Embed Size (px)
Citation preview

Eur. J. Biochem. 153,275-287 (1985) FEBS 1985
Bovine serum brevin Purification by hydrophobic chromatography and properties
Zohra SOUA, FranGoise PORTE, Marie-Cede HARRICANE, Jeanne FEINBERG and Jean-Paul CAPONY
Centre de Recherches de Biochimie Macromoleculaire du Centre National de la Recherche Scientifique et Unite 249 Institut National de la Sante et de la Recherche Medicale, Montpellier
(Received June 11/August 28, 1985) - EJB 850638
Brevin, an actin-severing protein present in serum from numerous mammals, has been purified to homogeneity from bovine serum, using hydrophobic chromatography as the last purification step. The physicochemical parameters of brevin have been established and some of them studied in the absence and presence of Ca2+. Brevin exhibits an apparent Stokes radius, R,, of 3.4 nm, an intrinsic sedimentation coefficient s&,,, of 4.8 S and 4.4 S in the absence and presence of Ca2 + respectively, indicative of calcium-induced conformational change. The native molecular mass of brevin was found to be 68 kDa and the hydrodynamic data suggest that the protein is an asymmetric molecule. Sedimentation equilibrium studies demonstrated that Ca2 ’ affects the shape (asymmetry) of brevin without altering its molecular mass. Limited tryptic and chymotryptic digestion of brevin distinguishes the Ca2 -induced conformation from the EGTA one. No change in the electrophoretic migration of brevin was seen upon CaZ + addition. Several isoforms were detected by two-dimensional gel electrophoresis. Brevin increases the rate of nucleation of actin but decreases the rate of elongation of the filaments and the steady-state viscosity of F-actin in substo’ichiometric amounts, as measured by viscometric assays under high shear conditions. Electron microscopic examination documents these effects. Brevin produces shorter actin filaments and binds to the ‘barbed’ end of filaments to which monomers add preferentially during elongation, as demonstrated by indirect immunogold staining of antibodies against brevin. Filament elongation occurs only at the slowly growing end. An enzyme-linked immunosorbent assay was developed and used to detect and quantify brevin and related proteins in extracts of different bovine cells and tissues. Liver and smooth muscles were found to contain the highest amounts of the severing protein.
Recent years have seen rapid progress in studies on the composition of various filaments within cells. Their spatial organization and possible functions have focused much in- terest on the cytoskeleton, and particularly on the microfila- ment system.
Actin is a contractile and structural protein widely dis- tributed in all eukaryotic cells [l, 21. Besides its structural role and maintenance of cell shape, in non-muscle cells actin is constantly being reorganized according to the physiological state of the cell [3] and in response to functional demands. These dynamic events 11, 41 are related to a spatial and a temporal control of actin assembly and diassembly.
The study of the molecular mechanisms underlying these events has led to the discovery within various cells of numerous proteins which modulate the assembly state of actin. Three major classes of actin binding proteins have been identified [5] : (a) actin cross-linking proteins, (b) severing and capping proteins, (c) G-actin stabilizing proteins.
Several proteins which interact with F-actin in a calcium- regulated way have been identified in a wide variety of cell types: gelsolin from rabbit macrophages [6 - 81, fragmin from
Correspondence to Z. Soua, Centre de Recherches de Biochimie Macromoleculaire du Centre National de la Recherche Scientifique, BP 5051, Route de Mende, F-34033 Montpellier Cedex, France
Abbreviations. G-actin, globular actin; F-actin, filamentous actin; DNase I, deoxyribonuclease I; HMM, heavy meromyosin; SDS, sodium dodecyl sulfate; ELISA, enzyme-linked immunosorbent assay.
the acellular slime mold PhjcsarzNn polyrephalum [9, lo], severin from Dictyostelium discoi‘deum [l l] , 90-kDa protein from human platelets [12, 131, villin from chicken intestinal epithelial cells [14- 161 and actin-fragmenting protein from spinal cord and brain [7].
Furthermore, blood plasma and serum from various mammals appear to be a rich source of Ca’+-dependent F-actin destabilizing factor. This factor was first described and partially characterized in human serum [18 -201, then was isolated from pig plasma as a 92-kDa polypeptide, but exhibited Ca2 +-independent interaction with actin [21]. It was purified latter from human serum [22]. The rabbit serum protein was isolated and named brevin [23].
Brevin and other actin-binding proteins which strongly bind Ca2+ ions 18, 241 restrict filament length at sub- stoichimetric concentration, in a Ca2 + -dependent manner, producing short non-sedimentable actin oligomers. This severing activity probably results from binding to the fast- growing end of actin filaments preventing them from annealing. In addition, severing proteins enhance actin nucleation leading to the formation of shorter filaments.
This paper describes a rapid and convenient purification of brevin from bovine serum. Brevin is obtained in a highly purified form and we have studied some of its physical and chemical properties, together with its mode of interaction with actin. The production of rabbit antibodies against brevin is described, as well as the use of these antibodies in an enzyme- linked immunosorbent assay to detect and quantify the pro- tein in extracts of different cells and tissues.

276
MATERIALS AND METHODS
Materials
DEAE-Sepharose CL-6 B, Sephacryl S-200 and S-300, phenyl-Sepharose were from Pharmacia. Polyacrylamide gel electrophoresis reagents were from Bio-Rad and molecular mass markers from Pharmacia. All other chemicals were from Merck or analytical grade.
Actin purification
Actin was purified from rabbit skeletal muscle acetone powder by the modified method of Spudich and Watt [25]. After a polymerization-depolymerization cycle, actin was sonicated and dialyzed against buffer A (2 mM Tris/HCl, 0.2 mM ATP, 0.2 mM CaC12, 0.5 mM dithiothreitol pH 7.8). Purified actin was stored as G-actin in buffer A, at - 80°C and used for several months without loss of activity.
Isolation of brevin
Bovine serum was obtained from the local slaughterhouse after blood clotting at room temperature during 3 h, then at 4°C for 5 h. The clot was removed by centrifugation (5000 x g , 15 min) and serum was stored frozen at - 20 "C.
All purification steps were performed at 4°C. In order to minimize proteolysis by Ca2+-activated proteases present in serum, EGTA was added to all buffers.
Thawed serum (250 ml) was fractionated between 30% and 50% ammonium sulfate saturation at pH 7.3. The re- sulting pellet was subjected to chromatography on a DEAE- Sepharose CL-6 B column equilibrated with 20 mM Tris/HCl, 0.2 mM dithiothreitol, 2 mM EGTA, 80 mM NaCI, pH 7.8. A linear NaCl gradient (80 - 200 mM in 7 bed volumes) was applied to the column and brevin was eluted at about 130 mM, together with a major contamination by serum albumin. Fractions containing brevin were concentrated, then loaded onto a Sephacryl-S 200 column. Final purification was achieved by hydrophobic chromatography on a phenyl- Sepharose column. Brevin was loaded on the column at high ionic strength in the presence of CaC1, and was then eluted at low ionic strength in the presence of EGTA. The hydrophobic phase has to be recycled very carefully in order to preserve its binding capacity.
After each use, the phenyl-Sepharose column was washed with 6 M guanidinium hydrochloride (1 column volume) in order to remove strongly bound proteins, then with water (2 column volumes) and finally regenerated as described by Pharmacia.
Antiserum preparation The antiserum against bovine brevin was produced in New
Zealand rabbits by injection, at multiple intradermal sites, of 0.2 mg brevin in 1 ml of 10 mM Tris/HCl, pH 7.5, 0.2 mM dithiothreitol, 2 mM CaC12. For the first injection, brevin was emulsified with an equal volume of complete Freund's adjuvant. Two other injections were made 2 weeks apart. Booster injections were made every month and rabbits were bled 13 days after the last injection.
The antiserum against bovine brevin was purified on an affinity column of brevin-Sepharose. The antibody was eluted from the column with 0.2 M HCI adjusted at pH 2.8 with 2 M glycine. The eluate was neutralized immediately with 1 M Tris base.
Antibody concentration was determined using A:;;",,, = 1.46 for 1-mg/ml solution [26].
Prepuration of cell und tissue extracts
Bovine alveolar macrophages were obtained by tracheal lavage of lung. Polymorphonuclear leucocytes were isolated from fresh bovine blood anticoagulated with heparin (0.1 mg/ ml) as described in [27]. All cells and tissues were frozen in liquid nitrogen and finely ground in a mortar. Resulting powders were suspended in 10% (w/v) trichloroacetic acid, centrifuged at 10000 x g for 10 min, washed successively with diethyl ether/ethanol (95/5, v/v), diethyl ether/ethanol (98/2, v/v), finally with diethyl ether, and dried under a stream of nitrogen. Proteins were extracted at room temperature for 10 min with 9.3 M urea and 2% Nonidet P40 [28].
Enzyme-linked irnmunosorbent assay (ELISA)
Brevin was quantified in various cells and tissues by using an indirect competitive ELISA [29]. To avoid interaction be- tween antibodies and brevin present in the antiserum, anti- bodies purified on a brevin-Sepharose affinity column were used throughout. To determine the optimal concentrations of antigen and antibody to be used in the assay, enzyme activity was studied as a function of antibody concentration for dif- ferent amounts of coated brevin (0.2 - 0.5 yg/ml).
Brevin was diluted in 50 mM carbonate/bicarbonate buff- er pH 9.6; 150 yl was added to each well in a flat-bottomed microtitre plate and incubated overnight at 4°C. Residual absorption sites on the plastic surface were saturated by treat- ment with lo% bovine serum albumin in NaCl/Pi (140 mM NaC1, 3 mM KCI, 10 mM potassium phosphate, pH 7.4), 0.050/0 Tween 20, for 2 h at room temperature.
The plate was then washed five times with NaCl/Pi containing 0.05% Tween 20,2 mM EGTA, and incubated for 2 h at room temperature with diluted antiserum (1 50 pl/well) which had been preincubated overnight at 4°C with or without the antigen-containing samples. Triplicate determina- tions were performed. After washing five times, the alkaline- phosphatase-conjugated antibody (Miles-Yeda Ltd) diluted 1 : 3000 with NaCl/Pi containing 0.05% Tween 20, was added and left 2 h at room temperature. The substrate, p-nitrophenyl phosphate, was dissolved (1 mg/ml) in 10% diethanolamine, 1 mM MgCl,, 0.02% NaN3, adjusted to pH 9.8 with 1 M HCl, and 150 pl was added to each well. The reaction devel- oped in 60 - 180 min and its extent was measured at 405 nm in a Titertek Multiskan spectrophotometer (Flow Laboratories, Meckenheim, FRG). All values were corrected for the blank values obtained in wells without antigen. Data were analyzed by program I described by Ritchie et al. [30].
Assay of brevin activity
The severing activity of brevin was followed by viscometric assays. Viscometry was carried out in a Cannon-Manning/ Semi-micro E 50 viscometer at 25 "C, under high shear conditions. The observed viscosity depends then only on poly- mer concentration and length distribution. The standard reac- tion mixture contained 0.5 mg/ml (12 yM) actin in buffer A. Polymerization was initiated by addition of 100 mM KC1, 2 mM MgC1, and 10 mM Tris/HCl pH 7.5 in a total volume of 0.3 ml. Specific viscosity is defined as flow time of the

277
sample solution divided by flow time of the corresponding buffer minus one. Generally buffer flow time was 150 - 156 s.
Deternzination of Stokes radius
Analytical gel filtration experiments were performed at room temperature by high-performance liquid chromatog- raphy on TSK G 3000 SW, (LKB) column (7.5 x 600 mm) equilibrated in 50 mM Tris/HCl, pH 7.5, 150 mM NaC1, 0 2 mM dithiothreitol and 2 mM EGTA. Elution volumes were reproducible to within 0.44 ml. Calibration of the column was made as in [31]. The void volume (V,) and total included volume ( V,) were identified with thyroglobulin and glucosamin respectively.
Brevin solution was run individually and proteins markers in two groups. The Stokes radius of brevin was determined as described by Siege1 and Monty [32].
Analytical ultracentrifugation
Ultracentrifugation studies were carried out in an MSE analytical ultracentrifuge equipped with an ultraviolet scanner to trace the protein boundary at 280 nm. Sedimentation coefficients were determined both with and without Ca2+ at a brevin concentration of 0.8 mg/ml in 10 niM Tris/HCl pH 7.5, 0.2 niM dithiothreitol, 0.1 M KCl, 2 mM CaZ+ or EGTA, simultaneously in a six-hole rotor. The molecular mass of native brevin was determined by the sedimentation equilibrium method of Yphantis [33], at an initial protein concentration of 0.25 mg/ml in 2 mM EGTA.
Amino m i d analysis
Brevin was lyophilized and hydrolyzed in constant-boiling HCl in evacuated sealed tubes at 110°C for 24 h, 48 h and 72 h using norleucine as internal standard. Amino acid ana- lyses were performed on a Beckman Multichrom analyzer model 4255 according to Moore and Stein [34]. Phospho- amino acid determination after partial acid hydrolysis was performed as in Capony and Demaille [35].
Gel electrophoresis
Sodium dodecyl sulfate/polyacrylamide gel electro- phoresis was used to identify brevin during purification steps and to determine its molecular mass. Protein samples were submitted to electrophoresis according to Laemmli [36] on 1.5-mm-thick slab gels consisting of 0.1 YO SDS/(5-20%) polyacrylamide gradient. The molecular mass of native brevin was determined using polyacrylamide gradient gel electrophoresis PAA 4/30 (Pharmacia). Two-dimensional polyacrylamide gel electrophoresis were performed according to O'Farrell [28] using a pH gradient of 5 - 7.
Peptide mapping
Limited digestions of brevin were carried out with chymotrypsin and trypsin in the presence or absence of Ca2+, using phenylmethylsulfonyl fluoride and soybean trypsin in- hibitor, respectively, to stop the protein digestion.
Peptide maps of brevin were obtained by SDS/ polyacrylamide gel electrophoresis using the gel system de- scribed above.
Electron microscopy
Actin filaments were examined under electron microscopy after negative staining. Samples were mounted on Formwar/ carbon-coated and glow-discharged grids, fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate, pH 7, then washed with 0.1 M ammonium acetate, pH 7, and negatively stained with 1% uranyl acetate [37]. Grids were observed with a JEM 200-CX electron microscope at an accelerating voltage of 80 kV.
Skeletal muscle actin (3 1M) was polymerized for 30 min as previously described. F-actin was incubated with brevin (at the indicated molar ratio) in the presence of 2 mM Caz+ for 15 min.
To observe the growth of actin filaments in the presence of brevin, shortened filaments were mixed with heavy meromyosin (HMM, 1 mg/ml) in 10 mM imidazole, pH 7.2, 40 mM KC1 and 5 mM MgC12, at an actin/HMM weight ratio of 1.7 for 10 min at 25°C. Decorated actin filaments showed the characteristic arrowhead structure under electron micros- copy. They were then glutaraldehyde-fixed according to Cooper and Pollard [38], incubated with two parts of G-actin (1 30 pg/ml), and further polymerization was initiated by 20 mM KC1. After 6 min at 25 "C, samples were processed for electron microscopy.
Brevin molecules were localized on HMM-decorated actin filaments using the indirect immunogold staining method. The brevin-antibrevin complex was obtained by preincubation for 10 min at weight ratio of 1 :2, then mixed with F-actin at brevin/actin ratio of 1 : 20, in the presence of 2 mM Caz+. Antibrevin IgG were labeled in a following incubation with 20-nm gold/goat anti-(rabbit-IgG) conjugates (GAR-G 20) (Janssen Pharmaceutica, Belgium) for 20 min at a 1/20th dilu- tion. The sample was processed for negative staining as pre- viously described.
For every experimental condition, an average of 350 filaments were counted to measure actin filament lengths. Length distribution of F-actin was calculated in the presence and absence of brevin.
Miscellaneous
Density measurements of buffer solutions were carried out at 25°C with a digital densitometer (DMA 40 Paar, Graz, Austria). Orthophosphate was measured by the Malachite green method [39]. Total Ca2+ was measured by atomic absorption spectrophotometry by using a Varian model 11 50 apparatus. EGTA was used for Ca2+ buffer as described in Haiech et al. [40]. Experimental points were fitted to the following theoretical curve:
where qmax is the specific viscosity of F-actin; r] is the specific viscosity of F-actin at different concentrations of CaCI,; Zmax is the maximal inhibition brought by brevin; [Ca] is the free CaZ + concentration; Kd is the apparent dissociation constant; n is the minimal number of Ca2+ ions involved in the activity of brevin. An SAS package was used to analyze the data. Protein determination was carried out by the Coomassie blue technique I411 with bovine immunoglobulins as standard (0.1 5 mgiml). Actin concentrations were measured using the
278
specific absorbance at 290nm (Az9,, = 0.63 Cm-' mg-' ml). Heavy meromyosin was prepared by limited digestion of rabbit skeletal muscle myosin according to Weeds and Pope [42]. Automatic Edman degradation was carried out in a Socosi liquid-phase sequenator according to Edman and Begg [431.
RESULTS
Purification o j hrevin
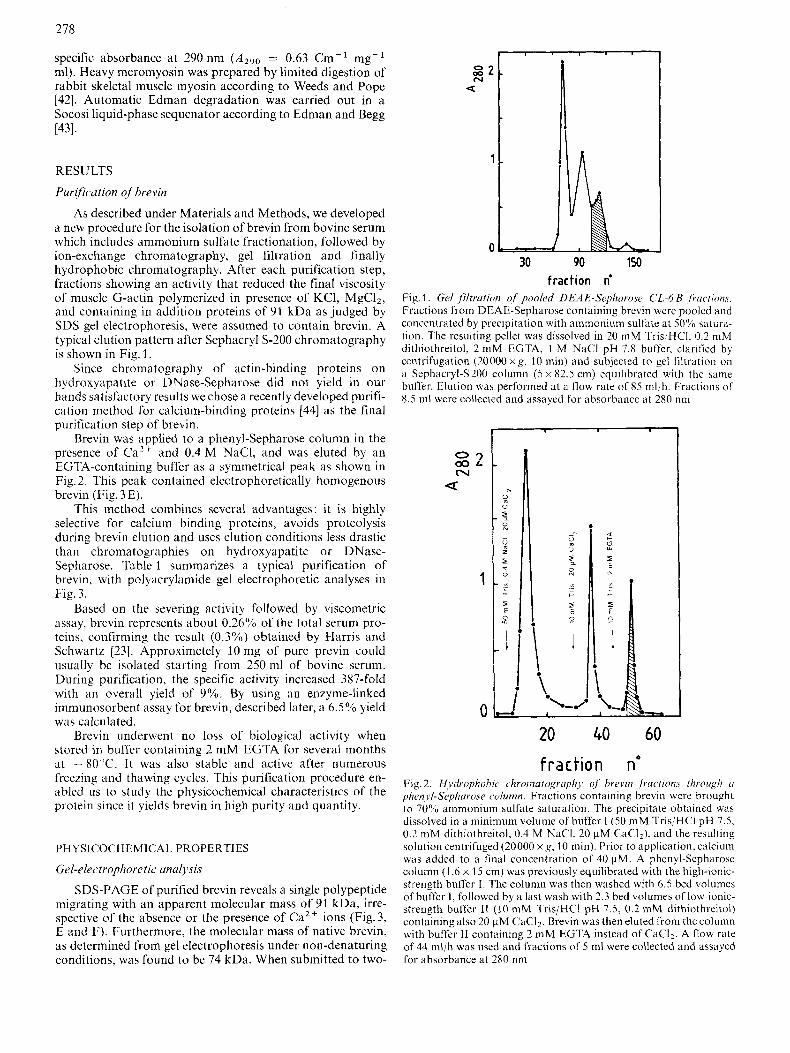
As described under Materials and Methods, we developed a new procedure for the isolation of brevin from bovine serum which includes ammonium sulfate fractionation, followed by ion-exchange chromatography, gel filtration and finally hydrophobic chromatography. After each purification step, fractions showing an activity that reduced the final viscosity of muscle G-actin polymerized in presence of KCI, MgCI2, and containing in addition proteins of 91 kDa as judged by SDS gel electrophoresis, were assumed to contain brevin. A typical elution pattern after Sephacryl S-200 chromatography is shown in Fig. 1.
Since chromatography of actin-binding proteins on hydroxyapatite or DNase-Sepharose did not yield in our hands satisfactory results we chose a recently developed purifi- cation method for calcium-binding proteins [44] as the final purification step of brevin.
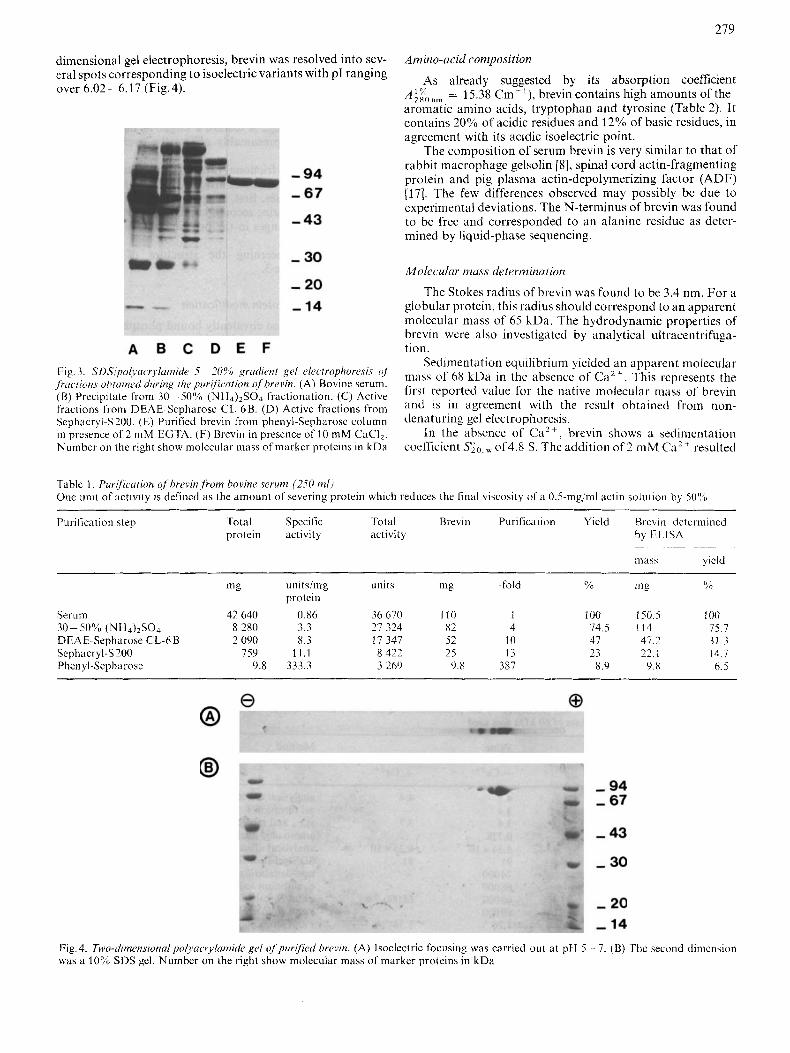
Rrevin was applied to a phenyl-Sepharose column in the presence of C a Z t and 0.4 M NaCl, and was eluted by an EGTA-containing buffer as a symmetrical peak as shown in Fig. 2. This peak contained electrophoretically homogenous brevin (Fig. 3 E).
This method combines several advantages: it is highly selective for calcium binding proteins, avoids proteolysis during brevin elution and uses elution conditions less drastic than chromatographies on hydroxyapatite or DNase- Sepharose. Table 1 summarizes a typical purification of brevin, with polyacrylamide gel electrophoretic analyses in Fig. 3.
Based on the severing activity followed by viscometric assay, brevin represents about 0.26% of the total serum pro- teins, confirming the result (0.3%) obtained by Harris and Schwartz [23]. Approximetely 10 mg of pure previn could usually bc isolated starting from 250 ml of bovinc serum. During purification, the specific activity increased 387-fold with an overall yield of 9%. By using an enzyme-linked imniunosorbent assay for brevin, described later, a 6.5% yield was calculated.
Brevin underwent no loss of biological activity when stored in buffcr containing 2 m M EGTA for several months at - 80 'C. It was also stable and active after numerous freezing and thawing cycles. This purification procedure en- abled us to study the physicochemical characteristics of the protein since it yields brevin in high purity and quantity.
PH Y SI COCHEMICAL PROPERTIES
CPI-electro~hovetir~ analysis
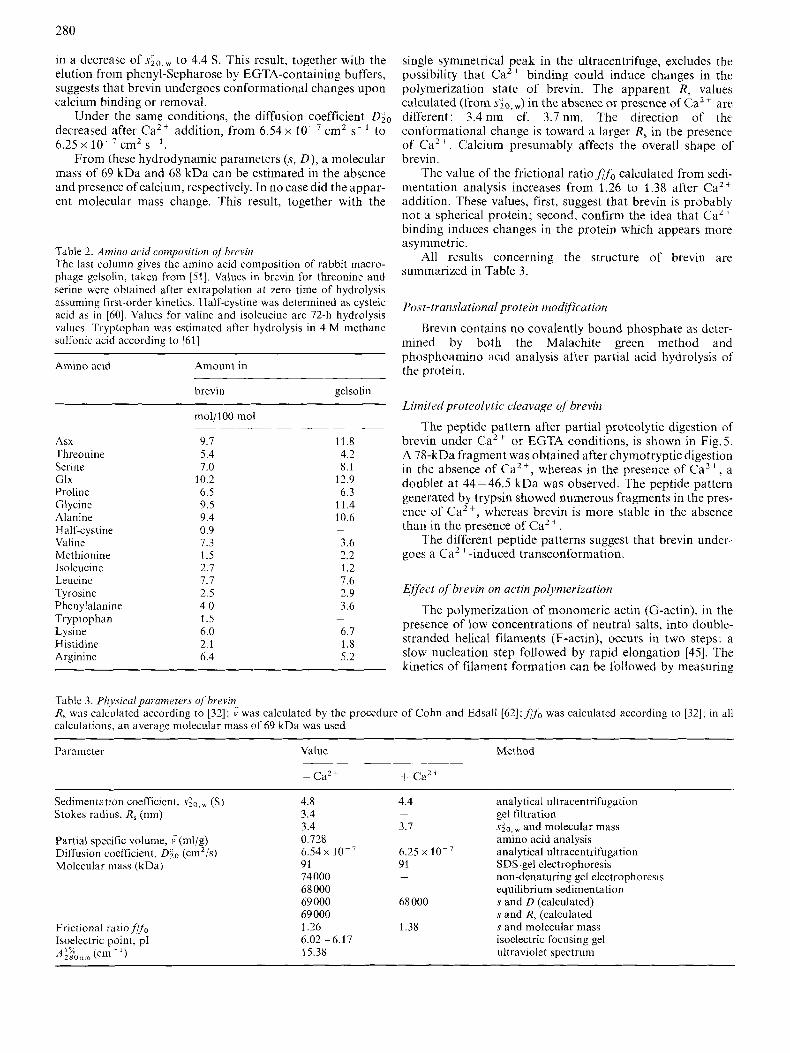
SDS-PAGE of purified brevin reveals a single polypeptide migrating with an apparent molecular mass of 91 kDa, irre- spective of the absence or the presence of Ca2+ ions (Fig.3, E and F). Furthermore, the molecular mass of native brevin, as determined from gel electrophoresis under non-denaturing conditions. was found to be 74 kDa. When submitted to two-
I
0 00 N
U
30 90 150 fraction n'
Fig. 1. Gel filtration of' pooled DEA E-Sephnvose CL-6 B fructions. Fractions from DEAE-Sepharose containing brevin were pooled and concentrated by precipitation with ammonium sulhte at 50% satura- tion. The resulting pellet was dissolved in 20 mM TrisjHCI, 0.2 mM dithiothreitol, 2 mM EGTA, 1 M NaCl pH 7.8 buffer, clarified by centrifugation (20000 x g , 10 min) and subjected to gel !iltrdtion on a Sephacryl-S200 column ( 5 x 82.5 cm) equilibrated with the same buffer. Elution was performed at a flow rate of 85 mbh. Fractions of 8.5 ml were collected and assayed for absorbance at 280 nm
0 N a02
Q
1
0 20 40 60 fraction no
Fig. 2. Hydrophobic chromutogrciphj of brevin ,fractions through ( I
phc,n.l.l-S~~phavose column. Fractions containing brevin were brought to 70% ammonium sulfate saturation. The precipitate obtained was dissolved i n a minimum volume of buffer I (50 mM Tris/HCI pH 7.5. 0.2 mM dithiothreitol, 0.4 M NaCI. 20 pM CaCI,), and the resulting solution centrifuged (20000 xg, 10 min). Prior to application, calcium was added to a final concentration of 40 pM. A phenyl-Sepharose column (1.6 x 15 cm) was previously equilibrated with the high-ionic- strength buffer I. The column was then washed with 6.5 bed volumes of buffer I, followed by a last wash with 2.3 bed volumes of low-ionic- strength buffer I1 (10 mM Tris/HCl pH 7.5, 0.2 mM dithiothreitol) containing also 20 pM CaC12. Brevin was then eluted from the column with buffer 11 containing 2 mM EGTA instead of CaCI,. A flow rate of 44 ml/h was used and fractions of 5 ml were collected and assayed for absorbance at 280 nm
279
dimensional gel electrophoresis, brevin was resolved into sev- era1 spots corresponding to isoelectric variants with PI ranging over 6.02-6.17 (Fig.4).
Amino-acid composition
As already suggested by its absorption coefficient AiFo,,m = 1.5.3s Cm-'), brevin contains high amounts of the aromatic amino acids, tryptophan and tyrosine (Table 2). It contains 20% of acidic residues and 12% of basic residues, in agreement with its acidic isoelectric point.
The composition of serum brevin is very similar to that of rabbit macrophage gelsolin [S], spinal cord actin-fragmenting protein and pig plasma actin-depolymerizing factor (ADF) [17]. The few differences observed may possibly be due to experimental deviations. The N-terminus of brevin was found to be free and corresponded to an alanine residue as deter- mined by liquid-phase sequencing.
Moleculur lyiuss deterniination
The Stokes radius of brevin was found to be 3.4 nm. For a globular protein, this radius should correspond to an apparent molecular mass of 6.5 kDa. The hydrodynamic properties of brevin were also investigated by analytical ultracentrifuga- tion.
Sedimentation equilibrium yielded an apparent molecular
first reported value for the native molecular mass of brevin and is in agreement with the result obtained from non- denaturi11g gel electrophoresis.
In the absence of Cazf , brevin shows it sedimentation coefficient s';o,w of4.8 S. The addition of2 mM Ca2+ resulted
Fig.3. SL)S lpO l~Ucr .~ lU tn i (~~~ 5-?0% gI'(idknt gel electrophoresis ( J f 68 kDa in the absence of ca2 t , hi^ represents the .Jrat~tiuiis ohrained during he pztrjfl'cation ojhrevin. (A) Bovine serum. (B) Precipitate from 30- jO,yo (NH4)2S04 Fractionation. (c) Active fract,ons fro,,, D E A E - S ~ ~ ~ ~ ~ ~ ~ ~ C L . ~ B , (D) ~~~i~~ fractions from Sephacryl-S 200. (E) Purified brevin from phenyl-Sepharosc column in presence of 2 mM EGTA. (F) Brevin in prcsence of 10 mM CaCI,. Number on thc right show molecular mass ofmarker proteins in kDa
Table 1 . Purification ofhrevin,frorn bovine serum (250 ml) Onc unit of activity is defined as the amount of severing protein which reduces the linal viscosily of R 0.5-mg/ml actin solution by 5036
Puri fica ti o ti step Total Specilic Total Brevin Purification Yicld Hrcvin determined protein activity activity by ELISA
~. . ~~~
niass yield
Serum 42 640 0.86 36 670 110 1 100 150.5 1 00 30-50%, (NI-14)2S04 8 280 3.3 21 324 82 4 14.5 114 75.7 DEAE-Sepharose CL-6B 2 090 8.3 I7 347 52 I 0 47 47.2 31.3
Phen yl-Sepharose 9.8 333.3 3 269 9.8 387 8.9 9.8 6.5 Sephacryl-S 200 759 11.1 8 422 25 13 23 22.1 14.7
Fig.4. Two-Ji~~ensiona/poll.acrl;lu~i~c gel ojpurj f id brevin. (A) Isoelectric focusing was carried o u t at pH 5 - 7. (B) The second dimension was a 10% SDS gel. Number on the right show molecular mass of marker proteins in kDa
280
in a decrease of . s ; ~ , ~ to 4.4 S. This result, together with the elution from phenyl-Sepharose by EGTA-containing buffers, suggests that brevin undergoes conformational changes upon calcium binding or removal.
decreased after Ca2+ addition, from 6.54 x cm2 s- l to 6.25 x cm2 s - ' .
From these hydrodynamic parameters (s, D ) , a molecular mass of 69 kDa and 68 kDa can be estimated in the absence and presence of calcium, respectively. In no case did the appar- ent molecular mass change. This result, together with the
Under the same conditions, the diffusion coefficient DZ0
Table 2. Amino acid composition c?f'hrevin The last column gives the amino acid composition of rabbit macro- phage gelsolin, taken from [51]. Values in brevin for threonine and serine wcre obtained after extrapolation a t zero time of hydrolysis assuming first-order kinetics. Half-cystine was determined as cysteic acid as in [60]. Values for valine and isoleucine are 72-h hydrolysis values. Tryptophan was cstimated after hydrolysis in 4 M methane sulfonic acid according to [61]
Amino acid Amount in
brevin gelsolin
mol/lOO mol
Asx Threoninc Scrine Glx Proline Glycine Alaninc Half-cystine Valine Methionine Isoleucine Leucinc Tvrosine
9.7 5.4 7.0
10.2 6.5 9.5 9.4 0.9 1.3 1.5 2.7 7.7 2.5
11.8 4.2 8.1
12.9 6.3
11.4 10.6 -
3.6 2.2 1.2
single symmetrical peak in the ultracentrifuge, excludes the possibility that Ca2+ binding could induce changes in the polymerization state of brevin. The apparent R, values calculated (from &, w) in the absence or presence of Ca2' are different: 3.4 nm cf. 3.7 nm. The direction of the, conformational change is toward a larger R, in the presence of Ca2+. Calcium presumably affects the overall shape of' brevin.
The value of the frictional ratio.ffo calculated from sedi- mentation analysis increases from 1.26 to 1.38 after Ca2+ addition. These values, first, suggest that brevin is probably not a spherical protein; second, confirm the idea that Ca" binding induces changes in the protein which appears more asymmetric.
All results concerning the structure of brevin are summarized in Table 3.
Post-translationul protein modification
Brevin contains no covalently bound phosphate as deter- mined by both the Malachite green method and phosphoamino acid analysis after partial acid hydrolysis of the protein.
Limited proteol-vtic cleavage o j brevin
The peptide pattern after partial proteolytic digestion of brevin under Ca2+ or EGTA conditions, is shown in Fig. 5. A 78-kDa fragment was obtained after chymotryptic digestion in the absence of Ca2+, whereas in the presence of Ca2+, a doublet at 44-46.5 kDa was observed. The peptide pattern generated by trypsin showed numerous fragments in the pres- ence of Ca2+, whereas brevin is more stable in the absence than in the presence of Ca2+.
The different peptide patterns suggest that brevin under- goes a Ca2 +-induced transconformation.
7.6 2.9 Effect of'brevin on actin po1,vmerizution
Phen ylalanine 4.0 3.6 The polymerization of monomeric actin (G-actin), in the presence of low concentrations of neutral salts, into double- Tryptophan 1.5
Lysine 6.0 6.1 stranded helical filaments (F-actin), occurs in two steps: a Histidine 2.1 1.8 Argininc 6.4 5.2 slow nucleation step followed by rapid elongation [45]. The
kinetics of filament formation can be followed by measuring
-
Table 3. Physical parameters of brevin R, was calculated according to [32]; calculations. an average molecular mass of 69 kDa was used
was calculated by the procedure of Cohn and Edsall [62];,flf0 was calculated according to [32]; in all
Parameter Value Method
Sedimentation coefficient, (S) Slokes radius, R, (nm)
Partial specific volume, i(m\/g) Diffusion coefficient, D;o (cm2/s) Molecular mass (kDa)
Frictional ratio,flfo Isoelectric point, pI A izo nm (cm ~ I )
4.8 3.4 3.4 0.728
91 74000 68 000 69 000 69 000 I .26
15.38
6.54x 10-7
6.02 - 6.17
4.4
3.7
6.25 x 91
-
-
68 000
1.38
analytical ultracentrifugation gel filtration sz0, and molecular mass amino acid analysis analytical ultracentrifugation SDS-gel electrophoresis non-denaturing gel electrophoresis equilibrium sedimentation s and D (calculated) s and R, (calculated s and molecular mass isoelectric focusing gel ultraviolet spectrum
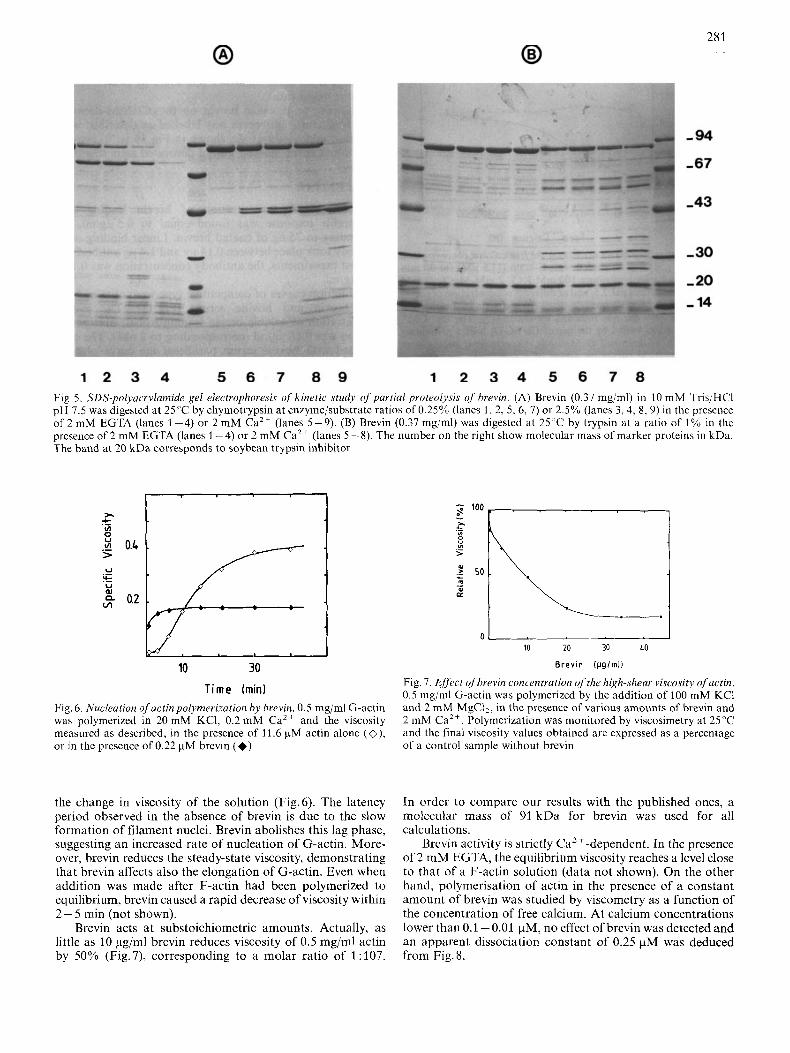
Fig. 5. SDS-polyarrylamide gel electrophoresis of kinetic study of purlial proteolysis of' brevin. (A) Brevin (0.37 mg/ml) in 10 mM Tris/HCI pH 7.5 was digested at 25°C by chymotrypsin at enzyme/substrate ratios of 0.25% (lanes 1, 2, 5, 6, 7) or 2.5% (lanes 3,4, 8 ,9) in the presence of 2 mM EGTA (lanes 1 -4) or 2 mM Ca2+ (lanes 5-9). (B) Brevin (0.37 mg/ml) was digested at 25°C by trypsin at a ratio of 1% in the presence of 2 mM EGTA (lanes 1-4) or 2 mM Ca2+ (lanes 5-8). The number on the right show molecular mass of marker proteins in kDa. The band at 20 kDa corresponds to soybean trypsin inhibitor
10 30 T i m e ( m i d
Fig. 6. Nucleation of'actin polyrnerization by hrevin. 0.5 mg/ml G-actin was polymerized in 20 mM KCI, 0.2 mM Ca2+ and the viscosity measured as described, in thc presence of 11.6 pM actin alone (O) , or in thc presence of 0.22 pM brevin (+)
the change in viscosity of the solution (Fig.6). The latency period observed in the absence of brevin is due to the slow formation of filament nuclei. Brevin abolishes this lag phase, suggesting an increased rate of nucleation of G-actin. More- over, brevin reduces the steady-state viscosity, demonstrating that brevin affects also the elongation of G-actin. Even when addition was made after F-actin had been polymerized to equilibrium, brevin caused a rapid decrease of viscosity within 2 - 5 min (not shown).
Brevin acts at substoichiometric amounts. Actually, as little as 10 pg/ml brevin reduces viscosity of 0.5 mg/ml actin by 50% (Fig. 7), corresponding to a molar ratio of 1 : 107.
10 20 30 40
Brevin lpgirnli
Fig. 7. Effert ofhrevin roncentrurion ofthe high-shenr visrosity ofactin. 0.5 mg/ml G-actin was polymcrized by the addition of 100 mM KC1 and 2 mM MgCl,, in thc presence or various amounts of brevin and 2 mM Ca2+. Polymerization was monitored by viscosimetry at 25°C and the final viscosity values obtaincd are expressed as a percentage of a control sample without brevin
In order to compare our results with the published ones, a molecular mass of 91 kDa for brevin was used for all calculations.
Brevin activity is strictly Caz+-dependent. In the presence of 2 mM EGTA, the equilibrium viscosity reaches a level close to that of a F-actin solution (data not shown). On the other hand, polymerisation of actin in the presence of a constant amount of brevin was studied by viscometry as a function of the concentration of free calcium. At calcium concentrations lower than 0.1 - 0.01 pM, no effect of brevin was detected and an apparent dissociation constant of 0.25 pM was deduced from Fig.8.
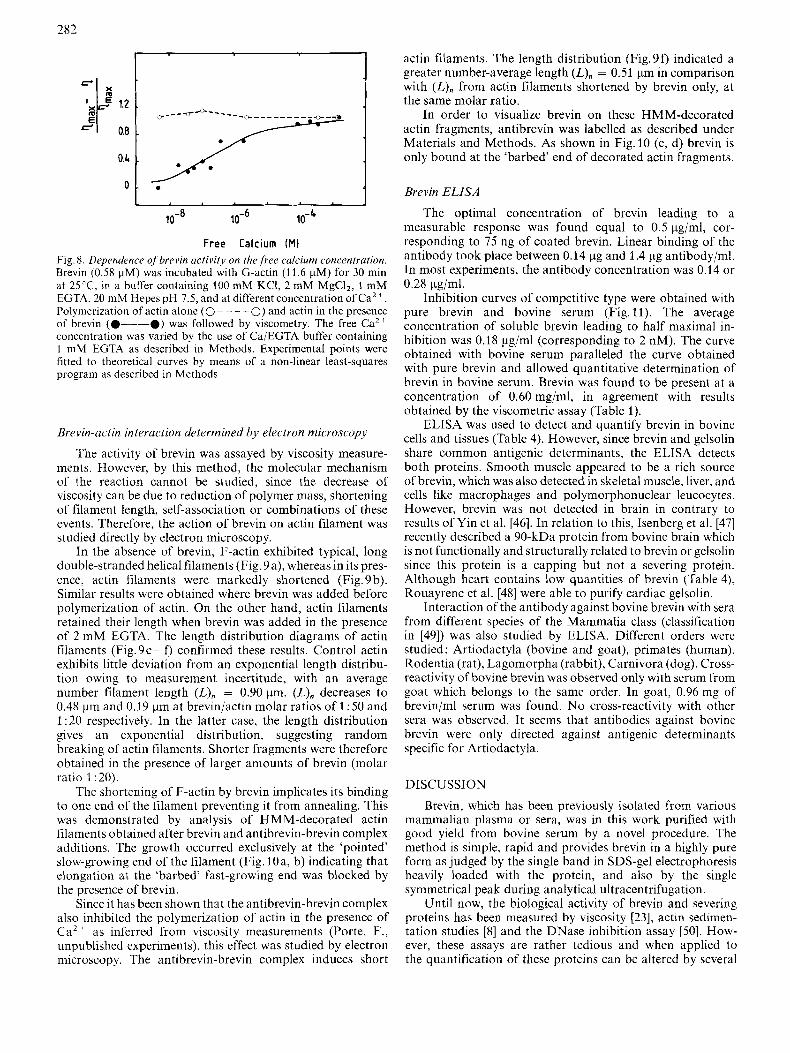
282
e
1.2
0.8
0.4
m E
0 .
1 -
.
lo-&
Free Calcium (MI Fig. 8. Dependence ojbrevin activity on the,free calcium concentration. Brevin (0.58 pM) was incubated with G-actin (1 1.6 pM) for 30 min at 25 'C, in a buffer containing 100 mM KCI, 2 mM MgC12, 1 mM EGTA, 20 mM Hepes pH 7.5, and at different concentration of Caz+. Polymerization of actin alone (0- - - - 0) and actin in the presence of brevin (0-0) was followed by viscomelry. The free Ca" conccntration was varied by thc use of Ca/EGTA buffer containing 1 mM EGTA as described in Methods. Experimental points were fitted to theoretical curves by means of a non-linear least-squares program as described in Methods
Brevin-actin interaction determined by electron microscopy
The activity of brevin was assayed by viscosity measure- ments. However, by this method, the molecular mechanism of the reaction cannot be studied, since the decrease of viscosity can be due to reduction of polymer mass, shortening of filament length, self-association or combinations of these events. Therefore, the action of brevin on actin filament was studied directly by electron microscopy.
In the absence of brevin, F-actin exhibited typical, long double-stranded helical filaments (Fig. 9a), whereas in its pres- ence, actin filaments were markedly shortened (Fig. 9b). Similar results were obtained where brevin was added before polymerization of actin. On the other hand, actin filaments retained their length when brevin was added in the presence of 2 mM EGTA. The length distribution diagrams of actin filaments (Fig. 9c- f , confirmed these results. Control actin exhibits little deviation from an exponential length distribu- tion owing to measurement incertitude, with an average number filament length (L), = 0.90 pm. (L),, decreases to 0.48 pm and 0.19 pm at brevin/actin molar ratios of 1 : 50 and 1 : 20 respectively. In the latter case, the length distribution gives an exponential distribution, suggesting random breaking of actin filaments. Shorter fragments were therefore obtained in the presence of larger amounts of brevin (molar ratio 1 :20).
The shortening of F-actin by brevin implicates its binding to one end of the filament preventing it from annealing. This was demonstrated by analysis of HMM-decorated actin filaments obtained after brevin and antibrevin-brevin complex additions. The growth occurred exclusively at the 'pointed' slow-growing end of the filament (Fig. 10a, b) indicating that elongation at the 'barbed' fast-growing end was blocked by the presence of brevin.
Since it has been shown that the antibrevin-brevin complex also inhibited the polymerization of actin in the presence of CaZ + as inferred from viscosity measurements (Porte, F., unpublished experiments), this effect was studied by electron microscopy. The antibrevin-brevin complex induces short
actin filaments. The length distribution (Fig. 9f) indicated a greater number-average length (I,),, = 0.51 pm in comparison with (L) , from actin filaments shortened by brevin only, at the same molar ratio.
In order to visualize brevin on these HMM-decorated actin fragments, antibrevin was labelled as described under Materials and Methods. As shown in Fig. 10 (c, d) brevin is only bound at the 'barbed' end of decorated actin fragments.
Brevin ELISA
The optimal concentration of brevin leading to a measurable response was found equal to 0.5 pg/ml, cor- responding to 75 ng of coated brevin. Linear binding of the antibody took place between 0.14 pg and 1.4 pg antibody/ml. In most experiments, the antibody concentration was 0.14 or 0.28 pg/ml.
Inhibition curves of competitive type were obtained with pure brevin and bovine serum (Fig. 11). The average concentration of soluble brevin leading to half maximal in- hibition was 0.18 pg/ml (corresponding to 2 nM). The curve obtained with bovine serum paralleled the curve obtained with pure brevin and allowed quantitative determination of brevin in bovine serum. Brevin was found to be present at a concentration of 0.60 mg/ml, in agreement with results obtained by the viscometric assay (Table 1).
ELISA was used to detect and quantify brevin in bovine cells and tissues (Table 4). However, since brevin and gelsolin share common antigenic determinants, the ELISA detects both proteins. Smooth muscle appeared to be a rich source of brevin, which was also detected in skeletal muscle, liver, and cells like macrophages and polymorphonuclear leucocytes. However, brevin was not detected in brain in contrary to results of Yin et al. [46]. In relation to this, Isenberg et al. [47] recently described a 90-kDa protein from bovine brain which is not functionally and structurally related to brevin or gelsolin since this protein is a capping but not a severing protein. Although heart contains low quantities of brevin (Table 4), Rouayrenc et al. [4X] were able to purify cardiac gelsolin.
Interaction of the antibody against bovine brevin with sera from different species of the Mammalia class (classification in [49]) was also studied by ELISA. Different orders were studied: Artiodactyla (bovine and goat), primates (human), Rodentia (rat), Lagomorpha (rabbit), Carnivora (dog). Cross- reactivity of bovine brevin was observed only with serum from goat which belongs to the same order. In goat, 0.96mg of brevin/ml serum was found. No cross-reactivity with other sera was observed. It seems that antibodies against bovine brevin were only directed against antigenic determinants specific for Artiodactyla.
DISCUSSION
Brevin, which has been previously isolated from various mammalian plasma or sera, was in this work purified with good yield from bovine serum by a novel procedure. The method is simple, rapid and provides brevin in a highly pure form as judged by the single band in SDS-gel electrophoresis heavily loaded with the protein, and also by the single symmetrical peak during analytical ultracentrifugation.
Until now, the biological activity of brevin and severing proteins has been measured by viscosity [23], actin sedimen- tation studies [ X I and the DNase inhibition assay [50]. How- ever, these assays are rather tedious and when applied to the quantification of these proteins can be altered by several
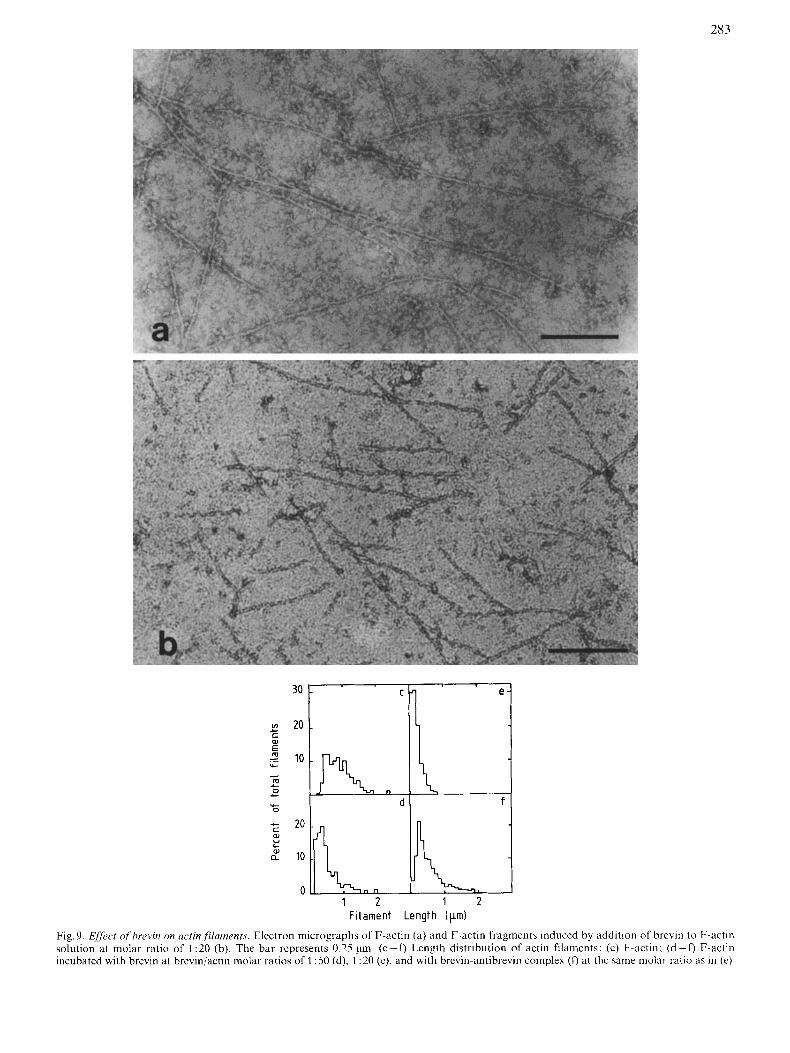
283
VI c 01
c
- E ._ v- A
m + 0 L
v- 0
+ c W Y L
W a
1 2 1 2 F i I ame nt Length ( pm)
Fig. 9. Efir,ct of'f'hrevin on actin fYamrnfs. Elcctron micrographs of F-actin (a) and F-actin fragment5 induced by addition of brevin to F-actin solution at molar ratio of 1 :20 (b). The bar represents 0.25 pm. ( c - 0 Length distribution of actin filaments: (c) F-actin: (d-f) F-actin incubated with brevin at breviniactin molar ratios of 1 :50 (d), 1 :20 (c), and with brcvin-antibrevin complex (0 at thc same molar ratio as in (e)

284
Fig. 10. Binding qfbrcvin to the ‘harhed’ end of acfin fikaments. (a, b) F-actin shortened by brevin a t molar ratio of 1 :20 in the presence of Ca2+, decorated with HMM and elongated by addition of G-actin (3 pM) in 20 mM KCl. (c, d) F-actin shortened by brevin-antibrevin complex. decorated with H M M and subsequently incubated with GAR G20. The bar represents 0.25 pm
factors, especially interaction of actin with other proteins in total extracts. We have therefore developed an ELISA which represents a convenient and rapid quantitative method for severing proteins and which was used to follow purification of bovine brevin in addition to the viscometric assay.
Our results yield some information concerning the structure of the molecule. Purified brevin has the properties of a monomeric protein with a native molecular mass of 68 kDa, but behaves as a non-spherical molecule according
to the analysis of its hydrodynamic properties. The presence of isoelectric variants may be due to a post-translational protein modification (e. g. glycosylation).
Alanine was found as the N-terminal residue of bovine brevin. Human plasma brevin [51] and bovine brevin have therefore identical N-termini.
Calcium-induced conformational changes in brevin are supported by several pieces of evidence including: binding on phenyl-Sepharose, sedimentation velocity analysis, pro-
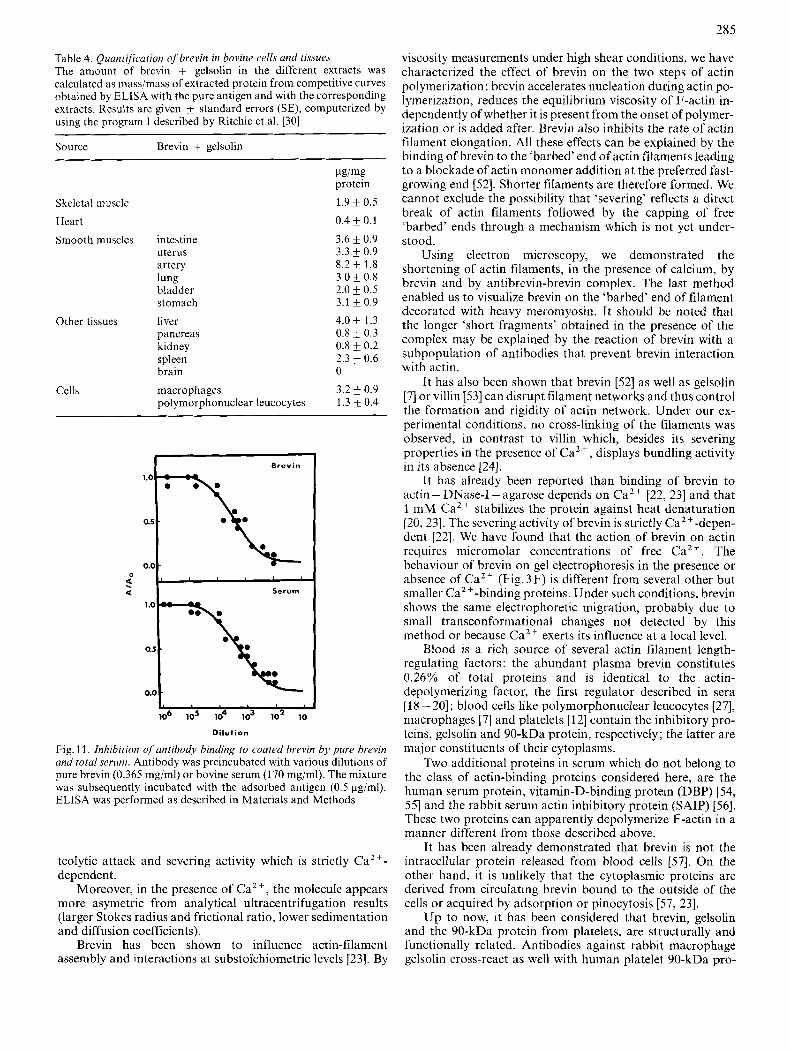
285
Table 4. Quantification o f brevin in bovine cells and tissues The amount of brevin + gelsolin in the different extracts was calculated as mass/mass of extracted protein from competitive curves obtained by ELISA with the pure antigen and with the corresponding extracts. Results are given k standard errors (SE), computerized by using the program 1 described by Ritchie et a]. [30]
Source Brevin + gelsolin
Izg/mg protein
Skeletal muscle 1.9 _+ 0.5
Heart 0.4 0.1
Smooth muscles intestine 3.6 k 0.9 uterus 3.3 f 0.9 artery 8.2 f 1.8 lung 3.0 & 0.8 bladder 2.0 f 0.5 stomach 3.1 k 0.9
Other tissues liver 4.0 k 1.3 pancreas 0.8 f 0.3 kidney 0.8 _+ 0.2 spleen 2.3 f 0.6 brain 0
Cells macrophages 3.2 f 0.9 polymorphonuclear leucocytes 1.3 k 0.4
I Brevin
0
Serum 2 a
0.5
0.0
L 106 105 lo4 lo3 lo2 10
Dilution
Fig. 1 1. Inhibition of antibody binding to coated hrevin by pure brevin and total serum. Antibody was preincubated with various dilutions of pure brevin (0.365 mg/ml) or bovine serum (1 70 mgiml). The mixture was subsequently incubated with the adsorbed antigen (0.5 pg/ml). ELISA was performed as described in Materials and Methods
teolytic attack and severing activity which is strictly Ca2+- dependent.
Moreover, in the presence of Ca2+, the molecule appears more asymetric from analytical ultracentrifugation results (larger Stokes radius and frictional ratio, lower sedimentation and diffusion coefficients).
Brevin has been shown to influence actin-filament assembly and interactions at substoi‘chiometric levels [23]. By
viscosity measurements under high shear conditions, we have characterized the effect of brevin on the two steps of actin polymerization: brevin accelerates nucleation during actin po- lymerization, reduces the equilibrium viscosity of F-actin in- dependently ofwhether it is present from the onset of polymer- ization or is added after. Brevin also inhibits the rate of actin filament elongation. All these effects can be explained by the binding of brevin to the ‘barbed’ end of actin filaments leading to a blockade of actin monomer addition at the preferred fast- growing end [52]. Shorter filaments are therefore formed. We cannot exclude the possibility that ‘severing’ reflects a direct break of actin filaments followed by the capping of free ‘barbed’ ends through a mechanism which is not yet under- stood.
Using electron microscopy, we demonstrated the shortening of actin filaments, in the presence of calcium, by brevin and by antibrevin-brevin complex. The last method enabled us to visualize brevin on the ‘barbed’ end of filament decorated with heavy meromyosin. It should be noted that the longer ‘short fragments’ obtained in the presence of the complex may be explained by the reaction of brevin with a subpopulation of antibodies that prevent brevin interaction with actin.
It has also been shown that brevin [52] as well as gelsolin [7] or villin [53] can disrupt filament networks and thus control the formation and rigidity of actin network. Under our ex- perimental conditions, no cross-linking of the filaments was observed, in contrast to villin which, besides its severing properties in the presence of Ca2 + , displays bundling activity in its absence [24].
It has already been reported than binding of brevin to actin- DNase-I -agarose depends on Ca2+ [22,23] and that 1 mM Ca2+ stabilizes the protein against heat denaturation [20, 231. The severing activity of brevin is strictly Ca2 ‘-depen- dent [22]. We have found that the action of brevin on actin requires micromolar concentrations of free Ca”. The behaviour of brevin on gel electrophoresis in the presence or absence of Ca2+ (Fig. 3 F) is different from several other but smaller Ca2+-binding proteins. Under such conditions, brevin shows the same electrophoretic migration, probably due to small transconformational changes not detected by this method or because Ca2’ exerts its influence at a local level.
Blood is a rich source of several actin filament length- regulating factors : the abundant plasma brevin constitutes 0.26% of total proteins and is identical to the actin- depolymerizing factor, the first regulator described in sera [18 - 201; blood cells like polymorphonuclear leucocytes [27], macrophages [7] and platelets [12] contain the inhibitory pro- teins, gelsolin and 90-kDa protein, respectively; the latter are major constituents of their cytoplasms.
Two additional proteins in serum which do not belong to the class of actin-binding proteins considered here, are the human serum protein, vitamin-D-binding protein (DBP) [54, 551 and the rabbit serum actin inhibitory protein (SAIP) [56]. These two proteins can apparently depolymerize F-actin in a manner different from those described above.
I t has been already demonstrated that brevin is not the intracellular protein released from blood cells [57]. On the other hand, it is unlikely that the cytoplasmic proteins are derived from circulating brevin bound to the outside of the cells or acquired by adsorption or pinocytosis [57, 231.
Up to now, it has been considered that brevin, gelsolin and the 90-kDa protein from platelets, are structurally and functionally related. Antibodies against rabbit macrophage gelsolin cross-react as well with human platelet YO-kDa pro-

286
tein [58] as with brevin from human serum [22] or rabbit serum (data not shown). Furthermore, a cross-reactivity between different species (e.g. rabbit and human) was reported [46]. However, a species specificity is found for bovine brevin: in fact, no activity was detected in sera from different species with the present ELISA, using antibodies against bovine brevin.
These differences may be explained by immunogenicity differences of specific epitopes. Bovine brevin could have highly immunogenic epitopes specific for the bovine species. Therefore cross-reactivity of proteins with cells and tissues derived from other species must be interpreted cautiously.
However, brevin could be quantified by ELISA in bovine cells and tissues. As brevin and gelsolin share common anti- genic determinants, the amount obtained probably represents the sum of brevin plus gelsolin. Therefore, these proteins are detected in almost all types of cells and tissues.
Recently, Yin et al. [51] reported that brevin, referred to as plasma gelsolin, is structurally similar but not identical to cytoplasmic gelsolin. Recent work also suggests both Caz+- dependent and independent interaction of actin-depoly- merizing factor [59] and the platelet 90-kDa protein [5X] with actin.
All these results reflect the multiple actions of brevin and related proteins on actin, in vitro. But there is no experimental evidence about their function in the cell, and their physiologi- cal role in vivo is not clearly understood at the present time. Gelsolin controls the Ca2 +-sensitive gel-sol transformation of cell cytoplasm. Because it is an extracellular protein, however, brevin may not be involved in the regulation of intracellular motility, but rather in a scavenging function [22, 231: after cell lysis, large amounts of microfilaments are liberated and eliminated rapidly and efficiently from the circulation, pre- sumably via brevin. The other actin-severing proteins de- scribed above, secreted on cell lysis, might cooperate in this 'cleaning operation'.
We are gratcful to J. Derancourt and R. Jeanneau for running the liquid-phase sequencer and the analytical ultracentrifuge. Helpful and stimulating discussions with Prof. J. G. Demaille are gratefully acknowledged. The cxpert editorial assistance of C. Bernie and S. Camalon is also gratefully acknowledged. Z. S. was supported partially by the Muscular Dystrophy Association of America (conven- tion 887).
REFERENCES 1. Pollard, T. D. & Weihing, R. R. (1974) CRC Crit. Rev. Biochem.
2. Korn, E. D. (1978) Proc. Natl Acad. Sci. USA 75, 588-5599, 3. Carlsson, L., Markey, F., Blikstad, I., Person, T. & Lindberg,
U. (1979) Proc. Natl Acad. Sci. USA 76,6376-6380. 4. Clarke, M. & Spudich, J . A. (1977) Annu. Rev. Biochem. 46,797-
822. 5. Weeds, A. (1982) Nature (Lond.) 296, 811 -816. 6. Yin, H. L. & Stossel, T. P. (1979) Nature (Lond.) 281, 583-586. 7. Yin, H. L., Zaner, K. S. & Stossel, T. P. (1980) J . Bid. Chem.
8. Yin, H. L. & Stossel, T. P. (1980) J . Biol. Chem. 255,9490-9493. 9. Hasegawa, T., Tokayashi, S., Hayashi, H. & Hatano, S. (1980)
2, 1-65.
255,9494 - 9500.
Biochemistry 19, 2677 -2683. 10. Hinssen, H. (1982) Eur. J. Cell Biol. 23, 225-233. 11. Brown, S. S., Yamamoto, K. & Spudich, J. A. (1982) J . Cell Biol.
12. Wang, L. L. & Bryan, J. (1981) CeN25, 637-649. 13. Olomucki, A,, Huc, C., Lefebure, F. & Coue, M. (1984) FEBS
14. Bretscher, A. & Weber, K. (1980) CeN20, 839-847.
93,205 -210.
Lett. 174, 80-85.
15.
16.
17.
18.
19.
20.
21. 22.
23.
24.
25. 26.
27.
28. 29. 30.
31.
32.
33. 34. 35.
36. 37. 38.
39 40.
41, 42 43 44.
45.
46
47
48.
Mooseker, M. S., Graves, T. A,, Wharton, K. A., Flaco, N. &
Glenney, J. R., Kaulfus, P. & Weber, K. (1981) Cdl 24. 471 -
Petrucci, T. C., Thomas, C. & Bray, D. (1983) J . Neurochem. 40,
Norberg, R., Thorstensson, R., Utter, G. & Fagraeus, A. (1 979)
Chaponnier, C., Borgia, R., Rungger-Brandle, E., Weil, R. &
Harris, H . E.. Bamburg, J . R. &Weeds, A. G. (1980) FEBS Lett.
Harris, H. E. & Gooch, J. (1981) FEBS Lett. 123, 49-53. Thorstensson, R., Utter, G. & Norberg, R . (1982) Eur. .I. Biochem.
Harris, D. A. & Schwartz, J . H. (1981) Proc. Natl Acad. Sci. USA
Glenney, J. R., Bretscher, A,, Jr & Weber, K. (1980) Proc. Nurl
Spudich, J . A. &Watt, S. (1971) J . Biol. Chem. 245,4866-4871. Onoue, K., ?agi, Y., Grossberg, A. L. & Pressman, D. (1965)
Southwick, F. S. & Stossel, T. P. (1981)J. Biol. Chem. 256,3030-
O'Farrell, P. H. (1975) J . Biol. Chem. 250,4007-4021. Engvall, E. (1980) Methods Enzymol. 70, 419-439. Ritchie, D. G., Nickerson, J . M. & Fuller, G. M. (1983) Methods
Hesterberg, L. K. & Weber, K. (1983) J . Biol. Chem. 258, 359 -
Siege], L. M. & Monty, K . J. (1966) Biochim. Biophys. Acta 112,
Yphantis, D. A. (1964) Biochemistry 3, 297-317. Moore, S. & Stein, W. H. (1963) Methods Enzymol. 6, 819-831. Capony, J. P. & Demaille, J. G. (1983) Anal. Biochem. 128,206-
Laemmli, U. K. (1970) Nature (Lond.) 227, 680-685. Craig, R. & Megermann, J. (1977) J. CeUBiol. 75, 990-996. Cooper, I. J. A. & Pollard, T. D. 1982) Methods Enzymol. 75,
Itaya, K. & Michio, U. (1966) Clin. Chim. Acta 14, 361 -366. Haiech, J., Derancourt, J., Pechere, J. F. & Demaille, J. G. (1979)
Spector, T. (1978) Anal. Biochem. 86, 142-146. Weeds, A. G. &Pope, B. (1977) J . Mol. Biol. I l l , 129-157. Edman, P. & Begg, G. (1967) Eur. J . Biochem. 1 , 80-92. Gopalakrishna, R. & Anderson, W. B. (1982) Biochem. Biophys.
Oosawa, F. & Asakura, S. (1975) Thermodynamics of thepolymer-
Yin, H. L., Albrecht, J. H. & Fattoum, A. (1981) J . Cell Bid. 91,
Isenberg, G., Ohnheiser, R. & Maruta, H. (1983) FEBS Lett. 163,
Rouayrenc, J. F., Fattoum, A,, Gabrion, J., Audemard, E. &
Howe, C. L. (1980) J . Cell Biol. 87, 809 - 822.
480.
1507-1516.
Eur. J . Biochem. 100, 575 - 583.
Gabbiani, G. (1979) Experientia (Basel) 35. 1039- 1040.
121, 2 75 - 1 77.
126, 11 - 16.
78,6798 -6802.
Acud. Sci. USA 77,6458-6462.
Immunochemistry 2, 401 -41 5.
3036.
Enzymol. 92, 577 - 588.
364.
346 - 362.
212.
182- 210.
Biochemistry 18, 2752 - 2758.
Res. Commun. 104, 830-836.
ization of protein, Academic Press, London.
901 -906.
225-2229,
Kassab. R. (1984) FEBS Lett. 167. 52-58. 49. Colbert, E. M.'(1955) in Evolution ofthe vertebrates, John Wiley &
50. Harris, H . E., Bamburg, J. R., Bernstein, B. W. & Weeds, A. G.
51. Yin, H. L., Kwiatkowski, D. J., Mole, J. E. & Cole, F. S. (1984)
52. Wilkins, J. A,, Schwartz, J. H. & Harris, D. A. (1983) Cell B id .
53. Nunally, M. M., Powell, L. D. & Craig, S. W. (1981) J . Biol.
54. Van Baelen, H., Bouillon, R. & De Moor, P. (1980) J . Biol. Chem.
55 . Coue, M., Constans, J., Vian, M. &Olomucki, A. (1983) Biochim.
56. Vandekerckhove, J. S. & Sandoval, I. V. (1982) Biochemistry 21.
Sons, Chichester.
(1982) Anal. Biochem. 119, 102-114.
J . Biol. Chem. 259, 5271 - 5276.
Int. Rep. 7, 1097- 1104.
Chem. 256,2083 - 2086.
255,2270 - 2272.
Biophys. Acta 759, 137 - 145.
3983 - 3991.

287
57. Markey, F., Person, T. & Lindberg, U. (1982) Biochim. Biophys. 60. Hirs, C. H. W. (1967) Methods Enzymol. 1 I , 59-62. 61. Simpson, R. J., Neuberger, M. J. & Liu, T. Y. (1975) J . B i d .
62. Cohn, E. J . & Edsall J. T. (1943) Proteins, amino acids and pepplides, p. 375, Van Nostrand Reinhold Co., New York.
Acta 709, 122-133.
C'hem, 20895-10903.
2741.
58. Kurth, M. C., Wang, L. L., Dingus, J . & Bryan, J . (1983) J . Biol. Chem. 2.51, 1936-1940.
59. Harris, H. E. & Weeds. A. G. (1983) Biochemistry 22, 2728-





























