Embed Size (px)
Citation preview

Eur. J. Biochem. 161,571 -577 (1986) 0 FEBS 1986
Conformational studies of d(m5CpGpm5CpG) and d(CpGpCpG) by ‘H and ”P NMR Muriel DELEPIERRE I, Bdatrice LANGLOIS D’ESTAINTOT’, Jean IGOLEN and Bernard P. ROQUES
Departement de Chimie Organique, Unite Associke 498 Centre National de la Recherche Scientifique et Unit6 266 de 1’Institut National de la Santd et de la Recherche Mtdicale, Universite Rent Descartes, Paris Unite de Chimie Organique, Unite Associke 487 Centre National de la Recherche Scientifique, Institut Pasteur, Paris
(Received April 29/August 25, 1986) - EJB 86 0449
Exhaustive conformational studies of d(CpG)2 and ~ ( ~ ’ C P G ) ~ , two convenient targets for DNA bi- sintercalating drugs, have been carried out by ‘H and 31P NMR in low salt concentration and in the presence of 30% ethanol. Unambiguous ‘P assignments of the B form are obtained with low-power heteronuclear decoupling experiments, while ‘P assignments in the Z form are obtained by two-dimensional homonuclear chemical exchange experiments. The 31P chemical shifts and 3 J H 3 , ~ coupling constants studied at various temperatures in methylated and non-methylated tetranucleotides, are interpreted as resulting from conformational differences between the compounds. These features are corroborated by homonuclear proton nuclear Overhauser effect experiments showing the steric role of the 5-methylcytosine in the induction of an alternating B form in ~ ( ~ ’ C P G ) ~ .
Ditercalinium, i.e. 2,2’-[(4,4‘-bipiperidine)-l ,l’-diyldi-2,1- ethane diyl]bis(l0-methoxy-7H-pyrido[4,3-c]carbazolium) te- tramethanesulfonate (NSC 366241), is a potent antitumor dimeric compound presently under clinical trial [ 11. The major interest of this compound, which behaves as a DNA bi- sintercalator, lies in its mechanism of action which is quite different from that of other antitumor agents [2]. Moreover the pharmacological activity of ditercalinium seems to be crucially related to the structure of its complex with DNA since the lengthening of the linking chain or an increase in its flexibility leads to an almost complete loss of cytotoxic- ity [3].
Structural information on complexes between DNA and monomeric or dimeric compounds can be obtained from NMR studies using self-complementary oligonucleotides [4]. Thus both ‘H- and 31P-NMR spectra provide information on the geometry of base-base interactions and backbone con- formations. Sequences of Z forms were found in natural DNA [5] and we have recently shown that ethidium dimer can in- tercalate in Z DNA[6]. It would therefore be of major interest to determine the structure of the complexes of ditercalinium with these various forms of DNA.
Owing to the symmetry of ditercalinium, the use of symmetric oligonucleotides such as d(CpGpCpG) and d(m’CpGpm’CpG) would simplify the analysis of the in- tercalation complexes. Moreover, in appropriate solvent conditions, the methylated tetranucleotide can form a minihelix of the Z type.
Correspondence to B. P. Roques, Departement de Chimie Organique, UA 498 CNRS et U 266 INSERM, Universite Rent Descartes, 4, Avenue de I’Observatoire, F-75006 Paris, France
Abbreviations. NOE, nuclear Overhauser effect; COSY, chemical shift correlated spectroscopy; NOESY, two-dimensional nuclear Overhauser spectroscopy.
Structural investigations of ditercalinium complexed with DNA require a preliminary careful analysis of the conformational behaviour of the free d(CpGpCpG) and d(m’CpGpm’CpG) in B and Z forms. Although ‘H-NMR studies of various methylated nucleotides such as d(rn’C~G)~ [7], poly[d(G-m’C)] [8], d(m’CpGpCpGpm’CpG) [9] or non- methylated nucleotides poly[d(G-C)] [8], d(G-C)8 [lo], poly[d(C-G)] [ll] have already been reported, ‘H resonances in the Z forms were not completely assigned and 31P signals only tentatively attributed [7, 12, 131. Besides, detailed conformational features of methylated oligonucleotides in the B form remain unknown since crystallographic data have only been obtained for the Z form. Therefore all proton and phosphorus chemical shifts in B and Z forms of d(mSCpGpmSCpG), as well as the conformationally impor- tant 3JH3,p coupling constants, were measured at different temperatures and compared with values obtained for the non- methylated compound. The data are interpreted as resulting from conformational differences between the compounds.
MATERIALS AND METHODS
The deoxytetranucleoside trisphosphate d(m’CpGpm ’CpG) was synthesized in solution from dimers by a standard procedure [14]. Paramagnetic impurities were eliminated by both treatment with Chelex 100 and addition of a small amount of EDTA (about 3%) [15]. The tetranucleotide was then lyophilized from 99.95% D20 and diluted to 1 mM helix concentration with deuteroacetate buffer (pD = 5.5, uncorrected pH meter reading; 100 mM deuteroacetic acid/ sodium deuteroacetate, 0.030 mM sodium EDTA, 0.05 mM sodium 4,4-dimethyl4-silapentane sulfonate).
‘H-NMR spectra were recorded at 400 MHz on a Bruker AM 400. The assignment of the proton spectrum was achieved by two-dimensional chemical shift correlated spectroscopy at
572
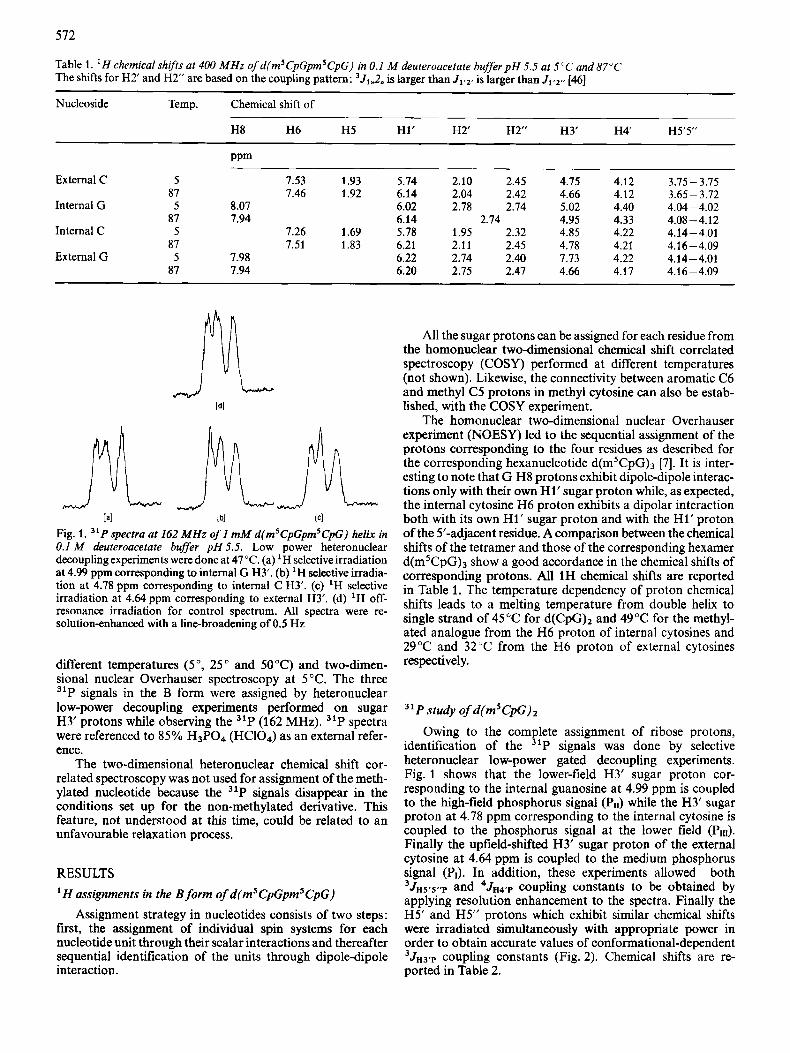
Table 1. ' H chemical shifts at 400 MHz of d(m5CpGpmsCpG) in 0.1 M deuteroacetate buffer p H 5.5 at 5°C and 87°C The shifts for H2' and H2' are based on the coupling pattern: 351,,2,, is larger than J1,2, is larger than J1,2- [46]
Nucleoside Temp. Chemical shift of
H8 H6 H5 H1' H2' H2" H3' H 4 H5'5"
PPm
External C 5 7.53 1.93 5.14 2.10 2.45 4.75 4.12 3.75- 3.75 87 7.46 1.92 6.14 2.04 2.42 4.66 4.12 3.65- 3.72
Internal G 5 8.07 6.02 2.78 2.74 5.02 4.40 4.04-4.02 87 7.94 6.14 2.74 4.95 4.33 4.08 -4.12
Internal C 5 7.26 1.69 5.78 1.95 2.32 4.85 4.22 4.14- 4.01 87 7.51 1.83 6.21 2.11 2.45 4.78 4.21 4.16 -4.09
External G 5 7.98 6.22 2.74 2.40 1.73 4.22 4.14- 4.01 87 7.94 6.20 2.75 2.47 4.66 4.17 4.16 -4.09
[a1 [ bl lcl Fig. 1. "P spectra at 162 MHz of 1 mM d(msCpGpmsCpG) helix in 0.1 M deuteroacetate buffer pH 5.5. Low power heteronuclear decoupling experiments were done at 47°C. (a) 'H selective irradiation at 4.99 ppm corresponding to internal G H3'. (b) 'H selective irradia- tion at 4.78 pprn corresponding to internal C H3'. (c) 'H selective irradiation at 4.64 pprn corresponding to external H3'. (d) 'H off- resonance irradiation for control spectrum. All spectra were re- solution-enhanced with a line-broadening of 0.5 Hz
different temperatures (5", 25" and SOT) and two-dimen- sional nuclear Overhauser spectroscopy at 5 "C. The three 31P signals in the B form were assigned by heteronuclear low-power decoupling experiments performed on sugar H3' protons while observing the 31P (162 MHz). "P spectra were referenced to 85% H3P04 (HClO,) as an external refer- ence.
The two-dimensional heteronuclear chemical shift cor- related spectroscopy was not used for assignment of the meth- ylated nucleotide because the 31P signals disappear in the conditions set up for the non-methylated derivative. This feature, not understood at this time, could be related to an unfavourable relaxation process.
RESULTS ' H assignments in the B form of d(m5CpGpm5CpG)
Assignment strategy in nucleotides consists of two steps: first, the assignment of individual spin systems for each nucleotide unit through their scalar interactions and thereafter sequential identification of the units through dipole-dipole interaction.
All the sugar protons can be assigned for each residue from the homonuclear two-dimensional chemical shdt correlated spectroscopy (COSY) performed at different temperatures (not shown). Likewise, the connectivity between aromatic C6 and methyl C5 protons in methyl cytosine can also be estab- lished, with the COSY experiment.
The homonuclear two-dimensional nuclear Overhauser experiment (NOESY) led to the sequential assignment of the protons corresponding to the four residues as described for the corresponding hexanucleotide d ( r n T ~ G ) ~ [7]. It is inter- esting to note that G H8 protons exhibit dipole-dipole interac- tions only with their own HI' sugar proton while, as expected, the internal cytosine H6 proton exhibits a dipolar interaction both with its own HI' sugar proton and with the HI' proton of the 5'-adjacent residue. A comparison between the chemical shifts of the tetramer and those of the corresponding hexamer d(rn 'C~G)~ show a good accordance in the chemical shifts of corresponding protons. All 1H chemical shifts are reported in Table 1. The temperature dependency of proton chemical shifts leads to a melting temperature from double helix to single strand of 45°C for ~ ( C P G ) ~ and 49°C for the methyl- ated analogue from the H6 proton of internal cytosines and 29°C and 32°C from the H6 proton of external cytosines respectively.
3 1 P study of d(msCpG),
Owing to the complete assignment of ribose protons, identification of the 31P signals was done by selective heteronuclear low-power gated decoupling experiments. Fig. 1 shows that the lower-field H3' sugar proton cor- responding to the internal guanosine at 4.99 ppm is coupled to the high-field phosphorus signal (Pll) while the H3' sugar proton at 4.78 ppm corresponding to the internal cytosine is coupled to the phosphorus signal at the lower field (PII1). Finally the upfield-shifted H3' sugar proton of the external cytosine at 4.64 ppm is coupled to the medium phosphorus signal (PI). In addition, these experiments allowed both 3JH5.5..p and 4JH4'p coupling constants to be obtained by applying resolution enhancement to the spectra. Finally the H5' and H5" protons which exhibit similar chemical shifts were irradiated simultaneously with appropriate power in order to obtain accurate values of conformational-dependent 3JH3rp coupling constants (Fig. 2). Chemical shifts are re- ported in Table 2.
513
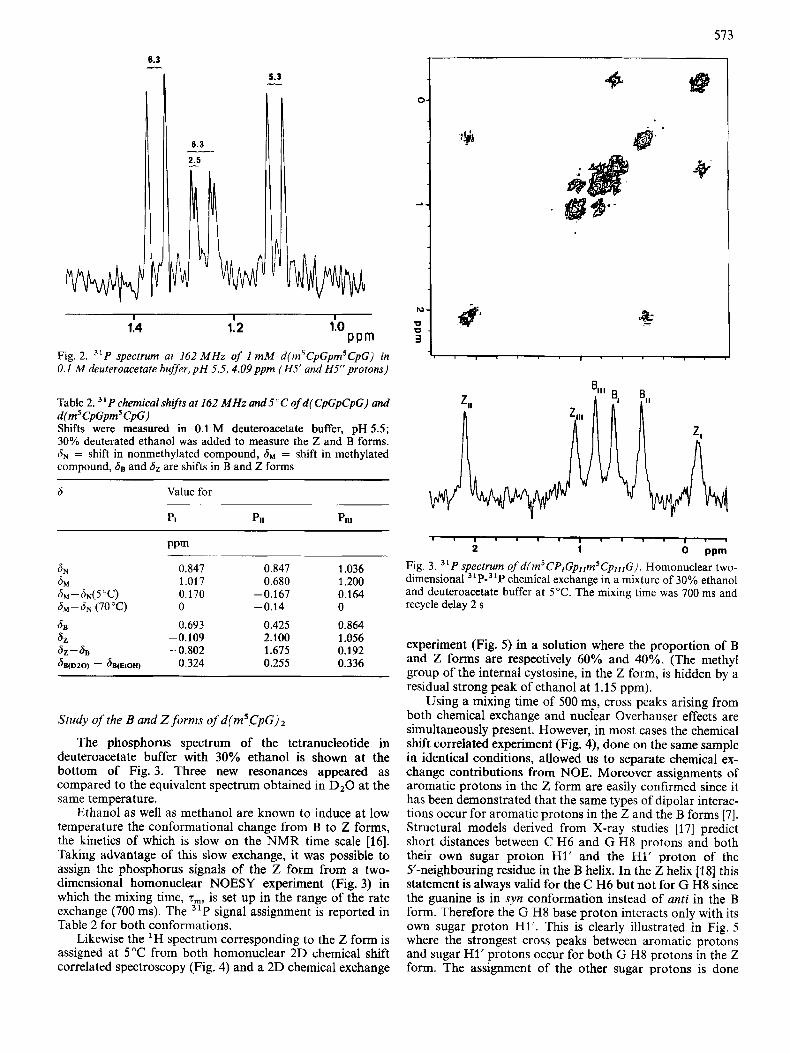
6.3 _L
2.5
5.3 -
1:4 1: 2 110 PPm
Fig. 2. 31P spectrum at 162 MHz of 1 mM d(m’CpGpm5CpG) in 0.1 M deuteroacetate buger, p H 5.5,4.09ppm (HS’ and H5“protons)
Table 2. 31 P chemical shijts at 162 MHz and 5°C of d ( CpGpCpG) and d(rn5CpGpm5CpC) Shifts were measured in 0.1 M deuteroacetate buffer, pH 5.5; 30% deuterated ethanol was added to measure the Z and B forms. SN = shift in nonmethylated compound, hM = shift in methylated compound, 88 and Sz are shifts in B and Z forms
6 Value for
SN 0.847 0.847 1.036 6M 1.01 7 0.680 1.200 SM-SN(5OC) 0.170 -0.167 0.164 8 M - S N (70°C) 0 -0.14 0
68 0.693 0.425 0.864 62 -0.109 2.100 1.056 62 - dB -0.802 1.675 0.192 ~ B ( D Z O ) - ~ B ( E ~ O H ) 0.324 0.255 0.336
Study of the B and Z forms of d(m5,CpG)2
The phosphorus spectrum of the tetranucleotide in deuteroacetate buffer with 30% ethanol is shown at the bottom of Fig. 3. Three new resonances appeared as compared to the equivalent spectrum obtained in D20 at the same temperature.
Ethanol as well as methanol are known to induce at low temperature the conformational change from B to Z forms, the kinetics of which is slow on the NMR time scale [16]. Taking advantage of this slow exchange, it was possible to assign the phosphorus signals of the 2 form from a two- dimensional homonuclear NOESY experiment (Fig. 3) in which the mixing time, z,, is set up in the range of the rate exchange (700 ms). The 31P signal assignment is reported in Table 2 for both conformations.
Likewise the ‘H spectrum corresponding to the Z form is assigned at 5°C from both homonuclear 2D chemical shift correlated spectroscopy (Fig. 4) and a 2D chemical exchange
“1 -+
i I , . I , , , , , , , , ,
- l , l . , , , l , , , . l . ,
1 0 PPm 2
Fig. 3. 31P spectrum of d(mSCPrGpl,m5CpIIIG). Homonuclear two- dimensional ”P-”P chemical exchange in a mixture of 30% ethanol and deuteroacetate buffer at 5°C. The mixing time was 700 ms and recycle delay 2 s
experiment (Fig. 5 ) in a solution where the proportion of B and Z forms are respectively 60% and 40%. (The methyl group of the internal cystosine, in the Z form, is hidden by a residual strong peak of ethanol at 1.15 ppm).
Using a mixing time of 500 m!, cross peaks arising from both chemical exchange and nuclear Overhauser effects are simultaneously present. However, in most cases the chemical shift correlated experiment (Fig. 4), done on the same sample in identical conditions, allowed us to separate chemical ex- change contributions from NOE. Moreover assignments of aromatic protons in the 2 form are easily confirmed since it has been demonstrated that the same types of dipolar interac- tions occur for aromatic protons in the Z and the B forms [7]. Structural models derived from X-ray studies [17] predict short distances between C H6 and G H8 protons and both their own sugar proton H1’ and the HI’ proton of the 5’-neighbouring residue in the B helix. In the Z helix [lS] this statement is always valid for the C H6 but not for G H8 since the guanine is in syn conformation instead of anti in the B form. Therefore the G H8 base proton interacts only with its own sugar proton Hl’. This is clearly illustrated in Fig. 5 where the strongest cross peaks between aromatic protons and sugar H1’ protons occur for both G H8 protons in the Z form. The assignment of the other sugar protons is done
574
I I I I I
6 5 4 3 2 PPm
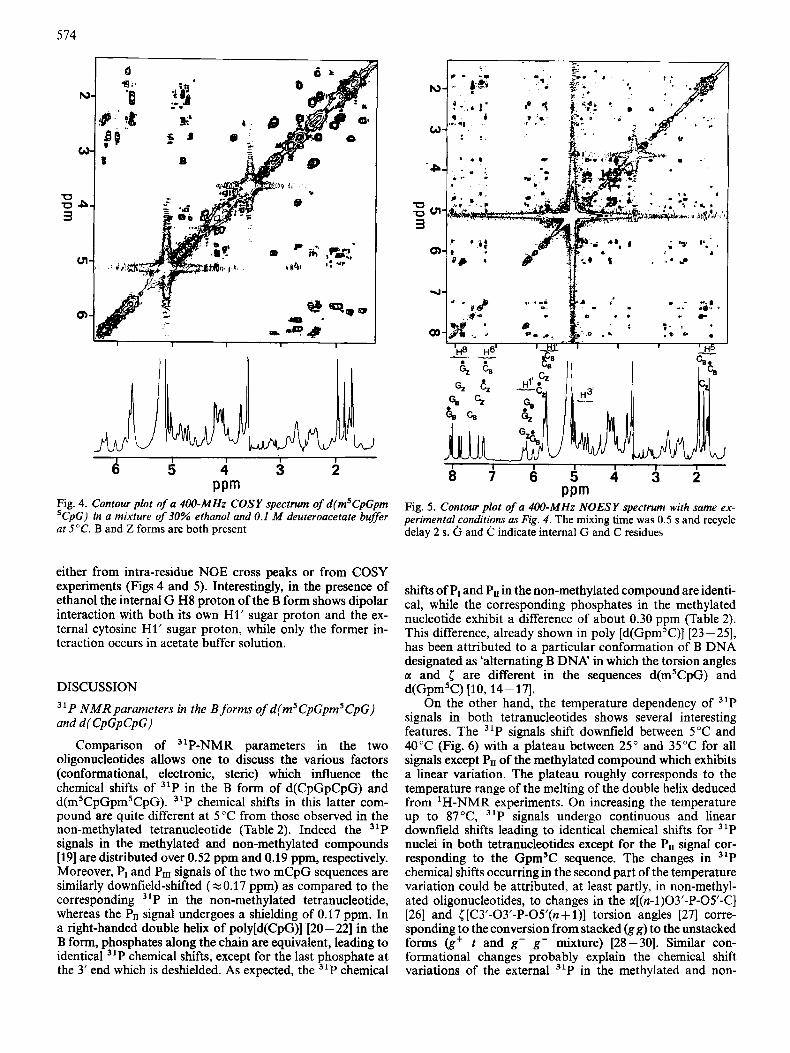
Fig. 4. Contour plot of a 400-MHz COSY spectrum of d(m5CpGpm ’CpG) in a mixture of 30% ethanol and 0.1 M deuteroacetate buffer at 5°C. B and Z forms are both present
either from intra-residue NOE cross peaks or from COSY experiments (Figs 4 and 5). Interestingly, in the presence of ethanol the internal G H8 proton of the B form shows dipolar interaction with both its own H1’ sugar proton and the ex- ternal cytosine H1’ sugar proton, while only the former in- teraction occurs in acetate buffer solution.
DISCUSSION
31P NMRparameters in the B forms of d(m’CpGpm’CpG) and d( CpGpCpG)
Comparison of ”P-NMR parameters in the two oligonucleotides allows one to discuss the various factors (conformational, electronic, steric) which influence the chemical shifts of 31P in the B form of d(CpGpCpG) and d(m5CpGpmSCpG). 31P chemical shifts in this latter com- pound are quite different at 5°C from those observed in the non-methylated tetranucleotide (Table 2). Indeed the ”P signals in the methylated and non-methylated compounds [19] are distributed over 0.52 ppm and 0.19 ppm, respectively. Moreover, PI and PIIl signals of the two mCpG sequences are similarly downfield-shifted (xO.17 ppm) as compared to the corresponding 31P in the non-methylated tetranucleotide, whereas the PI, signal undergoes a shielding of 0.17 ppm. In a right-handed double helix of poly[d(CpG)] [20 - 221 in the B form, phosphates along the chain are equivalent, leading to identical ”P chemical shifts, except for the last phosphate at the 3’ end which is deshielded. As expected, the 31P chemical
I . . .* 1
I z -
I I I I I I I
a 7 6 5 4 3 2 PPm
Fig. 5. Contour plot of a 4W-MHz NOESY spectrum with same ex- perimental conditions as Fig. 4. The mixing time was 0.5 s and recycle delay 2 s. and C indicate internal G and C residues
shifts of PI and Pn in the non-methylated compound are identi- cal, while the corresponding phosphates in the methylated nucleotide exhibit a difference of about 0.30 ppm (Table 2). This difference, already shown in poly [d(Gpm5C)] [23 - 251, has been attributed to a particular conformation of B DNA designated as ‘alternating B DNA’ in which the torsion angles a and [ are different in the sequences d(m’CpG) and d(Gpm’C) [lo, 14-17].
On the other hand, the temperature dependency of 31P signals in both tetranucleotides shows several interesting features. The 31P signals shift downfield between 5°C and 40°C (Fig. 6) with a plateau between 25” and 35°C for all signals except Pn of the methylated compound which exhibits a linear variation. The plateau roughly corresponds to the temperature range of the melting of the double helix deduced from ‘H-NMR experiments. On increasing the temperature up to 87”C, 31P signals undergo continuous and linear downfield shifts leading to identical chemical shifts for 31P nuclei in both tetranucleotides except for the PII signal cor- responding to the Gpm’C sequence. The changes in 31P chemical shifts occurring in the second part of the temperature variation could be attributed, at least partly, in non-methyl- ated oligonucleotides, to changes in the a[(n-l)03’-P-OY-C] [26] and [ [C3’-03’-P-O5’(n + l)] torsion angles [27] corre- sponding to the conversion from stacked (gg) to the unstacked forms (g’ t and g- g- mixture) [28-301. Similar con- formational changes probably explain the chemical shift variations of the external 31P in the methylated and non-
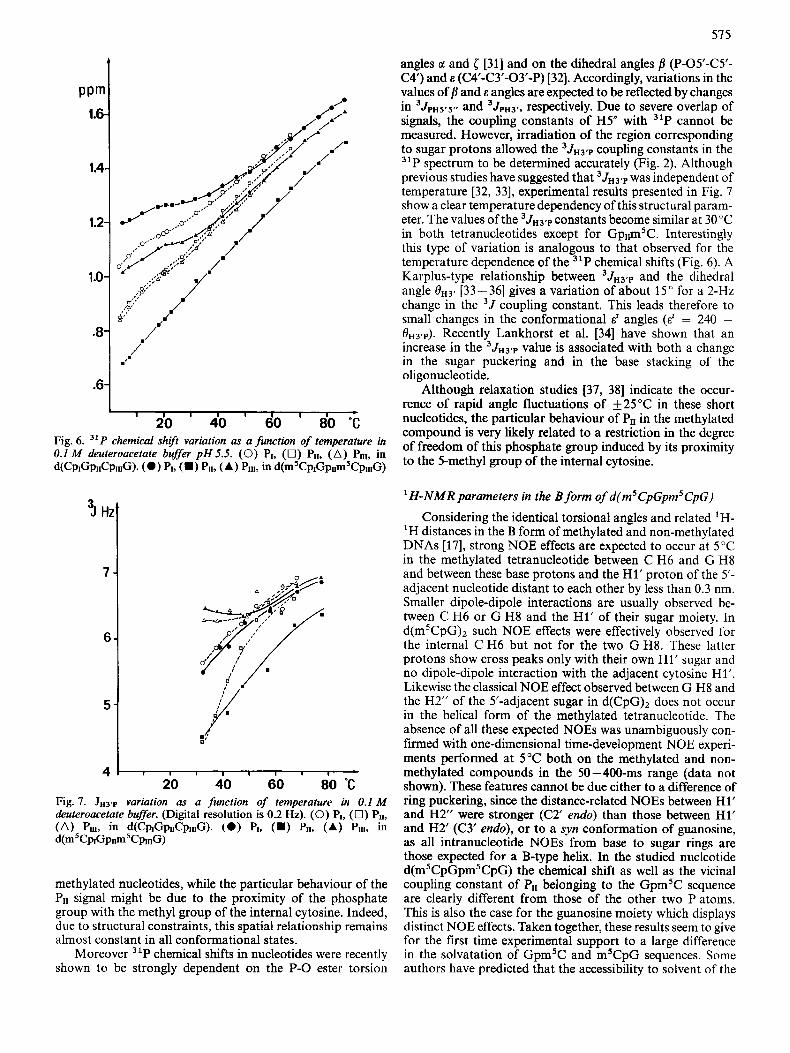
57 5
P Pm 1
1.2
1.0
.y ,./‘ i .6
I I I I I
20 40 do I 80 “C Fig. 6. 31P chemical shift variation as a function of temperature in 0.1 M deuteroacetate buffer p H 5.5. (0) PI, (0) Pu, (A) Pul, in d(CpK3&pIIIG). (0 ) PI, (W PII, (A) PIII, in ~ ( ~ ’ C P I G P I I ~ ~ C P W G )
!I Hz
7 -
6 -
5 -
4 20 40 60 80 ‘C
Fig. I. J H ~ T variation as a fwtction of temperature in 0.1 M deuteroacetate buffer. (Digital resolution is 0.2 Hz). (0) PI, (0) Pu, (A) PIU, in d(CplGplICpI1IG). (0 ) PI, (W Pu, (A) PLLL, in d(m5CnGp~~msCp~~~G)
methylated nucleotides, while the particular behaviour of the PII signal might be due to the proximity of the phosphate group with the methyl group of the internal cytosine. Indeed, due to structural constraints, this spatial relationship remains almost constant in all conformational states.
Moreover 31P chemical shifts in nucleotides were recently shown to be strongly dependent on the P-0 ester torsion
angles c1 and ( [31] and on the dihedral angles fl (P-05’-C5’- C4‘) and E (W-C3’-03’-P) [32]. Accordingly, variations in the values of j? and E angles are expected to be reflected by changes in 3JpH5r5r, and 3JpH3., respectively. Due to severe overlap of signals, the coupling constants of H5” with 31P cannot be measured. However, irradiation of the region corresponding to sugar protons allowed the 3JH3,p coupling constants in the 31P spectrum to be determined accurately (Fig. 2). Although previous studies have suggested that 3JH3.p was independent of temperature [32, 331, experimental results presented in Fig. 7 show a clear temperature dependency of this structural param- eter. The values of the 3JH3,p constants become similar at 30°C in both tetranucleotides except for GpI,mSC. Interestingly this type of variation is analogous to that observed for the temperature dependence of the 31P chemical shifts (Fig. 6) . A Karplus-type relationship between 3JH3,p and the dihedral angle e H 3 ’ [33 - 361 gives a variation of about 15 O for a 2-Hz change in the 3J coupling constant. This leads therefore to small changes in the conformational E‘ angles (E’ = 240 - 0H3,P). Recently Lankhorst et al. [34] have shown that an increase in the 3JH3,p value is associated with both a change in the sugar puckering and in the base stacking of the oligonucleotide.
Although relaxation studies [37, 381 indicate the occur- rence of rapid angle fluctuations of f25”C in these short nucleotides, the particular behaviour of PI, in the methyhted compound is very likely related to a restriction in the degree of freedom of this phosphate group induced by its proximity to the 5-methyl group of the internal cytosine.
‘H-NMR parameters in the B form of d(m5CpGpm5CpG)
Considering the identical torsional angles and related ‘H- ‘H distances in the B form of methylated and non-methylated DNAs [17], strong NOE effects are expected to occur at 5°C in the methylated tetranucleotide between C H6 and G H8 and between these base protons and the H1’ proton of the 5’- adjacent nucleotide distant to each other by less than 0.3 nm. Smaller dipole-dipole interactions are usually observed be- tween C H6 or G H8 and the H1’ of their sugar moiety. In d ( m ’ c ~ G ) ~ such NOE effects were effectively observed for the internal C H6 but not for the two G H8. These latter protons show cross peaks only with their own H1’ sugar and no dipole-dipole interaction with the adjacent cytosine Hl’. Likewise the classical NOE effect observed between G H8 and the H2” of the 5’-adjacent sugar in d(CpG)2 does not occur in the helical form of the methylated tetranucleotide. The absence of all these expected NOEs was unambiguously con- firmed with one-dimensional time-development NOE experi- ments performed at 5°C both on the methylated and non- methylated compounds in the 50 -400-ms range (data not shown). These features cannot be due either to a difference of ring puckering, since the distance-related NOES between H1’ and H2” were stronger (C2’ endo) than those between H1’ and H2’ (C3’ endo), or to a syn conformation of guanosine, as all intranucleotide NOEs from base to sugar rings are those expected for a B-type helix. In the studied nucleotide d(m5CpGpm5CpG) the chemical shift as well as the vicinal coupling constant of PII belonging to the Gpm’C sequence are clearly different from those of the other two P atoms. This is also the case for the guanosine moiety which displays distinct NOE effects. Taken together, these results seem to give for the first time experimental support to a large difference in the solvatation of Gpm’C and mSCpG sequences. Some authors have predicted that the accessibility to solvent of the

576
methyl group in B DNA should be lower in a Gpm5C than in a mSCpG sequence [18].
B S Z transition in the methylated tetranucleotide The addition of 30% ethanol in the solution of
d(m5CpGpm5CpG) at 5°C leads to the appearance of about 40% of the left-handed Z form and a small upfield shift of the 31P signals in the B form (Table 2). The well known in- fluence of organic solvents on the chemical shift of 31P is mainly related to their hydrogen-bond-donating ability and to the accessibility of the acceptor atom rather than to induced conformational changes [30, 391. The shielding induced on PI and P1lI in the B form by such a concentration of ethanol agrees with those reported in the literature [40]. As discussed before, the smaller perturbation observed for Pn is probably the consequence of a lesser accessibility to the solvent induced by the presence of the cytosine methyl group on the stacked cytosine.
31P chemical shifts in the Z form, reported in Table2, show opposite effects on PII (1.6-ppm deshielding) and on PI (0.8-ppm shielding), while the position of PIIl signal is only slightly downfield-shifted. The same pattern of 31P reso- nances occurs in the Z form of the hexanucleotide d(m5CpIGpIIm5CplIIGplvm5CpvG) [7]. Based on the unam- biguous assignment of ‘P resonances performed in the pre- sent study, the 31P spectrum of the hexanucleotide in the Z form can be assigned. Starting from low field to high field, the 31P resonances are: PII, PIv, Pv, PIIl and PI. These assignments and the induced shifts in methylated tetra- and hexanucleotides support the tentative attributions reported previously [7, 12, 13, 221 from conformational considerations
In contrast to Feigon et a1 [7], the unambiguous assign- ment of resonances in the B and Z forms of d ( m ’ c ~ G ) ~ makes it possible to evaluate a lower limit for the exchange rate between the two forms which are nearly equal. The smallest chemical shift difference is obtained for PIII (0.19 ppm at 162 MHz) leading to an exchange rate which must be lower than 193 s-’.
Using the ‘H-NMR spectrum we can evaluate the upper limit of the exchange rate between the B and Z forms. The smallest chemical shift difference between aromatic protons occurs for the H6 proton of the internal cytosine (8 Hz at 400 MHz), giving an upper limit of exchange of 58 s-’ which is in the same range as that derived from ‘P.
[41- 451.
CONCLUSIONS In this paper we have assessed a detailed compara-
tive study of the B forms of d(m5CpGpm5CpG) and d(CpGpCpG). The introduction of a methyl group at position C5 on the cytosine moieties leads to small but significant changes in the B conformation, reflected by differences in the ‘P chemical shifts and 3JH3,p coupling constants between
methylated and non-methylated nucleotides, and confirmed by a stronger NOE interaction between the G H8 proton and the H1’ proton of its own sugar moiety in the methylated compound. These results are interpreted by the occurrence, in the methylated nucleotide, of an alternating B form slightly different from the classical B form found in d(CpGpCpG) mainly induced by a large difference in the solvation of Gpm’C and m5CpG sequences.
These features characteristic of methylated oligo- nucleotides should serve as valuable probes to investigate
the structure of complexes of bis-intercalators with DNA (rnonintercalation, bis-intercalation with or without excluded site, external interaction etc.).
This research was supported by funds from the Fondation Pour la Recherche Medicale Frangaise, the Association pour la Recherche contre le Cancer and the Ligue Nationale Frangaise contre le Cancer. We wish to thank Dr C. Brevard for helpful discussions and A. Bouju for typing the manuscript.
REFERENCES
1. Roques, B. P., Pdaprat, D., Le Guen, I., Porcher, G., Gosse, C. & Le Pecq, J. B. (1979) Biochem. Pharmacol. 28, 1811 -1815.
2. Esnault, C., Roques, B. P., Jacquemin-Sablon, J. & Le Pecq, J. B. (1984) Cancer Res. 44,4355-4360.
3. Pklaprat, D., Delbarre, A., Le Guen, I., Le Pecq, J. B. & Roques, B. P. (1980) J . Med. Chem. 23, 1336-1342.
4. Krugh, T. R. & Nuss, M. E. (1979) in Biological applicarion of magnetic resonances (Schulman, R. G., ed.) pp. 113-178, Academic Press, New York.
5. Santella, R. M., Grunberger, D., Weinstein, I. B. & Rich, A. (1981) Proc. Nut1 Acad. Sci. USA 78,1451 -1455.
6. Markovits, J., Ramstein, J., Roques, B. P. & Le Pecq, J. B. (1985) Nucleic Acids Res. 13, 3373-3388.
7. Feigon, J., Wang, A. H. J., Van Der Marel, G. A., Van Boom, J. H. &Rich, A. (1984) Nucleic Acids. Res. 12, 1243-1263.
8. Patel, D. P., Kozlowski, S. A., Nordheim, A. & Rich, A. (1982) Proc. Natl Acad. Sci. USA 79, 1413- 1417.
9. Tran Dinh, S., Taboury, J., Neumann, J. M., Huynh Dinh, T., Genissel, R., Gouyette, C. & Igolen, J. (1983) FEBS Lett. 154,
10. Patel, D. J., Canuel, L. L. & Pohl, F. M. (1979) Proc. Nut1 Acad. Sci. USA 76,2508-2511.
11. Thamann, T. J., Lord, R. C., Wang, A. H. J. & Rich, A. (1981) Nucleic Acids Res. 9, 5443 - 5457.
12. Hartmann, B., Thuong, N., Pouyet, J., Ptak, M. & Leng, M. (1983) Nucleic Acids Res. 11,4453-4466.
13. Gesnest, D., Hartmann, B., Thuong, N., Ptak, M. & Leng, M. (1984) Biochem. Biophys. Res. Commun. 125, 803 - 81 1.
14. Sung, W. L. (1981) J . Chem. SOC. Chem. Commun., 1089. 15. Avedikian, A. M., Besserre, D. & Delepierre, M. (1983) J . Chim.
16. Pohl, F. M. (1976) Nature (Lond.) 260, 365-366. 17. Mellema, J. R., Van Kampen, P. N., Carlson, C. N., Bosshard,
H. E. & Altona, C. (1983) Nucleic Acids Res. 1 1 , 2893- 2905.
18. Fujii, S., Wang, A. H. J., Van Der Marel, G., Van Boom, J. H. & Rich, A. (1982) Nucleic Acids Res. 10, 7879 - 7982.
19. Cheng, D. M., Kan, L. S., Frechet, D., Ts’O, P. 0. P., Uesugi, S., Shida, T. & Ikehara (1984) Biopolymers 23,775-795.
20. Patel, D. J. & Canuel, L. L. (1979) Eur. J. Biochem. 96, 267- 276.
21. Simpson, R. T. & Shindo, H. (1979) Nucleic Acids Res. 7 , 481 - 492.
22. Cohen, J. S., Wooten, J. B. & Chatterjee, C. L. (1981) Biochemis- try 20, 3049 - 3055.
23. Shindo, H. & Zimmerman, S. B. (1980) Nature (Lond.) 283,
24. Shindo, H., Simpson, R. T. & Cohen, J. S. (1979) J . Biol. Chem.
25. Chen, C. W., Cohen, J. S. & Behe, M. (1983) Biochemistry 22,
26. Seeman, N. C., Rosenberg, J. M., Suddath, F. L., Park Kim, J. J. & Rich, A. (1976) J. Mol. Biol. 104, 142-143.
27. IUPAC-IUB Joint Commission on Biochemical Nomenclature (1983) Eur. J . Biochem. 131,9-15.
28. Patel, D. J. & Canuel, J. J. (1979) Eur. J . Biochem. 96,267-276. 29. Patel, D. J. (1976) Biopolymers IS, 533-558. 30. Patel, D. J. (1977) Biopolymers 16, 1635-1656.
407 - 41 0.
Phys. 80,831 -833.
690 - 691.
254,8125-8128.
21 36 - 21 42.

577
31. Gorenstein, D. G. & Kar, D. (1975) Biochem. Biophys. Res. Commun. 65,1073- 1080.
32. Giessner-Prettre, C., Pullman, B., Ribas-Prado, F., Cheng, D., Ivorno, V. & Ts’O, P. 0. P. (1984) Biopolymers 23,317-388.
33. Lown, J. W., Hanstock, C. C., Bleackley, R. C., Imbach, J. L., Rayner, B. & Vasseur, J. J. (1984) Nucleic Acids Res. 12,2519- 2535.
34. Lankhorst, P. P., Haasnoot, C. A. C., Erkelens, C., Westerink, H. P., Van Der Marel, G. A., Van Boom, J. H. & Altona, C. (1985) Nucleic Acid.y Res. 13, 927 -942.
35. Tran-Dinh, S., Neumann, J. M. & Borrel, J. (1981) Biochim. Biophys. Acta 655,167- 180.
36. Lce, C. H. & Sarma, R. H. (1976) J. Am. Chem. SOC. 98,3541 - 3548.
37. Hogan, M. E. & Jardetzky, 0. (1980) Biochemistry 19, 3460- 3468.
38. Keepers, J. W. & James, T. L. (1982) J. Am. Chem. SOC. 104,
39. Lerner, D. B., Becktel, W. J., Everett, R., Goodman, M. & 929 - 939.
Kearns, D. (1984) Biopolymers 23,2157-2172.
40. Lerner, D. B. & Kearns, D. R. (1980) J. Am. Chem. Soc. 102,
41. Wang, A. H. J., Quigley, G. J., Kolpack, F. J., Crawford, J. L., Van Boom, J. H., Van Der Marel, G. A. & Rich, A. (1979) Nature (Lond.) 283,743 - 145.
42. Wang, A. H. J., Quigley, G. J., Kolpack, F. J., Van Der Marel, G., Van Boom, J. H. & Rich, A. (1981) Sciences (Wash. DC)
43. Arnott, S., Chandrasekaran, R., Birdsall, D. L., Leslie, A. G. W. & Ratliff, R. L. (1980) Nature (Lond.) 283, 143- 745.
44. Cheng, D. M. & Sarma, R. (1977) J. Am. Chem. SOC. 99,7333- 7348.
45. Jovin, T. M., McIntosh, L. P., Arnol-Jovin, D. J., Zarling, D. A., Robert-Nicoud, M. & Van de Sande, J. H. (1983) J . Biomof. Struct. Dynamics I , 21 -57.
46. Davies, D. B. & Danyluk, S. S. (1974) Biochemistry 13, 4417- 4434.
7612- 761 3.
211,111 -174.





![NMR Hyperpolarization Techniques of GasesDevices for preparing HP noble gases may be grouped into two types: “stopped-flow” and “continuous-flow”. In a stopped-flow device,[44]](https://img.pdfslide.fr/doc/110x75/60ff24ddb23fee5a07037b1d/nmr-hyperpolarization-techniques-of-gases-devices-for-preparing-hp-noble-gases-may.jpg)