îlots de) 256Lansoprazole 166Laxatifs 168-175LDL 152Lécithines 20, 21Lèpre (traitement) 278Leu-enképhaline 208Leuconostoc mesenteroides 151Leucotriènes 194, 246, 314Leucovirine 294Leuproréline 240Lévétimide (énantiosélectivité) 62,63Lévomépromazine et vomissements 324Lévométhadone212Leydig (cellules de) 250LH 240,250,252Liaison(s)- amides 34- covalente 58- différents types 58Libération- pulsatile 241- des substances 9Lidocaïne 134,206- métabolisme 36Ligand 56Lin 168Lincomycine 274Lindane 288, 289Lipocortine 246Lipolyse 84Lipooxygénase 194Lipophilie 16,42- et élimination 42- et passage à travers les membranes 26

Index 367
Lipoprotéines 152P-Lipotropine 209Lisinopril 124Lisuride 186Lithium- et manie 232- et thyréotoxicose 244Lomustine 294Lopéramide 176,210Loratidine 114Lorazépam 227Lorcaïnide 136Lormétazépam 227- comme somnifère 220Lovastatine 154Low Density Lipoproteins 152LSD 126Lugol (solution) 244Lymphocytes 70Lymphokines 70Lynestrénol 252Lypressine 162Lysergique (acide) 126Lysosomes 26
MMacrolide 274Macromolécules- distribution dans l'organisme 28- et passage endothélial 24- propriétés colloïdales 150Macrophages 140Magnésium- empoisonnement par 164- hydroxyde 164- stéarate 16- sulfate 168- et transmission neuro-musculaire
180Mal de mer 106,324Maladie de Basedow 244Maladie de Parkinson 186Maladie sérique 72Malaria 290Manie (traitement) 232Mannitol- action diurétique 158,- action laxative 168, 169MAO 86, 88- B 186- inhibiteurs 88, 186,230Maprotiline 230
Marijuana 4, 238Masse (loi d'action) 56Mastocytes 72, 114, 320Maux de tête 197,316Mazindol 88Mébendazole 288,289Méclozine 114- et vomissements 324Médicaments- allergie aux 70- biotransformation (réactions de
phases I et II) 32, 34, 38- cinétique plasmatique 48- courbes de liaison 56- découverte 4- développement 6- et dilatation bronchique 126- dose et durée d'action 66- dose d'entretien 50- effets secondaires 68- effets sur des organes isolés 54- élimination 32- étude clinique (phases 1 à 4) 6- étude prospective 76- formes galéniques 8- interactions 32- liaison maximale/B,,,,, 56- liaison aux protéines plasmatiques 30- et malformations 74- mise sur le marché 6- relation dose-effet 52Médroxyprogestérone (acétate) 252Méfloquine 290Melphalan 294Membrane- étude de liaison 56- passage à travers 26- potentiel de 137- protéines intégrales 20- stabilisation 136Membrane basale 24- et glomérules 40Ménadione (vitamine K,) 144Menstruations et effet
des prostaglandines 194Mépéridine210Mépivacaïne 2076-Mercaptopurine 294Mérozoïtes 290Mésalazine 270Mescaline 116, 238MESNA 294Mestérolone 250

368 Index
Mestranol 252, 254Met-enképhaline 208Métabolisme 32- et liaison aux protéines
plasmatiques 30Métabolite 34Métamizol 196, 200Metamphétamine 86, 88Métaux- alcalins 232- lourds 113, 298Metendone 250Météorisme 178Metformine 260Méthadone 212- et accumulation 50Méthimazol 245Méthohexital218Méthotrexate 138, 294, 296, 314Méthoxyflurane 216N-méthyl-D-aspartique (acide)
NMDA 188, 218Méthylation 36Méthylcellulose 16Méthyldigoxine 132Méthyl-DOPA 96, 114- et hypertension 306- structure 97Méthyldopamine 96Méthylergométrine 126Méthyltestostérone 250, 251Méthylxanthine 320Méthysergide 114, 126- et migraine 316Métipranol 95Métoclopramide 114- et migraine 316- et vomissements 324, 325Métoprolol 94- et migraine 316Métronidazole 272Mexilétine 135Meziocilline 268Miansérine 230Miconazole 280Microcirculation 148Midazolam218,226Migraine (traitement) 316Milrinone 132Minéralocorticoïdes 246- et hypotension 308Minipilule 254Minocycline 276
Minoxidil 118- et hypertension 306Misoprostol 166Moclobémide 88, 230Molsidomine 120- et angine de poitrine 304Monoamine oxydase 82Monooxygénase 36Morphine 4, 208- hypersensibilité 68- test de hérissement de la queue 52Mucolytique 288Mucus bronchique et expectorants 318Multiplication virale 282Muqueuse (structure de l'épithélium) 22Muréine 266Muscarine 98Muscimol 238Mutagénicité 6Mycobactéries (traitements) 278Mycophénolate-mofétil 296Mycose 280- locale 280- systémique 280Mydriase 104Myorelaxants 182- et anesthésie 216- dépolarisants 184- non dépolarisants 182Myotonolyse 224Myotonolytiques 180
NNA voir NoradrenalineNa-, K-, Cl, cotransport 160Na-citrate et hypercalcémie 262Na (canal) 20, 136, 184,202- inactivation 174- Moqueurs 130, 134,202Na/Ca (échange) 130, 132Na-EDTA et hypercalcémie 262Na (influx) 137Na/glucose (cotransport) 20Na-K-ATPase 20, 130Naftifme 280Nalbuphine210Nalidixique (acide) 272Naloxone208,212Naltrexone212Nandrolone 250Naphazoline 84, 90- et allergie 320

Index 369
Naproxène 198Narcéine 4Narcose 214Narcotiques 214Naunyn, Bemhard 3Nédocromil 114,320Nématodes 288Néomycine 276- pommade 16Néostigmine 102- et plaque motrice 182Néphrite et allergie aux médicaments
72Néphron 158Nerf- facial 105- glossopharyngé 105- oculomoteur 105- vague 105Nétilmicine 276Neurohypophyse 240Neuroleptiques 114, 234- analgésie 214- régulation thermique 200- vomissements 324Neurones (pré- post-ganglionnaires) 82Neutropénie et cytostatiques 292Névrite et allergie aux médicaments 72Nicardipine 122Nicotiana tabacum 112Nicotine 98, 108- effet ganglionnaire 108- et fumée du tabac 112- et libération d'ADH 162- modification des fonctions
physiologiques 110- récepteurs 98, 108- structure 111Nicotinique (acide) 154Nifédipine 122- et angine de poitrine 304- et hypertension 306- structure 123Nimodipine 122Nisoldipine 122Nitrates 20, 116,118,122- et angine de poitrine 304- organiques 120- tolérance 120Nitrazépam 227- métabolisme 36- et somnifères 220, 224- structure 226
Nitrendipine 122Nitroglycérine/trinitrine 120, 304Nitro-imidazole 265, 272Nitroprussiate de sodium 118, 120- et hypertension 306Nitrosamine 113Nitrostigmine 102Niveau plasmatique 46N0100,120N,0, gaz hilarant 216Nocicepteurs 192Nœud atrio-ventriculaire 135Noradrenaline 82, 83- et anesthésiques locaux 204- et antidépresseurs 228- métabolisme 36- récepteurs et neuroleptiques 234- structure 85Nordiazépam 226, 227Noréthistérone 252Norfloxacine 272Noscapine4,210,318Nutrition parentérale 12Nystatine 280
0Obidoxime 300Occlusion 16Octréotide 240Ocytocine 126,240Œstrogènes 252, 254- antagonistes 254- conjugués 252- et ostéoporose 312- préparations contenant des 252Ofloxacine 272, 273Oméprazole 166Opioïdes 192, 208- et diarrhées 176- endogènes 208Opium 212- alcaloïdes 4, 208- teinture 4, 176Or (sels d') 314Orciprénaline 86, 87Organophosphores 102Ornipressine 162, 163Osmodiurétiques 156,158Ostéoblastes312Ostéoclaste 262Ostéomalacie 262Ostéoporose 312

370 Index
Ovocyte (maturation) 241Ovulation 252,254Ovule vaginal 12Oxacilline 268, 269Oxalate 142Oxatomide 114Oxazépam 226, 227Oxiconazol 280Oxydants (rôle désinfectant) 286Oxydase à fonction mixte 32Oxydation (reaction) 32, 36Oxymétazoline et allergie 320Oxypurinol 310
PP 450 32Paclitaxel 292Pamidronate312p-Amino-benzoïque (acide) 146Pancréas 256- structure des capillaires 24Pancréatozymine 172, 178Pancuronium 182Pantoprazole 166Papavérine 4, 208Paracelse 2, 68Paracétamol 196,200- empoisonnement 196- métabolisme 36- et migraine 316- et refroidissement 318Paraffine 16, 172Paralysie respiratoire 182Paraoxon 36, 102, 103Parasympathique 98,128- effets 105- récepteurs 104Parasympatholytiques 104,128, 132- effets hallucinogènes 238- et hypotension 308- et maladie de Parkinson 186- et régulation thermique 200- et rhume 318- et vomissements 324Parasympathomimétiques 102,104,
128- directs/indirects 102Parathion 102- métabolisme 36Parathormone 262Parkinson (maladie de) 186Paromomycine 276
Paroi cellulaire (inhibiteursde synthèse) 266
Paroxétine 230Pavot 4Peau- dermatologie 16- structure de l'épithélium 22Pectine 176Penbutolol 94Pénétration des virus 282Pénicillamine 314, 315Pénicilline (allergie à la) 72Pénicilline G 72, 266, 267Pénicilline V 268, 269Pénicillinase 268- inhibiteurs 268Pentazocine 204, 208- effet somnifère 211Pentétate 292Pentobarbital 221Pentoxifylline 148Peptidases 34Peptidyltransférase 274Perchlorate 244Pergolide 186Périndopril124Périneurium 204Période réfractaire 136Péristaltisme- et anti-diarrhéiques 176- et laxatifs 168Peroxydase (thyroïde) 244Peroxydes 286Péthidine 126, 208Petit mal (crise) 188Peyotl 238PGË2 126PGFzd 126Phagolysosomes 26Pharmacocinétique 6,44,48- fonction de Bateman 48Pharmacodynamie 4Pharmacogénétique 68Phase I, réactions de 32, 34Phase II, reactions de 32, 34,38Phénacétine (métabolisme) 36Phencyclidine 238Phéniramine 114Phénobarbital 188- et cellule hépatique 33- métabolisme 36Phénol 286Phénolphtaléine 172

Index 371
Phénothiazine 234Phénoxybenzamine 90Phénoxyméthylpénicilline 268Phenprocoumone 145- accumulation 50Phentolamine 90- et hypertension 306Phénylbutazone et crise de goutte 310Phénylmercure borate 287Phénylphénol 287Phénytoïne- propriétés anti-arythmiques 136- propriétés anti-épileptiques 188Phosphate (excrétion) 260Phosphatidylcholine 20, 21Phospatidylinositol 20Phosphatidylsérine 20Phosphodiestérases 66- inhibiteurs 20, 66, 128,132, 320Phosphokinase 84Phospholipase A, 194, 246Phospholipase C 66Phospholipides 142, 152, 194- et coagulation sanguine 142- et double membrane 20, 21- et lipoprotéines 152- rôle de barrière 22Phosphorique (acide) 20Physostigmine 102, 103Phytoménadione (vit. K,) 144Pilocarpine 102, 104Pilule
|/ - anticonceptionnelle 254- séquentielle 254- simultanée 254Pindolol 94, 95Pipécuronium 182Pipéracilline 268Pirenzépine 104, 164Pirétanide 160Piroxicam 198- et polyarthrite rhumatoïde 314Pizotifène et migraine 316pK 40,206Placebo (étude contrôlée contre) 76Placenta 126Plaque motrice 100, 186Plaquettes sanguines (thrombocytes) 142Plasma (substituts) 150Plasmine 146Plasminogène (activateur) 146Plasmodium 290Plathelminthes 288
Plicamycine et hypercalcémie 262Poison 2, 70Polarité 58- et solubilité 42Polonium (iodure) 244Polyarthrite rhumatoïde 314Polydocanol206Polyènes (antibiotiques) 280Polyéthylène glycol 16Polyglutamine (et acide folique) 138Polymyxine 264Polypeptides (antibiotiques) 264Polyvinyï pyn-olidone-iode 286Pores protéiques 136Post-charge 302Potassium- ions et douleur 192- permanganate 286Potentiel d'action 136- et cœur 128, 134- et muscle squelettique 180Potentiation (homéopathie) 76Poumon et métabolisme
des médicaments 42Poux (traitement contre) 288Pravastatine 154Prazépam 227Praziquantel 288Prazosine 90- et hypertension 306Prednisolone 246, 247- métabolisme 36Pregnandiol 253Pression artérielle 306Pression intra-oculaire 104Pression orthostatique et régulation 308PRH 240PRIH 240Prilocaïne 206, 207- métabolisme 34Primaquine 288, 290Primidone 189, 190Probénécide310- et élimination de la pénicilline 266Procaïnamide 134Procaïne 134, 135,206- métabolisme 34Procaïne-pénicilline G 266Prodynorphine 208Proenképhaline 208Progestérone 241, 252,253Proguanil 290Prolactine 240

372 Index
Prométhazine 114- spécificité d'action 70Pro-opiomélanocortine 208Propafénone 136Propofol218Propranolol 94- énantiosélectivité 62- métabolisme 36- et migraine 316Propylthio-uracile 245Propyphénazone 196Prostacycline 116, 148,194Prostaglandines 20, 192-196,246, 314- effets biologiques 194- inhibiteurs de synthèse 314- nomenclature 194- et régulation thermique 198- et ulcères 166Prostate (carcinome) 240,250Protamine 60- et héparine 146- et insuline 256Protéases 142Protéines- inhibiteurs de synthèse 274- synthèse 274Protéines G 64- récepteurs couplés aux 64Protéine kinase- A 66- C66Protéines plasmatiques- et élimination des substances 30- propriétés colloïdales 150Prothrombine 143, 144Protonation et élimination rénale 40Protozoaires 288Pseudo-allergie 196Pseudo-cholinestérase 184Psilocine 238Psilocybine 238Psychisme (médicaments du) 224Psychomimétiques 238Psychose 228Puces (traitement contre) 288Pupille (dilatation) 104Purine 270Purkinje (fibres de) 135Pyrazinamide 278Pyrazolidine et dérivés 196Pyridine méthanol 154Pyridostigmine 102Pyridoxine 278
Pyridylcarbinol 154Pyriméthamine 290Pyrogène 12, 200Pyrophosphate 312
QQuinalapril 124Quinidine 136- structure spatiale 62Quinine 62, 290Quinolone 272
RRachitisme 262Ramipril 124Ranitidine 165, 166Ranvier (nœud de) 204Rauwolfia 96Réactions- anaphylactiques 72- cytotoxiques 70- du métabolisme 34Récepteurs 58, 60- études de liaison 56- mécanismes de transduction 64
..- spécificité 69Récepteurs adrénergiques 82- a pré- post-synaptiques 90,96- sous-types 84Récepteurs a- post-synaptiques 96- pré-synaptiques 82- et transduction des signaux 66Récepteurs p 84Récepteurs cholinergiques 100- M, 104- M; 104- muscariniques 98, 100,104Récepteurs GABÂA 64, 180, 224Récepteurs GABAe 1805a-Réductase 250Réduction 36Refroidissement (traitement) 318Règle d'équivalence 76Réglisse 166Rein- circulation et action
des prostaglandines 194- diurétiques 156- élimination des médicaments 40- structure du glomérule 40

Index 373
REM (sommeil) 220Rémanence 286Rénine-angiotensine-aldostérone
(système) 118, 156Repolarisation 136Réserpine 96, 97, 114- et hypertension 306Résistance des bactéries 264Retard (produit) 10Réticulum endoplasmique 32Réticulum sarcoplasmique- et cœur 128- et muscle squelettique 180Reverse T, 245Reviparine 146Rhodanide synthétase 300Rhubarbe 164, 166Rhume 318Ribonucléique (acide), ARN 272,274,
282,294Ricin (huile) 172, 173Ricinoléique (acide) 172Ricinus communis 173Rifabutine 272Rifampicine 265, 272,278Rocuronium 182Roxithromycine 274
SSalazosulfapyridine 270Salbutamol 84, 86- et allergie 320Salicylique (acide) 199- glucuronidation 38- liaison aux protéines 30- structure 35Salmeterol 322Saliurétiques voir DiurétiquesSaquinavir 284Schistosomes 288Schizontes 290Schizophrénie (traitement) 234Schlemm (canal de) 105Schmiedeberg, Oswald 3Scopolamine 106- effet hallucinogène 238- et vomissements 324Sécrétion active 40Seigle (alcaloïdes de l'ergot) 126Sélégiline 186Sels métalliques 176Serine 20
Sérotonine 116- et antidépresseurs 228- et douleur 192- et hallucinogènes 238Sevrage 240- et prise de médicaments 72SIDA 282Signaux (transduction des) 66Silice 16Similitude (principe de) 76Simvastatine 154Sirop 8Site- d'action cellulaire 20- d'administration d'un médicament 18- de liaison 56P-Sitostérol 154Slow reacting substance of
anaphylaxis, SRS-A 194SNC (système nerveux central)- et structure des capillaires 24Sodium (Na)- carbonate acide, antiacide 164- hypochlorite 286
_- picosulfate 168, 171- sulfate 168- thiosulfate 120
••"- urate 310Somatomédine 241Somatostatine 240Sommeil- altérations 222- paradoxal 220- préparation au 222- seuil 222- stades 220Somnifères 220- dépendance 220Sorbitol- action diurétique 158- action laxative 168Sotalol 95Spasme coronaire 302Spasmolyse 126Spécificité d'organe
et effets secondairesdes médicaments 58,70,71
Spectre large/étroit et antibiotiques 264Spiramycine 274Spironolactone 162Sporozoïtes 290Stanozolol 250Staphylocoques 268

374 index
Status epilepticus 188Stérilisation 286Stéroïdes- et diabète 246- et transduction des signaux 66, voir
aussi Aldostérone, Androgènes,Glucocorticoïdes, Œstrogènes,Progestogènes
STH 240Streptocoques 147Streptokinase 146Streptomyces 272, 274,281Streptomyces griseus 139Streptomycine 274, 278g-Strophantine 132, 133Strychnine 180Substances psychédéliques 238Substantia nigra 186Succinylcholine 184Sucralfate 166, 167Sucre sanguin 258Suggestion (force de) 76Sulfacarbamide 271Sulfadoxine 290Sulfalène271Sulfaméthoxazole 270Sulfapyridine270,271Sulfasalazine 270Sulfate de calcium 8Sulfate et conjugaison 38Sulfisoxazole271Sulfones 278Sulfonamides 265, 270- et antidiabétiques oraux 260- liaison aux protéines 30Sulfotransférase 38Sulfoxide 36Sulfurique (acide) 39Sulprostone 126Sumatriptan 116Suppositoires 12Surdosage 70Suspension 8, 16Suxaméthonium 184Sympathectomie pharmacologique 96a-Sympatholytiques 90P-Sympatholytiques, P-bloquants 92Sympathomimétiques 128, 222- et allergie 320- directs 84-88- indirects 84-88- relation structure activité 86a-Sympathomimétiques 84
- et hypertension 306- et rhume 318P-Sympathomimétiques 84,118p2-Sympathomimétiques 126Synapse- adrénergique 82- cholinergique 100Synergie 60Syncythiotrophoblaste 74Système nerveux- autonome 80- somatique 80- végétatif 80Système réticulo-endothélial 140Système sympathique 80, 128- conséquences d'une activation 80- et régulation thermique 200- structure 82- transmetteur 82- types de récepteur 84
TÏ3 (triodothyronine) 242Ï4 (thyroxine) 242Tabac et épithélium bronchique 112Tachycardie sinusale 135Tachyphylaxie 88Tacrine 102Tacrolimus 296Talinolol 95Talc 16Tawara (réseau) 135Tazobactam 268Teinture 4Témazépam 227- action somnifère 220Téniposide 294Tensio-actifs (désinfectants) 286Tératogénicité 6,74,292Térazosine 90Terbutaline 84- et allergie 320Terfénadine 114Tertatolol 95Testostérone 250, 251- undécanoate 34Tête polaire 20Tétracaïne 206Tétracyclines 265, 274, 276- et grossesse 74Tétraéthylammonium 108Tétrahydrocannabinol 238

Index 375.
Tétrahydrofolique (acide) 138,294- inhibiteurs de synthèse 271- structure 250, 270Tétryzoline 84, 90- et allergie 320Thalamus 193Thalidomide 74Thallium 294, 300Thé 176, 320Théine 176, 320Théophylline 118,126,222, 320Thermorégulation 200Thiamazol 245Thiazides 160- et hypertension 306Thiocyanate 300Thiopental218Thiothepa 294Thiourée 244Thioxanthène 236Thrombine 143, 146Thrombocytes 142- inhibiteurs de l'agrégation 148Thrombocytopénie et médicaments 72Thrombokinase tissulaire 142Thrombopénie et cytostatiques 292Thrombose 142Thromboxane A; 148Thymérétiques 228Thymidine 282Thymidine kinase 282,283Thymine 282Thymol 287Thymoleptique 228Thyréoglobuline 244Thyréostatique 244Thyrothricine 244Thyroxine 242, 243Thyroxine binding globulin, TBG 30Tiagabine 188Ticarcilline 268Ticlopidine 148Tight junction 22Timolol 94Tinidazole 272Tinzaparine 146Titane (oxyde) 16Tobramycine 275,276Tocaïnide 136Tocolyse 84, 126Tolbutamide 260, 261Tolérance 210Tolonium (chlorure de) 300
Toluidine 35Topoisomérase n 272, 294Tout ou rien (principe du) et effets des
médicaments 52Toux 318Toxicité 6Toxicologie 6Toxine- botulinique 180- tétanique 180tPA (tissue plasndwgen activator) 146Traduction 274Traitement palliatif 292Tramadol 208Trandolapril 124Tranexamique (acide) 146, 147Transcobalamine 138Transcortine 30Transcriptase inverse 282,284Transcription 272Transferrine 30, 140Transmetteurs (seconds messagers)
20,66Transmission neuromusculaire 180Transpeptidase 266, 268Transport- énantiosélectivité 62- à travers les membranes 26- mucociliaire 14- - et fumée du tabac 112- protéine de 20- systèmes de et rein 40- vésiculaire 26Tranylcypromine 88,230Trématodes 288TRH 240, 242Triamcinolone 246, 247Triamtérène 162Triazolam 227- action somnifère 220, 222- structure 221Trichinella spiralis 288Trichlorméthiazide 160Trichomonas vaginalis 272Triflupromazine 234- structure 237Trifluridine 283Triglycérides 152Triiodothyronine (t,) 242,243Trimétaphan 98,108Triméthoprime 265, 270Triptoreline 240Tropicamide 104

376 Index
Tropisétron 116,324TSH 240, 242Tuberculose et traitement 278d-Tubocurarine 98, 182Tubule- distal 158- trans verse 128Tyrosine 82, 97, 242Tyrosine kinase 64Tyrothricine 244, 264
UUlcère gastrique (traitement) 164,166Uracile281Uricosurique 310Urine primitive 158Urique (acide) 160,310Urokinase 146Ursodésoxycholique (acide) 178Urticaire 72Utérus, action des prostaglandines 194
VValproïque (acide) 188,189Vancomycine 265, 268Van der Waals (liaisons) 58Vanylmandélique (acide) 82Varicosité 82Vasculitis 72Vaseline 16Vasoconstricteur et anesthésie
locale 204Vasodilatateurs 118Vasopressine 162, 163- et anesthésie locale 206Vécuronium 182Veine porte et hypophyse 240Vérapamil 122- et angine de poitrine 304- anti-arythmique 134,136- et hypertension 306Vers- plats 288- ronds 288Very low density lipoproteins, VLDL
152Vésicule biliaire 32Vidarabine 282, 283Vigabatrine 188
Vinblastine 292Vinca rosea 292Vincristine 292Viomycine 278Virus (lutte contre) 282Virustatiques282,318Vitamine A (acide rétinoïque)
et grossesse 74Vitamine- B,,278- B,2138,139- - carence 138- C140- D 262, 263- K 144- - et céphalosporines 268- K, 144, 145- K; 144,145- K:3 144, 145Voie- buccale 18- intramusculaire 18- intraveineuse 18- perlinguale 18- rectale 18- sous-cutanée 18Volume sanguin 28Vomissement (centre du) 324
WWarfarine 145Wepfer, Johann Jakob 3
XXanthine 310- nicotinate 154Xanthine oxydase 310Xylométazoline 84, 90
ZZidovudine/AZT 284Zinc- et insuline 256- oxyde 16Zollinger-Ellison (syndrome de) 166Zolpidem 220Zonula occludens 22, 24Zopiclone 220

Composition : PFC-Préface, Dole
Achevé d'imprimer par i^t^ Corlet, Imprimeur, S.A.14110 Condé-sur-Noireau (France)
° d'Éditeur : 10545 - N° d'Imprimeur : 52168 - Dépôt légal : janvier 2001Imprimé en U Ë.



























![Atlas de Poche Microbiologie-[Www.worldmediafiles.com]](https://img.pdfslide.fr/doc/110x75/55721316497959fc0b91917c/atlas-de-poche-microbiologie-wwwworldmediafilescom.jpg)

![Atlas de Poche de Pharmacologie [Flammarion]](https://img.pdfslide.fr/doc/110x75/55cf9699550346d0338c9852/atlas-de-poche-de-pharmacologie-flammarion.jpg)
