Embed Size (px)
Citation preview

Materials Science and Engineering A239–240 (1997) 777–783
Computer simulation of ‘order-order’ kinetics inL12 superstructure
P. Oramus a,*, R. Kozubski a, M.C. Cadeville b, V. Pierron-Bohnes b, W. Pfeiler c
a Institute of Physics, Jagellonian Uni6ersity, Reymonta 4, 30-048 Krakow, Polandb Institut de Physique et Chimie des Materiaux de Strasbourg, 23, rue du Loess, 67037 Strasbourg, France
c Institut fur Materialphysik, Uni6ersitat Wien, Strudlhofgasse 4, 1090 Wien, Austria
Abstract
Monte Carlo simulations of ‘order-order’ relaxations in AB3 systems with L12 superstructure were carried out assuming vacancymechanism of atomic jumps with constant atomic pair-interaction energy parameters and the saddle-point energies assigned tojumping atoms. The study was focused on relaxations following an increase of temperature. The simulated long-range order(LRO) relaxation curves fitted weighted sums of two exponentials strongly differing in relaxation times, the longer ones of whichobeyed the Arrhenius law. The contribution (weight) of the fast process was gradually reduced when the activation barrier energyfor B-atom jumps was increased in relation to that for A-atoms. It was found out that the initial stages of LRO relaxations weredominated by A-atom jumps between different sublattices. In conclusion, it is proposed that the fast component of the disorderingrelaxations in L12 superstructure is due to highly correlated pairs of A-atom and B-atom jumps, which occur provided themajority B-atoms are sufficiently mobile. © 1997 Elsevier Science S.A.
Keywords: Computer simulation; ‘Order-order’; L12 superstructure
1. Introduction
Experimental investigations of ‘order-order’ kineticsin the intermetallic compound Ni3Al were performed bymeans of resistometry [1,2]—the technique sufficientlysensitive to ultra-fine changes of the degree of long-range order (LRO).
The results are summarised as follows:� the measured activation energy for the ‘order-order’
kinetics equals 4.6 eV, i.e. is substantially higherthan the one for Ni diffusion in Ni3Al oscillatingaround 3 eV,
� the high activation energy is predominantly due tothe activation energy for vacancy formation reachingthe value of 2.9 eV
� ‘order-order’ relaxations involve two parallel pro-cesses strongly differing in rates, but showing closeactivation energies.Some of the above experimental results have been
partially explained by modelling the process:
� by first-principle calculations it was shown that inbinary Ni3Al the energy for Al-vacancy formation issubstantially higher than the energy for Ni-vacancyformation and approximates the experimentally esti-mated activation energy for vacancy formation—ac-tive in the ‘order-order’ relaxations [3],
� hypothetical model for the origin of two parallelrelaxation processes was proposed [1,4,5].The initiated Monte Carlo simulations aimed at the
reproduction of experimentally monitored LRO relax-ation curves and the explanation of the origin of twoparallel relaxation processes.
2. Principles of the model
2.1. Description of the simulated system
The software used in the present study was a newversion of one described in the previous paper on LROkinetics in B2 and DO3 superstructure [6]. The threedimensional L12 superlattice containing 403 superlatticecells was simulated. Two kinds of atom (A and B) inpurely stoichiometric proportion 1:3 were then intro-* Corresponding author.
0921-5093/97/$17.00 © 1997 Elsevier Science S.A. All rights reserved.
PII S0921 -5093 (97 )00666 -7
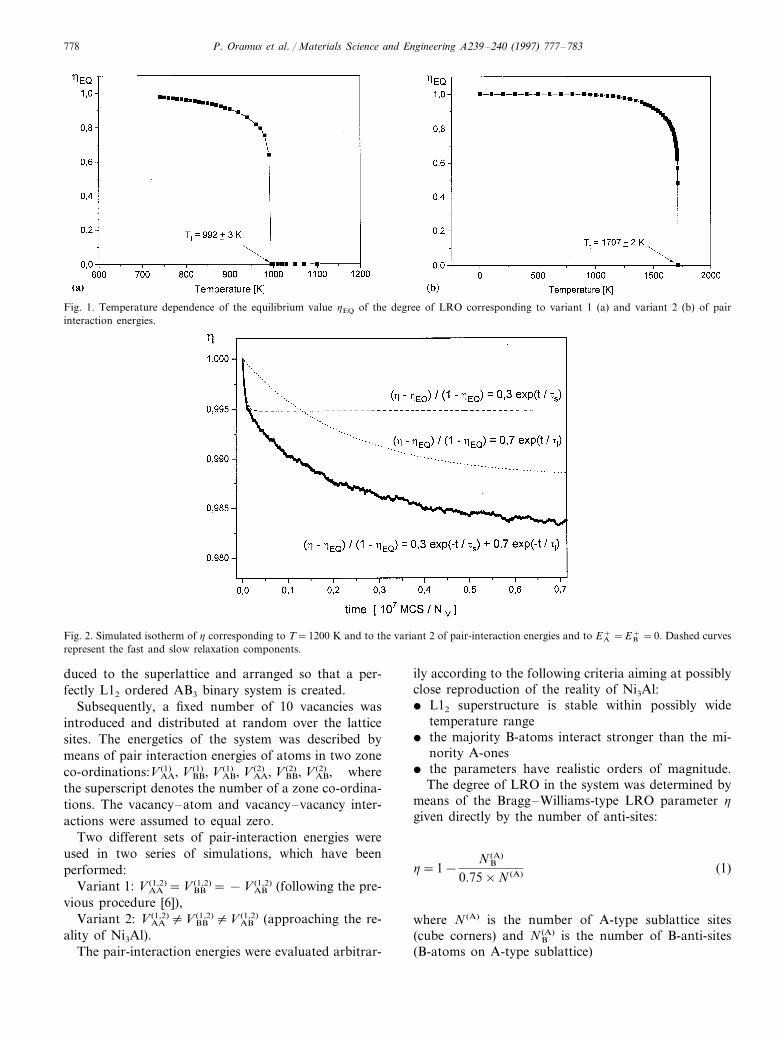
P. Oramus et al. / Materials Science and Engineering A239–240 (1997) 777–783778
Fig. 1. Temperature dependence of the equilibrium value hEQ of the degree of LRO corresponding to variant 1 (a) and variant 2 (b) of pairinteraction energies.
Fig. 2. Simulated isotherm of h corresponding to T=1200 K and to the variant 2 of pair-interaction energies and to E+A =E+
B =0. Dashed curvesrepresent the fast and slow relaxation components.
duced to the superlattice and arranged so that a per-fectly L12 ordered AB3 binary system is created.
Subsequently, a fixed number of 10 vacancies wasintroduced and distributed at random over the latticesites. The energetics of the system was described bymeans of pair interaction energies of atoms in two zoneco-ordinations:V (1)
AA, V (1)BB, V (1)
AB, V (2)AA, V (2)
BB, V (2)AB, where
the superscript denotes the number of a zone co-ordina-tions. The vacancy–atom and vacancy–vacancy inter-actions were assumed to equal zero.
Two different sets of pair-interaction energies wereused in two series of simulations, which have beenperformed:
Variant 1: V (1,2)AA =V (1,2)
BB = −V (1,2)AB (following the pre-
vious procedure [6]),Variant 2: V (1,2)
AA "V (1,2)BB "V (1,2)
AB (approaching the re-ality of Ni3Al).
The pair-interaction energies were evaluated arbitrar-
ily according to the following criteria aiming at possiblyclose reproduction of the reality of Ni3Al:� L12 superstructure is stable within possibly wide
temperature range� the majority B-atoms interact stronger than the mi-
nority A-ones� the parameters have realistic orders of magnitude.
The degree of LRO in the system was determined bymeans of the Bragg–Williams-type LRO parameter h
given directly by the number of anti-sites:
h=1−N (A)
B
0.75×N (A) (1)
where N (A) is the number of A-type sublattice sites(cube corners) and NB
(A) is the number of B-anti-sites(B-atoms on A-type sublattice)
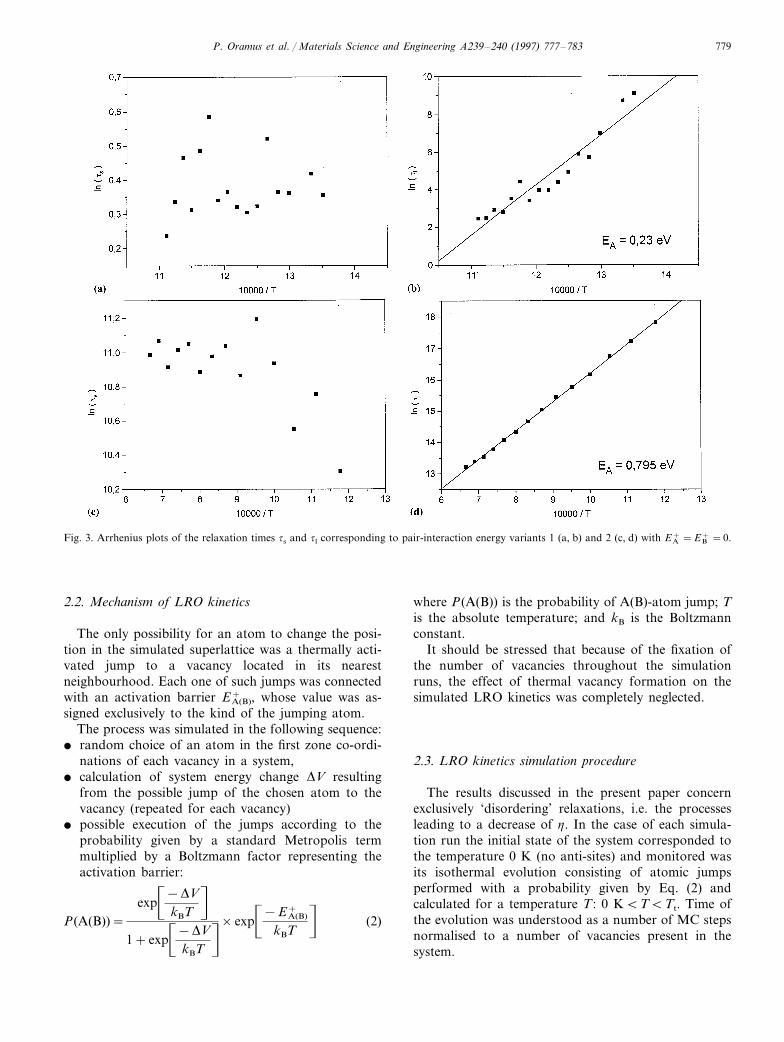
P. Oramus et al. / Materials Science and Engineering A239–240 (1997) 777–783 779
Fig. 3. Arrhenius plots of the relaxation times ts and tl corresponding to pair-interaction energy variants 1 (a, b) and 2 (c, d) with EA+ =EB
+ =0.
2.2. Mechanism of LRO kinetics
The only possibility for an atom to change the posi-tion in the simulated superlattice was a thermally acti-vated jump to a vacancy located in its nearestneighbourhood. Each one of such jumps was connectedwith an activation barrier E+
A(B), whose value was as-signed exclusively to the kind of the jumping atom.
The process was simulated in the following sequence:� random choice of an atom in the first zone co-ordi-
nations of each vacancy in a system,� calculation of system energy change DV resulting
from the possible jump of the chosen atom to thevacancy (repeated for each vacancy)
� possible execution of the jumps according to theprobability given by a standard Metropolis termmultiplied by a Boltzmann factor representing theactivation barrier:
P(A(B))=exp
�−DVkBT
n1+exp
�−DVkBT
n×exp�−E+
A(B)
kBTn
(2)
where P(A(B)) is the probability of A(B)-atom jump; Tis the absolute temperature; and kB is the Boltzmannconstant.
It should be stressed that because of the fixation ofthe number of vacancies throughout the simulationruns, the effect of thermal vacancy formation on thesimulated LRO kinetics was completely neglected.
2.3. LRO kinetics simulation procedure
The results discussed in the present paper concernexclusively ‘disordering’ relaxations, i.e. the processesleading to a decrease of h. In the case of each simula-tion run the initial state of the system corresponded tothe temperature 0 K (no anti-sites) and monitored wasits isothermal evolution consisting of atomic jumpsperformed with a probability given by Eq. (2) andcalculated for a temperature T : 0 KBTBTt. Time ofthe evolution was understood as a number of MC stepsnormalised to a number of vacancies present in thesystem.

P. Oramus et al. / Materials Science and Engineering A239–240 (1997) 777–783780
3. Results
The following values of pair-interaction energies havebeen applied:
Variant 1:
V (1)AA=V (1)
BB= −V (1)AB=0.034 eV
V (2)AA=V (2)
BB= −V (2)AB= −0.0042 eV
Variant 2:
V (1)AA=0.036 eV, V (1)
BB=0.072 eV,
V (1)AB= −0.056 eV
V (2)AA= −0.006 eV, V (2)
BB= −0.012 eV,
V (2)AB=0.008 eV
3.1. Results for EA(B)+ =0
Each isotherm of h saturated at an equilibrium levelhEQ corresponding to the current temperature T. Thetemperature dependencies of hEQ and the resultingL12 l A2 ‘order-disorder’ transition temperatures Tt
yielded by both series of the simulations are shown inFig. 1a, b and indicate the discontinuous character ofthe transformation-characteristic for the L12 super-structure.
A typical isotherm of h is shown in Fig. 2. All thecurves corresponding to both variants of the interactionenergies fitted weighted sums of single exponentials:
h−hEQ
1−hEQ
=C×exp�
−tts
n+ (1−C)×exp
�−
ttl
n(3)
The relaxation times ts and tl differed by about anorder of magnitude. Their Arrhenius plots shown inFig. 3a–d indicate that only the long one tl yielded adefinitely non-zero activation energy connected with theapplied pair-interaction energies. The unclear behaviourof the short one ts is currently a subject of furtherinvestigations.
3.2. Effect of the acti6ation barriers EA(B)+
Introduction of non-zero activation barriers EA(B)+
accounts for different mobilities of the A- and B-atoms.Their influence on the simulated LRO kinetics has upto now been studied at only one selected temperatureby imposing increasing barriers upon either A or Batoms. The results illustrated by Fig. 4a–c indicate thatimmobilisation of the majority B-atoms led to thedecay of the fast relaxation process, which, on the otherhand, showed up more pronounced if A-atoms wereimmobilised.
Detailed analysis of the curves indicated that (Fig. 5):� an increase of the absolute value of the difference
d+ between EB+ and EA
+ leads to a decrease of the
weight factor C in Eq. (3) (Fig. 5a)� the ratio ts/tl decreases when only EA
+ increases (d+
is negative and decreases),� no substantial change of ts/tl results from an in-
crease of EB+ (increase of positive d+).
3.3. Contribution of particular kinds of jumps to thesimulated LRO kinetics
In order to assess the role of particular kinds of theatomic jumps in the investigated process of LRO kinet-ics, the number of executed jumps of given kind perfixed number of MC steps was monitored. The resultsare displayed in Fig. 6a–f and the relaxation-like be-haviour of this quantity throughout the process is clear.In the case of both activation barriers equal to zero(Fig. 6c, d) it is visible that the initial stage of therelaxation was dominated by the jumps of the minorityA-atoms between A- and B-type sublattices. During theprocess the number of such jumps decreased by gradualrelaxation to a constant level. At the same time theB-atom jumped predominantly within the B-type sub-lattice. At the beginning of the process the number oftheir jumps between A- and B-type sublattices wasdefinitely lower, but then showed an increasing relax-ation. The presence of the activation barriers resultedonly in an overall reduction of the numbers of corre-sponding jumps, but the nature of the phenomenon(increasing or decreasing relaxations of the numbers ofcorresponding jumps) remained unaffected.
4. Discussion and conclusions
The following two results of the present study suggesta preliminary explanation of the origin of the twoparallel LRO relaxation processes observed both in thesimulated and experimental relaxation curves:� predominant contribution of the A-atom jumps be-
tween different sublattices to the initial stage of therelaxations
� the decay of the fast process when B-atoms areimmobilised.The number of A-atom jumps shown in Fig. 6a, c
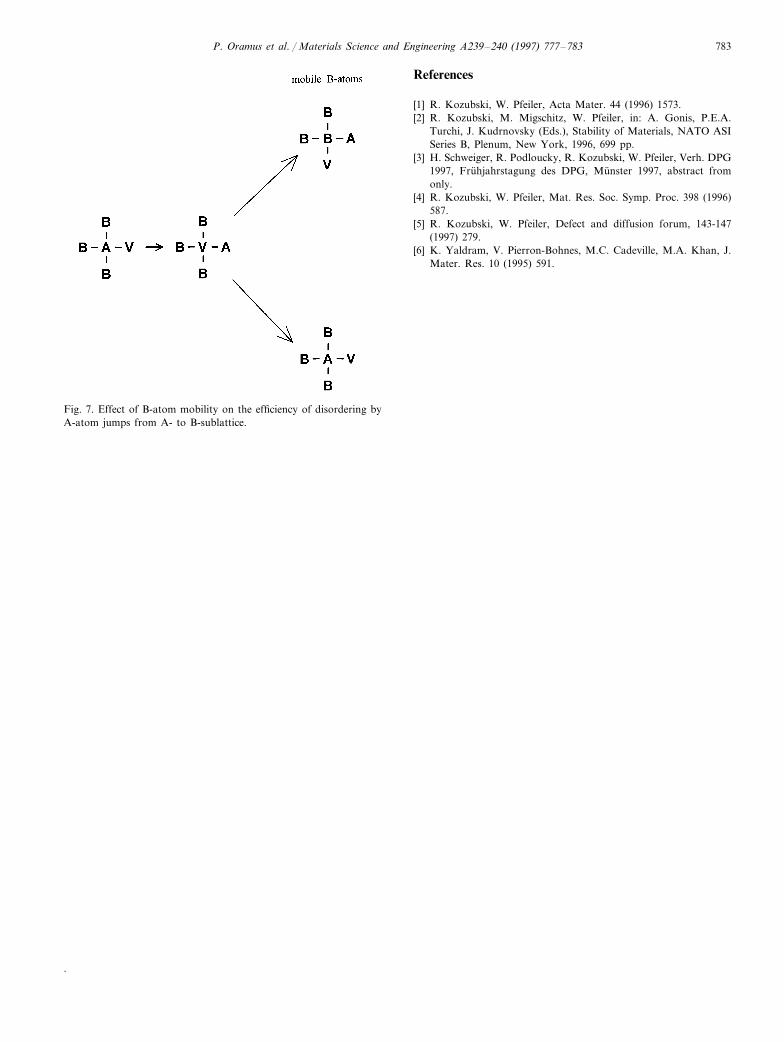
and e corresponds both to the ones from A- to B-sub-lattice (creating A-antisites) and the reversed ones backfrom B- to A-sublattice. The reversed jumps are highlyprobable, because of being energetically favourable. Ifthe number of reversed jumps nearly equals the numberof the original ones, the entire A-atom dynamics areinefficient in decreasing the degree of LRO. The effi-ciency is improved if a jump of an A-atom to a nnvacancy on the B-sublattice is immediately followed bya jump of a B-atom to this vacancy (see Fig. 7).Consequently, the reversed jump of the A-atom isblocked and a pair of A- and B-anti-sites is created. It
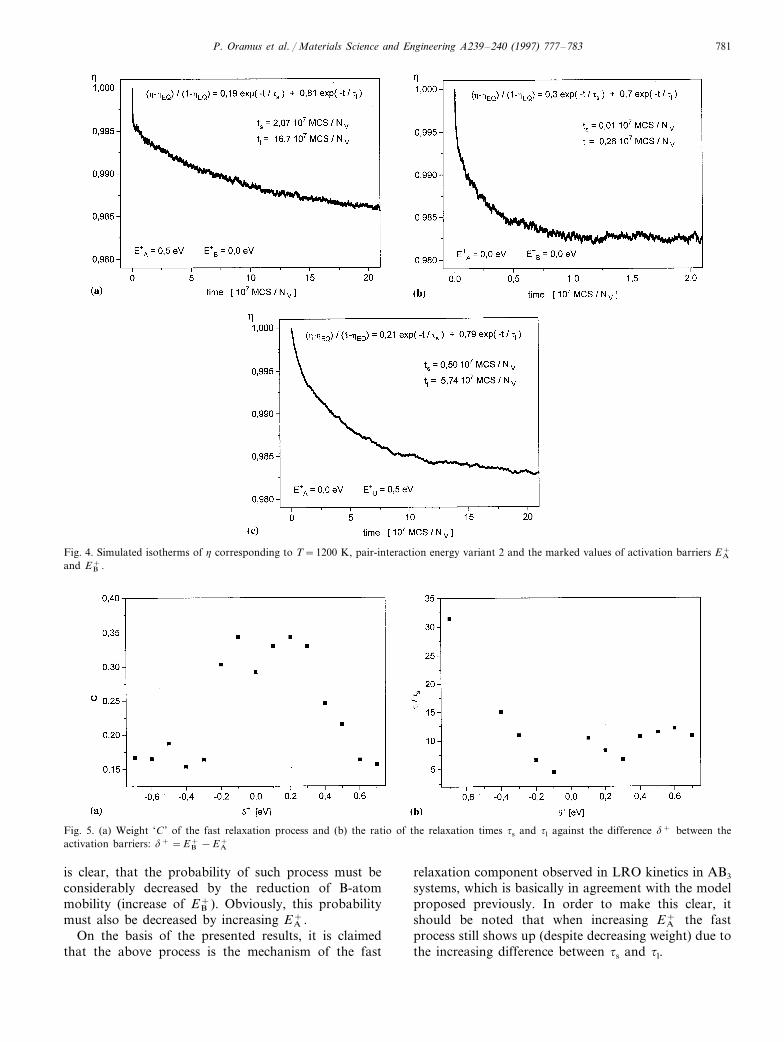
P. Oramus et al. / Materials Science and Engineering A239–240 (1997) 777–783 781
Fig. 4. Simulated isotherms of h corresponding to T=1200 K, pair-interaction energy variant 2 and the marked values of activation barriers EA+
and EB+.
Fig. 5. (a) Weight ‘C ’ of the fast relaxation process and (b) the ratio of the relaxation times ts and tl against the difference d+ between theactivation barriers: d+ =E+
B −E+A
is clear, that the probability of such process must beconsiderably decreased by the reduction of B-atommobility (increase of EB
+). Obviously, this probabilitymust also be decreased by increasing EA
+.On the basis of the presented results, it is claimed
that the above process is the mechanism of the fast
relaxation component observed in LRO kinetics in AB3
systems, which is basically in agreement with the modelproposed previously. In order to make this clear, itshould be noted that when increasing EA
+ the fastprocess still shows up (despite decreasing weight) due tothe increasing difference between ts and tl.
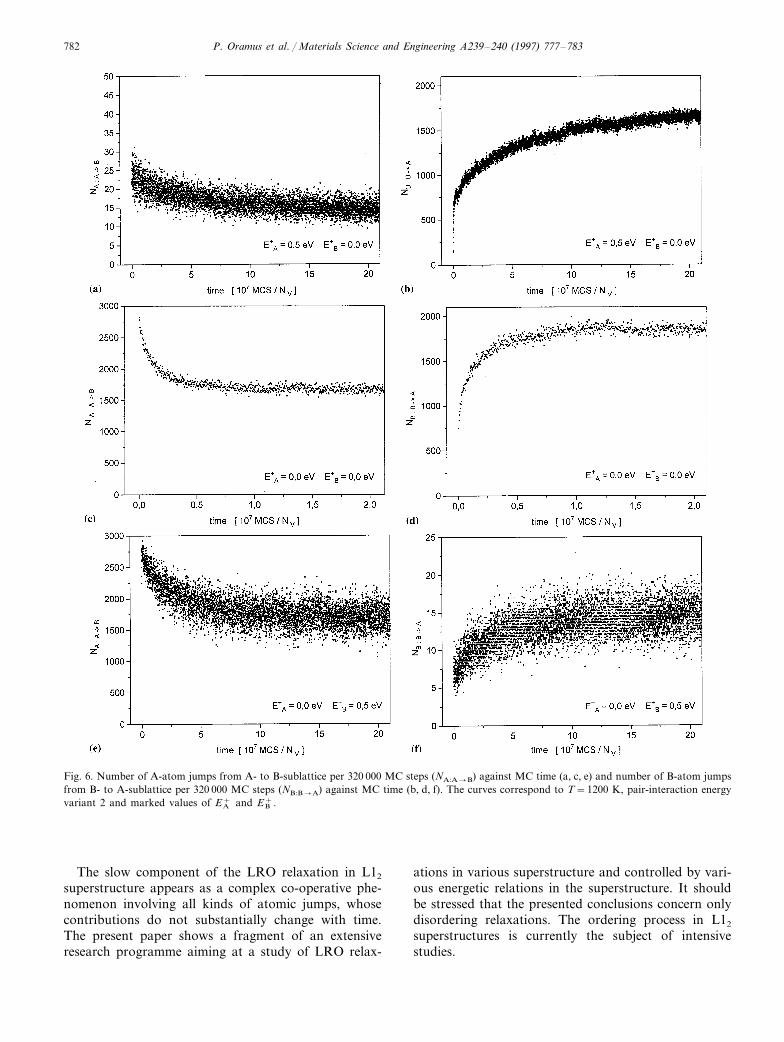
P. Oramus et al. / Materials Science and Engineering A239–240 (1997) 777–783782
Fig. 6. Number of A-atom jumps from A- to B-sublattice per 320 000 MC steps (NA:A�B) against MC time (a, c, e) and number of B-atom jumpsfrom B- to A-sublattice per 320 000 MC steps (NB:B�A) against MC time (b, d, f). The curves correspond to T=1200 K, pair-interaction energyvariant 2 and marked values of EA
+ and EB+.
The slow component of the LRO relaxation in L12
superstructure appears as a complex co-operative phe-nomenon involving all kinds of atomic jumps, whosecontributions do not substantially change with time.The present paper shows a fragment of an extensiveresearch programme aiming at a study of LRO relax-
ations in various superstructure and controlled by vari-ous energetic relations in the superstructure. It shouldbe stressed that the presented conclusions concern onlydisordering relaxations. The ordering process in L12
superstructures is currently the subject of intensivestudies.
P. Oramus et al. / Materials Science and Engineering A239–240 (1997) 777–783 783
Fig. 7. Effect of B-atom mobility on the efficiency of disordering byA-atom jumps from A- to B-sublattice.
References
[1] R. Kozubski, W. Pfeiler, Acta Mater. 44 (1996) 1573.[2] R. Kozubski, M. Migschitz, W. Pfeiler, in: A. Gonis, P.E.A.
Turchi, J. Kudrnovsky (Eds.), Stability of Materials, NATO ASISeries B, Plenum, New York, 1996, 699 pp.
[3] H. Schweiger, R. Podloucky, R. Kozubski, W. Pfeiler, Verh. DPG1997, Fruhjahrstagung des DPG, Munster 1997, abstract fromonly.
[4] R. Kozubski, W. Pfeiler, Mat. Res. Soc. Symp. Proc. 398 (1996)587.
[5] R. Kozubski, W. Pfeiler, Defect and diffusion forum, 143-147(1997) 279.
[6] K. Yaldram, V. Pierron-Bohnes, M.C. Cadeville, M.A. Khan, J.Mater. Res. 10 (1995) 591.
.





























