Embed Size (px)
Citation preview

Review ArticleContribution of In Vivo and Organotypic 3D Models toUnderstanding the Role of Macrophages and Neutrophils in thePathogenesis of Psoriasis
Isabelle Lorthois,1,2 Daniel Asselineau,3 Nathalie Seyler,4 and Roxane Pouliot1,2
1Centre LOEX de l’Université Laval, Génie Tissulaire et Régénération, Centre de Recherche FRQS du CHU de Québec,Axe Médecine Régénératrice, Québec, QC, Canada2Faculté de Pharmacie, Université Laval, Québec, QC, Canada3L’Oréal Research & Innovation, Aulnay-sous-Bois, France4Episkin Academy, Lyon, France
Correspondence should be addressed to Roxane Pouliot; [email protected]
Received 19 May 2017; Revised 15 September 2017; Accepted 2 October 2017; Published 8 November 2017
Academic Editor: Anna Balato
Copyright © 2017 Isabelle Lorthois et al. This is an open access article distributed under the Creative Commons AttributionLicense, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work isproperly cited.
Psoriasis, a common chronic immune-mediated skin disease, is histologically characterized by a rapid keratinocyte turnover anddifferentiation defects. Key insights favor the idea that T cells are not the only key actors involved in the inflammatory process.Innate immune cells, more precisely neutrophils and macrophages, provide specific signals involved in the initiation and themaintenance of the pathogenesis. Current data from animal models and, to a lesser extent, three-dimensional in vitro modelshave confirmed the interest in leaning towards other immune cell types as a potential new cellular target for the treatment of thedisease. Although these models do not mimic the complex phenotype nor all human features of psoriasis, their development isnecessary and essential to better understand reciprocal interactions between skin cells and innate immune cells and toemphasize the crucial importance of the local lesional microenvironment. In this review, through the use of in vivo and 3Dorganotypic models, we aim to shed light on the crosstalk between epithelial and immune components and to discuss the role ofsecreted inflammatory molecules in the development of this chronic skin disease.
1. Introduction
Psoriasis is a chronic autoimmune disease that affects 2-3%of the world’s population, characterized by hyperprolifera-tion and abnormal differentiation of epidermal keratinocytes[1–6]. Psoriatic skin lesions are also characterized byincreased permeability of lymphatic capillaries, increasedblood flow, and angioproliferation [7–10]. Eighty percent ofpatients suffer from mild to moderate forms of the disease,while 20% of patients develop moderate to severe psoriasis,affecting more than 5% of their body surface area [11]. Itis also known that patients with plaque psoriasis have anincreased risk of inflammatory diseases affecting noncuta-neous sites—including psoriatic arthritis, cardiovascular dis-ease, and inflammatory bowel disease—associated with
common pathophysiological mechanisms. These comorbidi-ties are multifactorial and in many cases related to inflamma-tion, induced by close pathogenic mechanisms related tocytokine dysregulation.
PSORS1 is the major susceptibility locus for psoriasisvulgaris and lies within an approximately 300 kb segment ofthe major histocompatibility complex on chromosome6p21.3 [12–14]. Several studies have indicated HLA-Cw6 asthe primary PSORS1 risk allele within the candidate region,coherent with the fact that MHC class I molecules play animportant role in the function of CD8+ T cells [15, 16].
More than 32 PSORS have been identified, containinggenes involved in inflammatory metabolic pathways andepidermal proliferation as well as skin barrier function, buthave not demonstrated their complete involvement in
HindawiMediators of InflammationVolume 2017, Article ID 7215072, 13 pageshttps://doi.org/10.1155/2017/7215072

pathology. Also, the variations in the number of copies of agene may be involved in the pathology. For example, betadefensins, antimicrobial peptides involved in innate immu-nity, are a good example of a gene known to be associatedwith psoriasis. Of the 8 defensins, the hBD-2, hBD-3, andhBD-4 proteins encoded, respectively, by DEFB2, DEFB3,and DEFB4 were linked to keratinocyte stimulation via pro-inflammatory interleukins 8, 18, and 20 [17]. The eightdefensin genes are linked on two different chromosomes,chromosome 20 as well as chromosome 8p23.1. Most of thedefensin genes encoded on chromosome 8p23.1 have longergene repeat units, which are highly variable in copy number.Several studies have attributed a relationship betweenpsoriasis and the number of gene copies of these defensins[18]. In 2012, a meta-GWAS (genome-wide associationstudies), which aims to identify SNPs (single nucleotidepolymorphisms) in DNA associated with a clinicallydefined disease (phenotype) by comparing the allele fre-quency of each SNP between a group of individuals withpsoriasis versus healthy patients, confirmed 21 SNPs, andidentified 15 new SNPs [19].
The current research tends to demonstrate that theprocess is initiated by an inflammatory immune reactionagainst autoantigens of the skin, in which dendritic cells, Tlymphocytes, macrophages, and neutrophils play a pivotalrole. Dendritic cells, antigen-presenting cells, are present ingreater numbers in psoriatic lesions. Dendritic cells oflymphoid origin, such as plasmacytoid dendritic cells, wouldbe involved in initiating lesions [20], recognizing autoanti-gens, and causing IFN-α secretion by these cells [21]. Thiswould follow the activation of innate immunity cells, suchas neutrophils or macrophages, and adaptive immune cells,such as T lymphocytes. Persistent activation of these cellswould lead to the chronicization of psoriatic lesions, suchas a vicious circle of inflammation [2, 22, 23].
Resident macrophages and dendritic cells are among thecells most involved in the “sensing” of danger signals. Theactivation of macrophages via the secretion of proinflamma-tory cytokines, such as IL-6 and TNF-α, but also ofchemokines, such as CXCL8 (also known as IL-8), CCL5,CXCL1, and CXCL2, promotes the recruitment of inflamma-tory cells, like neutrophils [24]. Multiple signals are likelyto trigger, via interaction with their receptor(s), a secretionof chemokines, thus attracting neutrophils to the inflam-matory site. Macrophages and neutrophils may act as Tlymphocyte-dependent effectors, as they are present atthe site of inflammation even before a specific immuneresponse has developed.
It is therefore evident that monocytes, macrophages, andneutrophils have a particular function in the early phases ofinflammation, and their role in driving and maintaining thisinflammatory process in the pathogenesis of psoriasis mustbe clarified. Here, we aim to discuss the specific role of innateimmune cells, such as neutrophils and macrophages, in theinitiation and the sustainability of chronic inflammatory skindiseases, such as psoriasis, through the use of organotypicmodels and mouse models. These models allow a betterunderstanding of cellular and molecular mechanisms withthe aim of identifying new potential therapeutic targets.
2. Macrophages
Monocytes can differentiate to become tissue-residentmacrophages or dendritic cells. Macrophages are phagocyticcells [25] within the dermis, important for tissue homeostasisand the regulation of lymphocyte activation and proliferation[26]. Some macrophages are long-term tissue residents andplay an important role in controlling the repair [27, 28] andregeneration of skin tissue [29]. Inflammatory macrophages,on the other hand, participate in the innate immuneresponse and play a dual role in the immune system asphagocytes and antigen-presenting cells capable of activatingT lymphocytes (Figure 1).
As early as the 1980s, some evidence of increased macro-phage activity in psoriasis [30] highlights their key role ininducing psoriasis-like skin disease.
In 2010, Fuentes-Duculan and colleagues observed that asubpopulation of CD163-positive macrophages was foundmostly in psoriatic lesions. The CD163 marker, a scavengerreceptor intervening in the elimination of the hemoglobin-haptoglobin complex, expressed both on the surface of themature tissue macrophages and on blood monocytes, is innormal human skin, a marker more assimilated to the so-called “alternative” macrophages or M2 [31]. However,CD163-positive macrophages also express IFN-γ-regulatedgenes (STAT1, CXCL9), and IFN-γ is known to be a “type1 cytokine.” These results suggest a great phenotypic plastic-ity of the macrophages in responding to their environmentand acquiring new properties and new markers, whilepreserving their original characteristics.
Other macrophage markers such as RFD7 [32], CD68[33, 34], CD107 [35, 36], MARCO [37], Stabilin-1 [38], andMS-1 [39, 40] are not generally expressed at the same timeon their surface, but their expression fluctuates according totheir location in the skin compartments. For example,CD68+ cells coexpress CD163 in the upper reticular dermiswhile they do not colocalize with CD163 near the dermoepi-dermal junction. It has also been shown that these markerscoexpress to some extent CD11c, a marker of myeloiddendritic cells, but CD163 has the weakest coexpression withCD11c, an ideal candidate for labeling macrophages inpsoriasis [31]. The cutaneous macrophages of lesional pla-ques probably do not have a single phenotype, M1, or M2but rather have a mixed, microenvironment-dependentphenotype, associated with the different roles they can playin a context of chronic inflammation.
Egawa et al. have also demonstrated that CCR2+ mono-cytes recruited at inflammatory sites had the potential toacquire an M2 phenotype in response to IL-4, thus exertingan anti-inflammatory function [41], while the expression ofCCR2 (MCP-1/CCL2 protein receptor) of the peripheralmonocytes of patients with psoriasis or atopic dermatitiswas increased compared to that of healthy patients [42]. Ithas also been demonstrated that macrophages M1 or M2have the ability to be repolarized by the cytokines of Th2 orTh1 lymphocytes, respectively [43].
The hypomorphic PL/J CD18 murine model is charac-terized by reduced expression (2–16% of the wild-typelevels) of the common integrin 2 (CD11/CD18) chain,
2 Mediators of Inflammation
leukocyte adhesion molecules required for cell-cell con-tacts. Transgenic mice generally develop psoriasiform skininflammation: erythema, scaling, abnormal keratinocyte pro-liferation/differentiation, subcorneal microabscesses, andincreased inflammatory infiltrate. The authors reported thatthe activation of cutaneous macrophages by recombinantMCP-1 or LPS alone was not sufficient to produce chronicpsoriasis-like inflammation as observed in vivo in humanskin lesions [44]. Nevertheless, the combined injection ofrecombinant murine MCP-1 and TNF-α in nonlesional skinarea of hypomorphic CD18 mice led, respectively, to theaccumulation and activation of macrophages, unlike CD4-positive T cells. Activated macrophages would secrete moreTNF-α, responsible for sustained activation of macrophages,thus causing a positive feedback loop. T cells, mast cells, andendothelial cells would participate directly in the chroniciza-tion of lesions via TNF-α secretion. Finally, depletion of skinmacrophages in this murine model via the injection ofclodronate liposomes (specific depletion of macrophages)and the neutralization of a single cytokine, TNF-α, attenuatesthe severity of skin inflammation, emphasizing the impor-tance of macrophages in the psoriasis physiopathology.
A specific deletion of the keratinocyte NF-κB kinaseinhibitor (IKK2) in mice results in an inflammatory andhyperproliferative cutaneous phenotype. The treatment oftransgenic mice with a TNF-neutralizing antibody abolished
the inflammatory phenotype and therefore improved theskin phenotype. The injection of clodronate liposomes in thismurine model restored the expression of early and late kera-tinocyte differentiation markers and reduced the number ofgranulocytes and T cells present, highlighting the importanceof macrophages in the accumulation of granulocytes andT cells in inflammatory skin areas [45]. Moreover, a K14-Cre-IKK2fl/fl murine model showed the upward expressionof gene coding for proteins regulated by IFN-γ. The presenceof IFN-γ receptor may accelerate the onset of the psoriasis-like inflammatory skin disease in K14-Cre-IKK2fl/fl micebut is not essential for it to develop. The authors demon-strated that the migration of macrophages to the lesional skinareas and their subsequent activation was a necessary keyfeature for the development of psoriatic inflammation. Theactivated macrophages were then able to initiate andmaintain psoriasiform skin inflammation. These investiga-tions were also commented in the Journal of Clinical Investi-gation by Clark and Kupper in 2006 [46].
The topical application model of imiquimod (IMQ), a 7/8TLR (toll-like receptor) agonist widely used in mice,activated immune cells, such as macrophages and plasmacy-toid dendritic cells. The mice displayed a hyperplasticcutaneous epithelial-squamous phenotype similar to humanpsoriasis. Imiquimod-treated mice, KO for CX3CR1 (frac-talkine/CX3CL1 receptor), developed minor inflammation
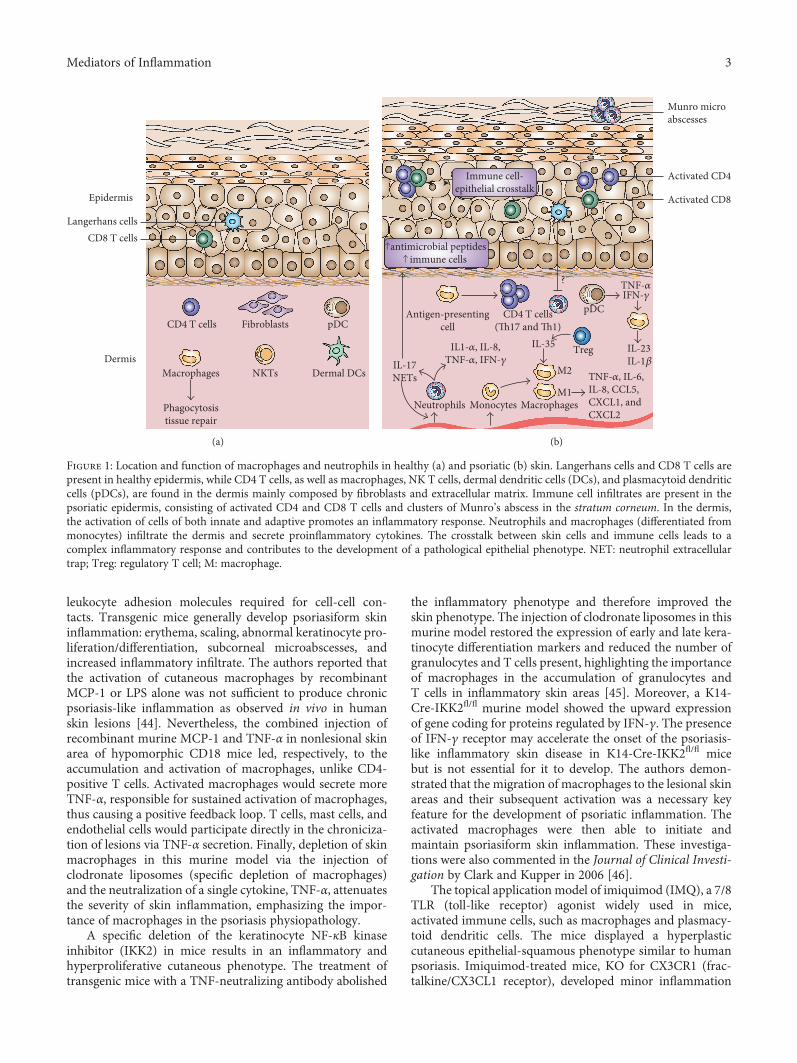
Dermis
Epidermis
CD8 T cellsLangerhans cells
Fibroblasts
Dermal DCsNKTsMacrophages
CD4 T cells
Phagocytosistissue repair
pDC
(a)
Activated CD8
Activated CD4
Munro microabscesses
CD4 T cells(Th17 and Th1)
TNF-�훼, IL-6,IL-8, CCL5,CXCL1, andCXCL2
MacrophagesMonocytesNeutrophils
TregIL-35
M2
M1
IL1-�훼, IL-8,TNF-�훼, IFN-�훾IL-17
NETs
antimicrobial peptidesimmune cells
TNF-�훼IFN-�훾
IL-23IL-1�훽
Immune cell-epithelial crosstalk
pDCAntigen-presentingcell
?
(b)
Figure 1: Location and function of macrophages and neutrophils in healthy (a) and psoriatic (b) skin. Langerhans cells and CD8 T cells arepresent in healthy epidermis, while CD4 T cells, as well as macrophages, NK T cells, dermal dendritic cells (DCs), and plasmacytoid dendriticcells (pDCs), are found in the dermis mainly composed by fibroblasts and extracellular matrix. Immune cell infiltrates are present in thepsoriatic epidermis, consisting of activated CD4 and CD8 T cells and clusters of Munro’s abscess in the stratum corneum. In the dermis,the activation of cells of both innate and adaptive promotes an inflammatory response. Neutrophils and macrophages (differentiated frommonocytes) infiltrate the dermis and secrete proinflammatory cytokines. The crosstalk between skin cells and immune cells leads to acomplex inflammatory response and contributes to the development of a pathological epithelial phenotype. NET: neutrophil extracellulartrap; Treg: regulatory T cell; M: macrophage.
3Mediators of Inflammation

compared toWTmice. In fact, the production of IL-12, IL-23,IL-17A, IL-22, IL-6, IL-1, TNF-α, and IL-36 cytokines wasdecreased in these mice. The macrophages of CX3CR1−/−
mice expressed CCR2, unlike WT mice treated with IMQ,and attenuated the inflammation generated, in part, byTh1/Th17 lymphocytes, following functional changes [47].The authors hypothesized that CCR2 could partially com-pensate for the loss of CX3CR1 by directing the migrationof resident macrophages. Surprisingly, CCR2−/− mice exhib-ited an exacerbation of inflammation despite altered recruit-ment of inflammatory monocytes to cutaneous sites.CX3CR1−/− mice expressed fewer M1 macrophage markerscompared to WT mice, suggesting that decreasing thenumber of M1 macrophages would contribute to decreasedinflammation resulting from CX3CR1 deficiency. Finally, atransfer of macrophages from WT mice to CX3CR1−/− micenormalized psoriasiform type inflammation induced byIMQ, emphasizing the importance of macrophages in theregulation of psoriatic inflammation.
The KC-Tie2-overexpressing mice developed a cutane-ous psoriasiform phenotype. These mice spontaneouslydeveloped characteristic hallmarks of human psoriasis,including acanthosis (hyperplasia of the epidermis),increases in dermal CD4+ T cells, infiltrating epidermalCD8+ T cells, dermal dendritic cells and macrophages, andincreased expression of cytokines and chemokines associatedwith psoriasis (IFN-γ, TNF-α, IL-1α, IL-6, IL-12, IL-22,IL-23, and IL-17). Cathelicidin, β-defensin, and S100A8/A9were also upregulated in the hyperproliferative skin [48].
The administration of clodronate liposomes in the skinof transgenic KC-Tie2 mice resulted in the elimination ofF4/80+ macrophages, CD11b+ myeloid cells, and CD11C+
dendritic cells. The eradication of these cells would result inthe disappearance of acanthosis, a decrease in the numberof T cells, and a significant reduction in the production ofTNF-α, IL-23, IL-1, IL-6, and S1008/9, stressing the impor-tance of myeloid cells and their cytokines in maintainingthe pathology [49].
The use of murine models, in particular the topicalapplication of imiquimod in mice, makes it possible to mimicthe dominance of monocytes, neutrophils, and dendritic cellsderived frommonocytes at an early lesion stage, and later, thedecreased number of neutrophils and monocytes and theparallel increase in the number of dermal macrophages andLangerhans cells [50], which are then depleted. Langerhanscells are found mainly in the spinous layer of the epidermis.It is estimated that they constitute 2 to 4% of the epidermalcell population [51]. Derived from the bone marrow, theyare transported by the blood to the epidermis [52]. After cap-turing the outer antigens, they migrate to the lymph nodeswhere they will initiate a specific immune response bydisplaying these antigens to the T lymphocytes. Langerhanscells are therefore antigen-presenting cells [52]. Similarly, inthe murine DKO model, whose Jun and JunB transcriptionfactors were deleted, resulting in psoriasiform type inflam-mation, the increase in the number of Langerhans cells(LCs) by proliferation followed by their subsequent decreasewould reproduce the presence of proliferative Langerhanscells in a human lesional context [53]. In this model, the
authors indicated that LCs would exert an immunoregula-tory role by increasing the expression of IL-10 and PD-L1.Without LCs, the absence of regulatory signals would resultin increased skin inflammation in these mice. The depletionof LCs did not alter the number of regulatory T cells in theskin, thus excluding the possibility that a reduced numberof regulatory T cells could be responsible for worsening ofthe pathology. In addition, genetic depletion of LCs duringthe inflammatory phase in mice treated with imiquimodcaused increased neutrophil infiltration and extension ofpustular plaques, suggesting an anti-inflammatory role forLCs during psoriatic inflammation [50].
Recently, Leite Dantas et al. focused on the contributionof macrophages and T cells in the development of psoriasi-form inflammation in a transgenic mouse model ihTNFtg(doxycycline- [Dox-] inducible human TNF-transgenicmouse line). In this murine model, the authors found thatonly macrophages (M1 and M2), Th1, and Treg were presentin large quantities. While depletion of macrophages greatlyreduced the development of the disease, Treg depletionincreased the infiltration of macrophages into psoriaticinflammatory areas, contributing to the worsening of thepathology. Adoptive transfer of Treg in RAG-1-deficientmice, without either mature B or T lymphocytes, or immuno-competent mice induced the opposite effect, attenuation ofsymptoms. Thus, Tregs would limit migration of macro-phages to injured areas, thereby reducing the harmful effecton tissue of macrophages in these transgenic mice [54].
Recently, IL-35, produced by regulatory T cells, demon-strated immunosuppressive effects in mouse models ofpsoriasis. Indeed, IL-35 may reduce the local infiltration ofmacrophages by reducing the levels of cutaneous expressionof the macrophages M1 while conversely increasing M2macrophages. Also, IL-35 may regulate the production ofproinflammatory CD4+ T cell cytokines and may decreaselocal lymphocytic infiltration of Th17 cells in K14-VEGF-A-Tg mice and in mouse models of imiquimod-inducedpsoriasis [55]. IL-37, expressed in macrophages, epithelialcells, and effector-memory cells, likewise demonstrated animmunosuppressive role in K14-VEGF-A transgenic miceby downregulating the production of proinflammatory cyto-kines, such as CXCL8, IL-6, and S100A7 [56]. IL-37 acted as anegative feedback inhibitor of inflammatory responses as thereduction of IL-37 protein synthesis in PBMCs with specificsiRNA increased the production of several proinflammatorymediators [57] (Table 1).
3. Neutrophils
Polymorphonuclear neutrophils (PMNs), or leukocytes, arephagocytic cells characterized by a segmented lobularnucleus and cytoplasmic granules filled with degradationenzymes. PMNs are the most abundant circulating whiteblood cells and are the first type of cells involved in acuteinflammatory reactions to bacterial infections [58]. Thesephagocytes ingest the microbes and release reactive oxygenspecies, antimicrobial peptides, proteases, and neutrophilextracellular traps [59, 60].
4 Mediators of Inflammation
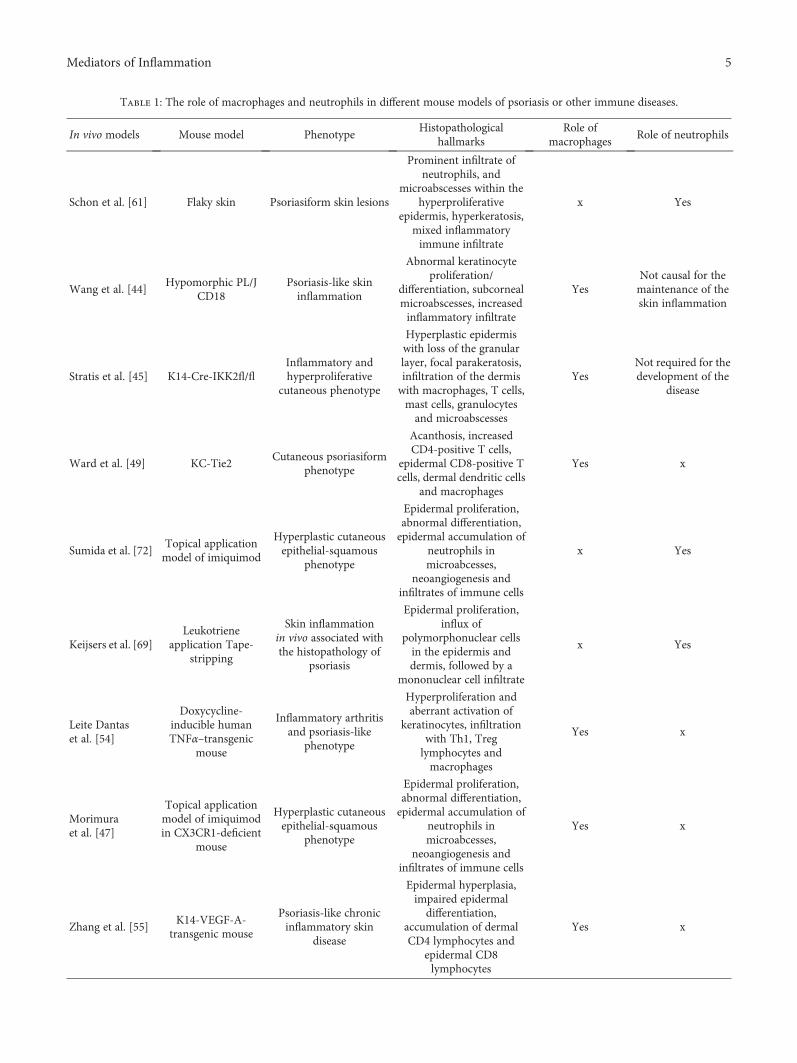
Table 1: The role of macrophages and neutrophils in different mouse models of psoriasis or other immune diseases.
In vivo models Mouse model PhenotypeHistopathological
hallmarksRole of
macrophagesRole of neutrophils
Schon et al. [61] Flaky skin Psoriasiform skin lesions
Prominent infiltrate ofneutrophils, and
microabscesses within thehyperproliferative
epidermis, hyperkeratosis,mixed inflammatoryimmune infiltrate
x Yes
Wang et al. [44]Hypomorphic PL/J
CD18Psoriasis-like skininflammation
Abnormal keratinocyteproliferation/
differentiation, subcornealmicroabscesses, increasedinflammatory infiltrate
YesNot causal for themaintenance of theskin inflammation
Stratis et al. [45] K14-Cre-IKK2fl/flInflammatory andhyperproliferative
cutaneous phenotype
Hyperplastic epidermiswith loss of the granularlayer, focal parakeratosis,infiltration of the dermiswith macrophages, T cells,mast cells, granulocytesand microabscesses
YesNot required for thedevelopment of the
disease
Ward et al. [49] KC-Tie2Cutaneous psoriasiform
phenotype
Acanthosis, increasedCD4-positive T cells,
epidermal CD8-positive Tcells, dermal dendritic cells
and macrophages
Yes x
Sumida et al. [72]Topical applicationmodel of imiquimod
Hyperplastic cutaneousepithelial-squamous
phenotype
Epidermal proliferation,abnormal differentiation,epidermal accumulation of
neutrophils inmicroabcesses,
neoangiogenesis andinfiltrates of immune cells
x Yes
Keijsers et al. [69]Leukotriene
application Tape-stripping
Skin inflammationin vivo associated withthe histopathology of
psoriasis
Epidermal proliferation,influx of
polymorphonuclear cellsin the epidermis anddermis, followed by a
mononuclear cell infiltrate
x Yes
Leite Dantaset al. [54]
Doxycycline-inducible humanTNFα–transgenic
mouse
Inflammatory arthritisand psoriasis-like
phenotype
Hyperproliferation andaberrant activation of
keratinocytes, infiltrationwith Th1, Treglymphocytes andmacrophages
Yes x
Morimuraet al. [47]
Topical applicationmodel of imiquimodin CX3CR1-deficient
mouse
Hyperplastic cutaneousepithelial-squamous
phenotype
Epidermal proliferation,abnormal differentiation,epidermal accumulation of
neutrophils inmicroabcesses,
neoangiogenesis andinfiltrates of immune cells
Yes x
Zhang et al. [55]K14-VEGF-A-
transgenic mouse
Psoriasis-like chronicinflammatory skin
disease
Epidermal hyperplasia,impaired epidermaldifferentiation,
accumulation of dermalCD4 lymphocytes and
epidermal CD8lymphocytes
Yes x
5Mediators of Inflammation

Neutrophils infiltrate psoriatic lesions early from bloodvessels within the dermis and form microabscesses, calledMunro abscesses in humans, due to their accumulation inthe form of microbial clusters in the thickened and paraker-atotic stratum corneum [61] (Figure 1). Neutrophils accumu-late in the skin, attracted by a gradient of chemotactic factors,which may be small induced secreted cytokines, such as IL-8,NAP-2, and NAP-3; membrane lipid derivatives such as leu-kotriene and platelet-activating factor (PAF); or substancesof bacterial origin (LPS) [62–65].
Schon et al. noted that neutrophil depletion in the flakyskin (fsn)/fsn mutant mouse model of psoriasis-like lesiondevelopment contributed to a decrease in epidermal thick-ness, neutrophilic infiltrate, epidermal microabscesses, andthe number of CD3-positive T cells, thus to an improvementof psoriasiform skin lesions [61].
Extravasation of neutrophils was made possible by thebinding of integrin αMβ2 (CD11b/CD18) to ICAM-1(CD54) of endothelial cells. ICAM-1 would be expressed denovo on the surface of hyperproliferative psoriatic keratino-cytes, thus contributing to the migration of neutrophilsexpressing αMβ2 to the epidermis [61].
Some neutrophil-derived T cell attractants, such asdefensin-1, defensin-2, or CAP37/azurocidin [66], mayexplain the reduction in the number of tissue T lympho-cytes upon in vivo depletion of neutrophils in SCID mice.However, the influx of neutrophils into the epidermalcompartment via chemoattractants seems to follow theinflux of lymphocytes.
Moreover, the depletion of neutrophils in patients withmoderate to severe generalized pustular psoriasis via anextracorporeal circulation therapy that selectively elimi-nates elevated myeloid lineage leukocytes resulted in adecrease in erythroderma, pustules, and edema up to 10weeks after therapy [67].
Neutrophils would represent a major source of IL-17[68–70], via the formation of extracellular traps [71], whoseproduction is defined as the ultimate stage in a process ofneutrophil polymorphonuclear activation. The topical appli-cation of leukotriene B4 (LTB4), found in high concentra-tions in psoriatic lesions, is a chemoattractant forneutrophils, eosinophils, monocytes, macrophages, mastcells, dendritic cells, and effector T cells. LTB4 induced arapid influx of polymorphonuclear cells into the epidermisand dermis, followed by an infiltrate of mononuclear cells[69]. Moreover, a recent study has demonstrated that LTB4receptor 1 (BLT1) and CXCR2 promoted the recruitmentof neutrophils at psoriatic lesional sites and that these cellswould secrete IL-1β, perpetuating psoriatic inflammation[72] (Table 1).
Moreover, the release of this mediator, IL-17, by NETosisamplified the accumulation of neutrophils [73] by increasingthe expression of CXCL1, CXCL2, and IL-8. IL-17 increasedthe expression of antimicrobial peptides—β-defensin-2(HBD-2), S100A7, S100A8, S100A9, and LL37—by keratino-cytes [74–76]. These antimicrobial peptides can stimulateimmune cell infiltration, and NET-derived DNA-LL37nucleic acid complexes promoted IFN-α secretion of plasma-cytoid dendritic cells [21]. IFN-α and TNF-αwould stimulate
the influx of inflammatory dendritic cells and macrophages,which would produce cytokines, including IL-23 and IL-1β,in the presence of IFN-γ.
Blocking IL-17A via a neutralizing antibody (secukinu-mab) reduced hyperkeratosis, acanthosis, and hyperproli-feration, significantly decreased the levels of geneexpression of chemokines derived from keratinocytes, suchas CXCL1 (GRO) and CXCL8 (IL-8), and triggered near-total elimination of IL-17-positive epidermal neutrophils.The authors suggested that the inhibition of IL-17A wouldindirectly block the influx of neutrophils due to the lack ofkeratinocyte response to IL-17A [70].
Furthermore, the specific deletion of the A-chain of IL-17receptor in mice contributed to the delay and attenuation ofpsoriatic inflammation in mice treated with imiquimod butdid not prevent its development. KO mice for IL-17RAshowed a delay and alteration of peripheral neutrophils atthe site of injury. The authors hypothesized that IL-6,strongly expressed in KO mice for IL-17RA and treated withimiquimod, may play a role in the development of pathologyin the absence of IL-17RA [77]. Nevertheless, there is nodoubt that compensatory mechanisms ensure the attractionof neutrophils to the inflammatory site in the absence ofthe IL-17RA signaling pathway.
The importance of IL-6 has also been demonstrated in amurine model in which IL-17A and GFP are coexpressed inkeratinocytes, resulting in the formation of psoriatic-likelesions. A blockage of IL-6 signaling would reduce the pathol-ogy in these mice by reducing the formation of neutrophilmicroabscesses in the epidermis and reducing the numberof myeloperoxidase-positive cells [78].
In 2011, Garcia-Romo et al. reported that NETs wouldproduce a greater amount of LL37 in response to PMA andIFN-γ in systemic lupus erythematosus [79]. The secretionof LL37 facilitated the uptake and recognition of DNA byplasmacytoid dendritic cells. The antimicrobial peptideLL37 was also overexpressed in the lesional psoriatic skinsand would participate in the activation of cells of innateimmunity [80].
Neutrophils participate in the secretion of inflammatorymediators, including IL-1α, IL-8, TNF-α, and IFN-γ cyto-kines [81–84]. The interaction of neutrophils and fibroblastsmay increase the secretion of IL-8 [85]. In addition, IL-12,expressed on the surface of mononuclear cells, such asepidermal neutrophils, is overexpressed in psoriatic lesions.Although fibroblasts do not secrete IL-12, the fibroblast-neutrophil interaction upregulates IL-12 secretion, highlight-ing the importance and necessity of considering cooperationbetween the different cells to better understand these interac-tions. Furthermore, IL-12 promoted the survival and growthof Th1 cells, as well as their differentiation, and inhibited theformation of Th2 cells.
The formation of neutrophil-containing microabscesseswould be dependent on IL-1R1. It has been demonstratedin the imiquimod-induced murine model that the signalingof IL-1 via IL-1R1 regulated constitutive and induced chemo-kine expression in response to imiquimod, involved in thein vivo recruitment of neutrophils. However, the deletion ofIL-1R1 did not block the formation of microabscesses,
6 Mediators of Inflammation

implying that other cytokines are involved in their formation[86]. CEACAM-1 expression in superficial keratinocytesfound in psoriatic lesions would also contribute to the persis-tence of neutrophils and to the underlying inflammation inpsoriatic patients [87].
Aldara creammodifies the immune response by stimulat-ing the body’s defenses that fight certain types of skinaffections. The topical application of Aldara in mice deficientin IL-17A, IL-17F, or IL-22 drastically reduced the severity ofpsoriasis. However, γδ T cell populations and innate RORγt-positive lymphocytes produced large amounts of theseinflammatory cytokines and were necessary and sufficientfor the formation of psoriatic lesion plaques in this murinemodel. A reverse RORγt agonist, developed by Janssen, hasdemonstrated its efficacy in murine models for psoriasisand inflammatory arthritis. The blockade of the Th17 differ-entiation led to the decrease of the production of IL-17A bythe memory T cells and reduced the production of IL-17Aand IL-22 by the cells NKT and γδ [88].
Furthermore, the combined action of mannan-activatedmacrophages and IL-17A from T cells provoked the infiltra-tion of neutrophils into skin compartments, leading tohistopathological features [89].
IL-17F would induce the secretion of IL-8 by thekeratinocytes and would favor the infiltration of neutrophilsinto the dermis. On the other hand, the blocking of neutro-phil infiltration by an anti-IL-8 antibody underlined theimportance of the IL-17F/IL-8 axis in the pathophysiologyof psoriasis [90].
At last, a treatment with ustekinumab (human monoclo-nal antibody directed against IL-12 and IL-23p40) or inflixi-mab (monoclonal chimeric antibody directed against TNF),both used to treat psoriasis, in severe psoriatic patientsappeared to decrease the activity of neutrophils and mono-cytes. Indeed, the expression of CD62L, a molecule of celladhesion, is restored in patients receiving biological therapy,while expressions of CD11b (also called integrin alpha M)and CD66b, another adhesion molecule, were decreased aftertreatment. Also, the ratio of activated CD14high monocyteswas normalized in patients receiving therapy [91], stressingonce again the importance of these immune cells in psoriasis.
In vivo models have demonstrated the complex butevident interrelationship between different immune cellsat the level of psoriatic lesions and provide a better under-standing of the influence that cells have on each other andthe possible modulating effect of cytokines and chemokineson the functioning of neighboring cells in the local microen-vironment (Figure 1).
4. 3D Organotypic Skin Models
At present, few organotypic models emphasize the impor-tance of macrophages or neutrophils in psoriasis, but theenthusiasm for such models could lead us to make newdiscoveries in the years to come.
Some models already demonstrated the role of T cells inthe pathogenesis of psoriasis. In the 1990s, transplants ofhuman psoriatic skins in immunodeficient mice [92] andinjected with autologous T cells, from either peripheral blood
or the lesion site, indicated that only the latter was able tomaintain the psoriatic phenotype in the grafted mice [93].In 2010, to counter the limitations associated with suchmodels, Guerrero-Aspizua et al. isolated both skin cells—ker-atinocytes and fibroblasts—and peripheral blood frompsoriatic patients and reproduced cutaneous equivalents bybioengineering [94]. These equivalents were then graftedonto immunodeficient mice. The authors indicated thatintraepidermal injection of activated human immunocytesinduced the formation of psoriatic lesion in the skin modelof xenotransplantation. The authors observed that thecombination of factors secreted by Th1 cells and cytokinesderived from Th17 cells was essential for the completedevelopment of a psoriatic phenotype, emphasizing theimportance of T cells in the pathology.
In 2002, Del Rio and colleagues performed a long-termfollow-up of gene-transferred bioengineered artificial humanskin based on a fibroblast-containing fibrin dermal substrateorthotopically grafted onto mice [95]. This preclinicalapproach, considered more clinically relevant and betterpredictive models of drug efficacy, will certainly identifynew therapeutic targets for psoriasis. Moreover, the topicalapplication of nanosomes containing siRNAs inhibiting theexpression of hBD-2 in such a mouse model improved thecutaneous phenotype and reduced the number and the sizeof blood vessels in the dermal compartment [96].
Modeling psoriatic inflammation requires paying closeattention to the immune component. Some organotypic 3Dmodels, with no immune component, attempt to mimicpsoriatic inflammation. In our lab, we have alreadydemonstrated that the generation of skin equivalents fromhuman psoriatic fibroblasts and keratinocytes produced bythe self-assembly method displayed major hallmarks of pso-riasis [97]. Also, although the addition of a cytokine cocktail[94, 98–100], or a single protein [101], to mimic psoriaticinflammation has demonstrated some histopathologicalaspects of psoriasis, it is unlikely that all mediators releasedby immune cells will be generated with a mixture of a fewinflammatory cytokines. Although essential, animal modelscannot reflect the etiology of psoriasis or represent the humancomplexity associated with pathology.
An alternative to animal experimentation is the develop-ment of equivalent three-dimensional models generatedfrom human skin cells. These organotypic models, whichare rapidly expanding, make it possible, by adding one ormore cutaneous components, to dissect the specific role ofeach cell type present. Conversely, current models, usuallywith only one immune component, cannot summarize allthe cellular and molecular interactions occurring in vivodue to the absence of other components, which are certainlyimportant to the general pathophysiology of psoriasis. More-over, deletion of an element (macrophages by clodronateliposomes, e.g.) very rarely results in a complete reversionof the phenotype, thus implying that other constituents areinvolved in the pathology development.
Thus far, too few in vitro three-dimensional modelsincluding the main cellular components involved in the path-ophysiology of autoimmune diseases, such as psoriasis oratopic dermatitis, are currently being studied to further
7Mediators of Inflammation
understand the role of the microenvironment in maintaininginflammation. The influence of the components on themodulation of the soluble and nonsoluble factors consti-tuting the local lesional microenvironment can be appre-hended by the generation of 3D models. However, thedifficulty of incorporating immune cells into skin models,along with the complexity involved in modeling a three-dimensional environment necessary and sufficient to main-tain cell viability and to respect the anatomical arrangementof cells, represents a major challenge.
In 2006, Dezutter-Dambuyant et al. optimized thedevelopment of a reconstructed skin model by incorporat-ing hematopoietic progenitor cells in an endothelializedskin equivalent. The team demonstrated that the differen-tiation of dendritic cell precursors into Langerhans cellsdepends on the state of differentiation of keratinocyte cells.In this case, the differentiation program of the Langerhanscells started only if keratinocytes were differentiated. Inter-action between fibroblasts and keratinocytes in a three-dimensional skin model supported the regulation of theirown differentiation and also that of anatomically closecells [102].
The encapsulation of dendritic cells in an agarose andfibronectin gel, compartmentalized between a layer of fibro-blasts and keratinocytes and treated with dichlorobenzenefor 24 hours, makes it possible to study the cellular interac-tions and mechanisms of skin sensitization [103]. In addition
to maintaining their viability and horizontal and verticalmigration, dendritic cells appeared to maintain an immaturephenotype in the presence of fibronectin, expressed higherlevels of endocytic receptors, and had a greater potential toinduce T cell activation.
In 2011, Bechetoille et al. developed a dermal equivalentmodel of bovine collagen, chitosan, chondroitin sulfate,fibroblasts, and dermal macrophages derived from mono-cytes [104]. Macrophages, with the “classical” fusiform mor-phology, expressed CD14, CD163, and DC-SIGN/CD209markers and produced large amounts of IL-10 in responseto LPS, but little TNF. While LPS stimulated immuneresponses by interacting with the CD14 membrane receptorand induced the secretion of proinflammatory cytokines,such as TNF-α, IL-1, and IL-6, a stimulation of LPS macro-phages in this in vitro model promoted their anti-inflammatory activity.
In 2014, van den Bogaard et. al developed a three-dimensional healthy in vitro skin substitute model in whichthey injected allogeneic healthy T lymphocytes into the der-mal compartment. The authors demonstrated that the polar-ization of T cells towards a Th1 or Th17 phenotype, theninjected in skin substitutes, induced the expression of molec-ular markers associated with psoriasis, although no hyper-proliferation or acanthosis was observed. The inflammatoryphenotype thus developed is similar to the psoriatic pheno-type observed in human lesional plaques [105].
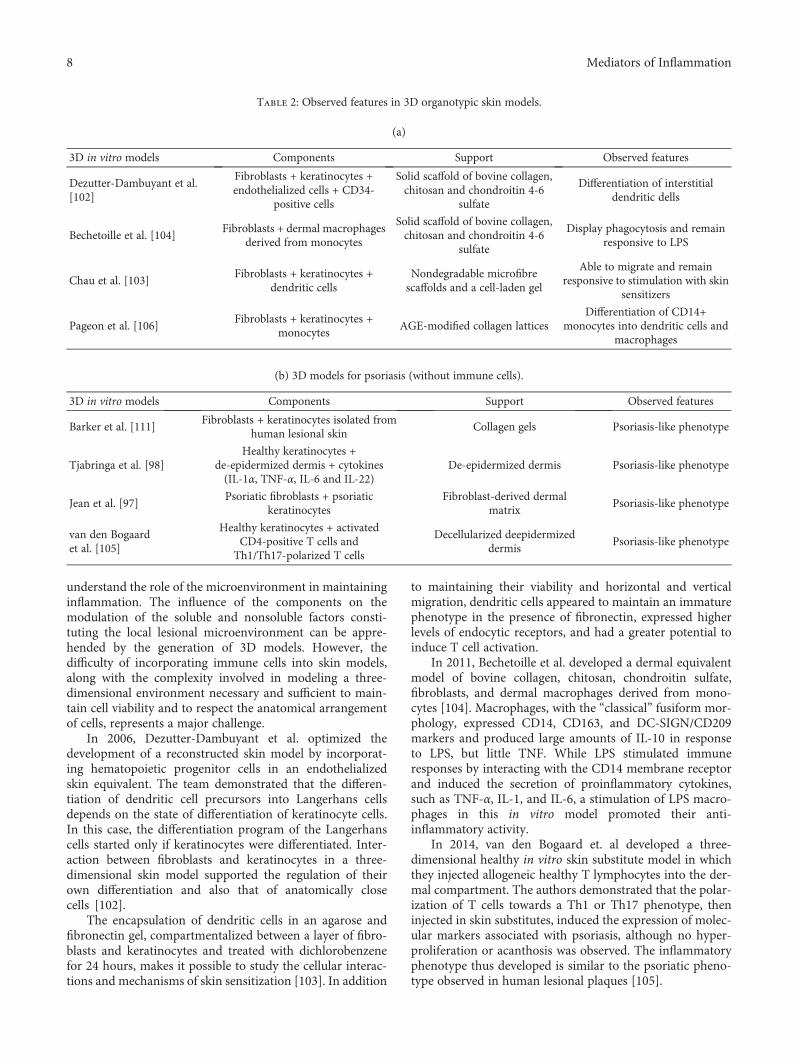
Table 2: Observed features in 3D organotypic skin models.
(a)
3D in vitro models Components Support Observed features
Dezutter-Dambuyant et al.[102]
Fibroblasts + keratinocytes +endothelialized cells + CD34-
positive cells
Solid scaffold of bovine collagen,chitosan and chondroitin 4-6
sulfate
Differentiation of interstitialdendritic dells
Bechetoille et al. [104]Fibroblasts + dermal macrophages
derived from monocytes
Solid scaffold of bovine collagen,chitosan and chondroitin 4-6
sulfate
Display phagocytosis and remainresponsive to LPS
Chau et al. [103]Fibroblasts + keratinocytes +
dendritic cellsNondegradable microfibrescaffolds and a cell-laden gel
Able to migrate and remainresponsive to stimulation with skin
sensitizers
Pageon et al. [106]Fibroblasts + keratinocytes +
monocytesAGE-modified collagen lattices
Differentiation of CD14+monocytes into dendritic cells and
macrophages
(b) 3D models for psoriasis (without immune cells).
3D in vitro models Components Support Observed features
Barker et al. [111]Fibroblasts + keratinocytes isolated from
human lesional skinCollagen gels Psoriasis-like phenotype
Tjabringa et al. [98]Healthy keratinocytes +
de-epidermized dermis + cytokines(IL-1α, TNF-α, IL-6 and IL-22)
De-epidermized dermis Psoriasis-like phenotype
Jean et al. [97]Psoriatic fibroblasts + psoriatic
keratinocytesFibroblast-derived dermal
matrixPsoriasis-like phenotype
van den Bogaardet al. [105]
Healthy keratinocytes + activatedCD4-positive T cells and
Th1/Th17-polarized T cells
Decellularized deepidermizeddermis
Psoriasis-like phenotype
8 Mediators of Inflammation

In another context, it has also been demonstrated that thegeneration of glycated cutaneous equivalents promoted thedifferentiation of monocytic cells towards a macrophage ordendritic cell phenotype. It would be interesting to character-ize the influence of these differentiated cells on the modula-tion of the secretory profile in glycated substitutes [106](Table 2).
5. Future Directions
The aim of this review was to analyze the importance ofimmune cells, and more particularly leukocyte cells, as neu-trophils and macrophages in the pathophysiology of psoria-sis. The emphasis is often on T cells, yet the influence ofother immune cells on lymphocytes and keratinocytes mustbe better characterized. The specific roles of each cell typesmust be dissected to better understand the cellular hierarchyestablished in psoriasis. We also discussed the need todevelop new in vitromodels with a three-dimensional micro-environment and appropriate and relevant cellular compo-nents to better mimic the pathology and improve ourunderstanding of it. Besides, it would also be advantageousto integrate other types of immune cells, such as dendriticcells, into human skin three-dimensional models, given theirrole in the presentation of antigens to T lymphocytes.Although there is a great temptation to develop a more com-plete model including immune, cutaneous, and endothelialcells to better reflect the actors involved in inflammation, itis often difficult to know if an observed effect is due to a singlecell type or an interaction between anatomically close cells.
The development of complex three-dimensional modelsis an important issue for research in pharmacology. Today,most of the available models reproduce only very partiallythe in vivo situation because their architecture does not con-sider the complexity of the tissue interfaces and the vascularperfusion. Recently, some companies have begun to developorgans or tissues as relevant tools to repetitively assess thepharmacological action of drugs. For instance, Organovo,based in the United States, commercialized its first livermodel in 2014, which incorporates hepatocytes, stellate cells,and endothelial cells, printed in a matrix. This model wouldbe more discriminating than 2D cultures and would makeit possible to mimic a patient’s response to a drug [107]. Itwould be wise to focus on these in vitro models, given theethical and financial constraints associated with the use ofanimal models.
Recently, new antipsoriatic therapies have emerged,such as apremilast (Otezla, Celgene), a phosphodiesterase 4(PDE4) inhibitor, which is also the first oral anti-inflammatory treatment for psoriasis in more than 20 years.PDE4 inhibition would also be a potential target for systemicsclerosis, as its blockage decreased dermal fibrosis throughthe downregulation of profibrotic mediators from M2macrophages [108].
Other promising drugs are currently being tested inphase III in the US, specifically targeting the inhibition ofphospholipase A2 (PLA2), which controls the biosynthesisof inflammatory mediators, such as leukotrienes and prosta-glandins. Other biological agents, such as etanercept (Enbrel,
Amgen), infliximab (Remicade, Merck & Co./Janssen Bio-tech), and adalimumab (Humira, AbbVie), which are tumornecrosis factor (TNF) antagonists, are more commonlyadministered, although some side effects are frequentlyobserved in patients (>10%): viral infection, dyspnea,migraine, and nausea. Since 2009, biological agents againstpsoriasis targeting IL-12, IL-17, and IL-23, cytokines thatplay a key role in inflammatory and immune responses, havebeen licensed on the market. Ustekinumab was the first drugspecifically designed to suppress inflammation by targetingthe signaling pathway of the cytokine family of interleukin-12 (IL-12) and interleukin-23 (IL-23).
Another therapeutic approach, the encapsulation ofantipsoriatic agents in nanoparticles [109, 110], is currentlyunder study with the aim of improving the efficacy, safety,and compliance of potential agents.
Advances in psoriasis research continue to lead to newtherapeutic strategies that promise better management of thiscomplex disease in the future. For sure, development ofin vitro models and mouse models will help to revolutionizethe care of psoriasis in the years to come.
Conflicts of Interest
The authors declare that there is no conflict of interestregarding the publication of this review.
Authors’ Contributions
Isabelle Lorthois wrote the manuscript, which has been cor-rected and revised by Daniel Asselineau, Nathalie Seyler, andRoxane Pouliot.
References
[1] F. O. Nestle, D. H. Kaplan, and J. Barker, “Psoriasis,” The NewEngland Journal of Medicine, vol. 361, no. 5, pp. 496–509,2009.
[2] C. E. Griffiths and J. N. Barker, “Pathogenesis and clinicalfeatures of psoriasis,” The Lancet, vol. 370, no. 9583,pp. 263–271, 2007.
[3] B. A. Bernard, S. M. Robinson, S. Vandaele, J. N. Mansbridge,and M. Darmon, “Abnormal maturation pathway of kerati-nocytes in psoriatic skin,” The British Journal of Dermatology,vol. 112, no. 6, pp. 647–653, 1985.
[4] B. A. Bernard, A. Reano, Y. M. Darmon, and J. Thivolet,“Precocious appearance of involucrin and epidermal trans-glutaminase during differentiation of psoriatic skin,” TheBritish Journal of Dermatology, vol. 114, no. 3, pp. 279–283,1986.
[5] B. A. Bernard, D. Asselineau, L. Schaffar-Deshayes, and M. Y.Darmon, “Abnormal sequence of expression of differentia-tion markers in psoriatic epidermis: inversion of two stepsin the differentiation program?,” The Journal of InvestigativeDermatology, vol. 90, no. 6, pp. 801–805, 1988.
[6] F. Bernerd, T. Magnaldo, and M. Darmon, “Delayed onsetof epidermal differentiation in psoriasis,” The Journal ofInvestigative Dermatology, vol. 98, no. 6, pp. 902–910,1992.
9Mediators of Inflammation

[7] M. Detmar, L. F. Brown, K. P. Claffey et al., “Overexpressionof vascular permeability factor/vascular endothelial growthfactor and its receptors in psoriasis,” The Journal of Experi-mental Medicine, vol. 180, no. 3, pp. 1141–1146, 1994.
[8] M. Verschoore, C. Kowalewski, M. J. Chorzelska, B. A.Bernard, and Y. M. Darmon, “Intraepidermal leakage ofplasma proteins after tape stripping of normal skin anduninvolved psoriatic skin,” The British Journal of Dermatol-ogy, vol. 122, no. 3, pp. 391–397, 1990.
[9] Meier TO, L. Kovacicova, R. Huggenberger, A. A. Navarini,G. Gitzelmann, and B. R. Amann-Vesti, “Increased perme-ability of cutaneous lymphatic capillaries and enhanced bloodflow in psoriatic plaques,” Dermatology, vol. 227, no. 2,pp. 118–125, 2013.
[10] D. Parent, B. A. Bernard, C. Desbas, M. Heenen, and M. Y.Darmon, “Spreading of psoriatic plaques: alteration ofepidermal differentiation precedes capillary leakiness andanomalies in vascular morphology,” The Journal of Investiga-tive Dermatology, vol. 95, no. 3, pp. 333–340, 1990.
[11] A. Menter, A. Gottlieb, S. R. Feldman et al., “Guidelines ofcare for the management of psoriasis and psoriatic arthri-tis: section 1. Overview of psoriasis and guidelines of carefor the treatment of psoriasis with biologics,” Journal ofthe American Academy of Dermatology, vol. 58, no. 5,pp. 826–850, 2008.
[12] R. C. Trembath, R. L. Clough, J. L. Rosbotham et al., “Identi-fication of a major susceptibility locus on chromosome 6pand evidence for further disease loci revealed by a two stagegenome-wide search in psoriasis,” Human MolecularGenetics, vol. 6, no. 5, pp. 813–820, 1997.
[13] R. P. Nair, T. Henseler, S. Jenisch et al., “Evidence for twopsoriasis susceptibility loci (HLA and 17q) and two novelcandidate regions (16q and 20p) by genome-wide scan,”HumanMolecular Genetics, vol. 6, no. 8, pp. 1349–1356, 1997.
[14] A. D. Burden, S. Javed, M. Bailey, M. Hodgins, M. Connor,and D. Tillman, “Genetics of psoriasis: paternal inheritanceand a locus on chromosome 6p,” The Journal of InvestigativeDermatology, vol. 110, no. 6, pp. 958–960, 1998.
[15] J. E. Gudjonsson, A. Johnston, H. Sigmundsdottir, andH. Valdimarsson, “Immunopathogenic mechanisms in psori-asis,” Clinical and Experimental Immunology, vol. 135, no. 1,pp. 1–8, 2004.
[16] J. T. Elder, R. P. Nair, S.W. Guo, T. Henseler, E. Christophers,and J. J. Voorhees, “The genetics of psoriasis,” Archives ofDermatology, vol. 130, no. 2, pp. 216–224, 1994.
[17] J. Jarczak, E. M. Kosciuczuk, P. Lisowski et al., “Defensins:natural component of human innate immunity,” HumanImmunology, vol. 74, no. 9, pp. 1069–1079, 2013.
[18] E. J. Hollox, U. Huffmeier, P. L. Zeeuwen et al., “Psoriasis isassociated with increased β-defensin genomic copy number,”Nature Genetics, vol. 40, no. 1, pp. 23–25, 2008.
[19] L. C. Tsoi, S. L. Spain, J. Knight et al., “Identification of 15new psoriasis susceptibility loci highlights the role of innateimmunity,” Nature Genetics, vol. 44, no. 12, pp. 1341–1348,2012.
[20] S. P. Jariwala, “The role of dendritic cells in the immuno-pathogenesis of psoriasis,” Archives of DermatologicalResearch, vol. 299, no. 8, pp. 359–366, 2007.
[21] R. Lande, J. Gregorio, V. Facchinetti et al., “Plasmacytoiddendritic cells sense self-DNA coupled with antimicrobialpeptide,” Nature, vol. 449, no. 7162, pp. 564–569, 2007.
[22] B. J. Nickoloff and F. O. Nestle, “Recent insights into theimmunopathogenesis of psoriasis provide new therapeuticopportunities,” The Journal of Clinical Investigation, vol. 113,no. 12, pp. 1664–1675, 2004.
[23] M. A. Lowes, A. M. Bowcock, and J. G. Krueger, “Pathogen-esis and therapy of psoriasis,” Nature, vol. 445, no. 7130,pp. 866–873, 2007.
[24] G. Arango Duque and A. Descoteaux, “Macrophage cyto-kines: involvement in immunity and infectious diseases,”Frontiers in Immunology, vol. 5, p. 491, 2014.
[25] M. L. Karnovsky, “Metchnikoff in Messina: a century of stud-ies on phagocytosis,” The New England Journal of Medicine,vol. 304, no. 19, pp. 1178–1180, 1981.
[26] T. M. Doherty, “T-cell regulation of macrophage function,”Current Opinion in Immunology, vol. 7, no. 3, pp. 400–404,1995.
[27] R. Mirza, L. A. DiPietro, and T. J. Koh, “Selective and specificmacrophage ablation is detrimental to wound healing inmice,” The American Journal of Pathology, vol. 175, no. 6,pp. 2454–2462, 2009.
[28] S. J. Leibovich and R. Ross, “The role of the macrophage inwound repair. A study with hydrocortisone and antima-crophage serum,” The American Journal of Pathology,vol. 78, no. 1, pp. 71–100, 1975.
[29] T. Lucas, A. Waisman, R. Ranjan et al., “Differential roles ofmacrophages in diverse phases of skin repair,” Journal ofImmunology, vol. 184, no. 7, pp. 3964–3977, 2010.
[30] M. S. Koh, B. B. Majewski, and E. L. Rhodes, “Increased mac-rophage activity in psoriasis,” Acta Dermato-Venereologica,vol. 65, no. 3, pp. 194–198, 1985.
[31] J. Fuentes-Duculan, M. Suarez-Farinas, L. C. Zaba et al., “Asubpopulation of CD163-positive macrophages is classicallyactivated in psoriasis,” The Journal of Investigative Dermatol-ogy, vol. 130, no. 10, pp. 2412–2422, 2010.
[32] L. S. Taams, L. W. Poulter, M. H. Rustin, and A. N. Akbar,“Phenotypic analysis of IL-10-treated macrophages usingthe monoclonal antibodies RFD1 and RFD7,” Pathobiology,vol. 67, no. 5-6, pp. 249–252, 2000.
[33] K. Micklem, E. Rigney, J. Cordell et al., “A humanmacrophage-associated antigen (CD68) detected by six dif-ferent monoclonal antibodies,” British Journal of Haematol-ogy, vol. 73, no. 1, pp. 6–11, 1989.
[34] K. Weber-Matthiesen and W. Sterry, “Organization of themonocyte/macrophage system of normal human skin,”The Journal of Investigative Dermatology, vol. 95, no. 1,pp. 83–89, 1990.
[35] B. K. Min, K. Suk, and W. H. Lee, “Stimulation of CD107affects LPS-induced cytokine secretion and cellular adhesionthrough the ERK signaling pathway in the humanmacrophage-like cell line, THP-1,” Cellular Immunology,vol. 281, no. 2, pp. 122–128, 2013.
[36] E. L. Eskelinen, “Roles of LAMP-1 and LAMP-2 in lysosomebiogenesis and autophagy,” Molecular Aspects of Medicine,vol. 27, no. 5-6, pp. 495–502, 2006.
[37] O. Elomaa, M. Kangas, C. Sahlberg et al., “Cloning of a novelbacteria-binding receptor structurally related to scavengerreceptors and expressed in a subset of macrophages,” Cell,vol. 80, no. 4, pp. 603–609, 1995.
[38] J. Kzhyshkowska, “Multifunctional receptor stabilin-1 inhomeostasis and disease,” Scientific World Journal, vol. 10,pp. 2039–2053, 2010.
10 Mediators of Inflammation

[39] S. Goerdt, R. Bhardwaj, and C. Sorg, “Inducible expressionof MS-1 high-molecular-weight protein by endothelial cellsof continuous origin and by dendritic cells/macrophagesin vivo and in vitro,” The American Journal of Pathology,vol. 142, no. 5, pp. 1409–1422, 1993.
[40] V. Kodelja and S. Goerdt, “Dissection of macrophagedifferentiation pathways in cutaneous macrophage disordersand in vitro,” Experimental Dermatology, vol. 3, no. 6,pp. 257–268, 1994.
[41] M. Egawa, K. Mukai, S. Yoshikawa et al., “Inflammatorymonocytes recruited to allergic skin acquire an anti-inflammatory M2 phenotype via basophil-derived interleu-kin-4,” Immunity, vol. 38, no. 3, pp. 570–80, 2013.
[42] C. Vestergaard, H. Just, J. Baumgartner Nielsen, K. Thestrup-Pedersen, and M. Deleuran, “Expression of CCR2 on mono-cytes and macrophages in chronically inflamed skin in atopicdermatitis and psoriasis,” Acta Dermato-Venereologica,vol. 84, no. 5, pp. 353–358, 2004.
[43] A. Gratchev, J. Kzhyshkowska, K. Kothe et al., “Mphi1 andMphi2 can be re-polarized by Th2 or Th1 cytokines,respectively, and respond to exogenous danger signals,”Immunobiology, vol. 211, no. 6–8, pp. 473–486, 2006.
[44] H. Wang, T. Peters, D. Kess et al., “Activated macrophagesare essential in a murine model for T cell-mediated chronicpsoriasiform skin inflammation,” The Journal of ClinicalInvestigation, vol. 116, no. 8, pp. 2105–2114, 2006.
[45] A. Stratis, M. Pasparakis, R. A. Rupec et al., “Pathogenic rolefor skin macrophages in a mouse model of keratinocyte-induced psoriasis-like skin inflammation,” The Journal ofClinical Investigation, vol. 116, no. 8, pp. 2094–2104, 2006.
[46] R. A. Clark and T. S. Kupper, “Misbehaving macrophages inthe pathogenesis of psoriasis,” The Journal of ClinicalInvestigation, vol. 116, no. 8, pp. 2084–2087, 2006.
[47] S. Morimura, T. Oka, M. Sugaya, and S. Sato, “CX3CR1deficiency attenuates imiquimod-induced psoriasis-likeskin inflammation with decreased M1 macrophages,” Jour-nal of Dermatological Science, vol. 82, no. 3, pp. 175–188,2016.
[48] J. A. Wolfram, D. Diaconu, D. A. Hatala et al., “Keratinocytebut not endothelial cell-specific overexpression of Tie2 leadsto the development of psoriasis,” The American Journal ofPathology, vol. 174, no. 4, pp. 1443–1458, 2009.
[49] N. L. Ward, C. M. Loyd, J. A. Wolfram, D. Diaconu, C. M.Michaels, and T. S. McCormick, “Depletion of antigen-presenting cells by clodronate liposomes reverses thepsoriatic skin phenotype in KC-Tie2 mice,” The BritishJournal of Dermatology, vol. 164, no. 4, pp. 750–758, 2011.
[50] D. Terhorst, R. Chelbi, C. Wohn et al., “Dynamics and tran-scriptomics of skin dendritic cells and macrophages in animiquimod-induced, biphasic mouse model of psoriasis,”Journal of Immunology, vol. 195, no. 10, pp. 4953–4961, 2015.
[51] A. B.Wysocki, “Skin anatomy, physiology, and pathophysiol-ogy,” The Nursing Clinics of North America, vol. 34, no. 4,pp. 777–797, 1999.
[52] J. Kanitakis, “Anatomy, histology and immunohistochemis-try of normal human skin,” European Journal of Dermatol-ogy, vol. 12, no. 4, pp. 390–399, 2002.
[53] E. Glitzner, A. Korosec, P. M. Brunner et al., “Specific rolesfor dendritic cell subsets during initiation and progressionof psoriasis,” EMBO Molecular Medicine, vol. 6, no. 10,pp. 1312–1327, 2014.
[54] R. Leite Dantas, D. Masemann, T. Schied et al., “Macrophage-mediated psoriasis can be suppressed by regulatory Tlymphocytes,” The Journal of Pathology, vol. 240, no. 3,pp. 366–377, 2016.
[55] J. Zhang, Y. Lin, C. Li et al., “IL-35 decelerates the inflamma-tory process by regulating inflammatory cytokine secretionandM1/M2macrophage ratio in psoriasis,” Journal of Immu-nology, vol. 197, no. 6, pp. 2131–2144, 2016.
[56] X. Teng, Z. Hu, X. Wei et al., “IL-37 ameliorates the inflam-matory process in psoriasis by suppressing proinflammatorycytokine production,” Journal of Immunology, vol. 192, no. 4,pp. 1815–1823, 2014.
[57] M. F. Nold, C. A. Nold-Petry, J. A. Zepp, B. E. Palmer,P. Bufler, and C. A. Dinarello, “IL-37 is a fundamental inhib-itor of innate immunity,”Nature Immunology, vol. 11, no. 11,pp. 1014–1022, 2010.
[58] E. Kolaczkowska and P. Kubes, “Neutrophil recruitment andfunction in health and inflammation,” Nature Reviews.Immunology, vol. 13, no. 3, pp. 159–175, 2013.
[59] G. B. Segel, M. W. Halterman, and M. A. Lichtman, “Theparadox of the neutrophil’s role in tissue injury,” Journal ofLeukocyte Biology, vol. 89, no. 3, pp. 359–372, 2011.
[60] B. G. Yipp and P. Kubes, “NETosis: how vital is it?,” Blood,vol. 122, no. 16, pp. 2784–2794, 2013.
[61] M. Schon, D. Denzer, R. C. Kubitza, T. Ruzicka, and M. P.Schon, “Critical role of neutrophils for the generation ofpsoriasiform skin lesions in flaky skin mice,” The Journalof Investigative Dermatology, vol. 114, no. 5, pp. 976–983,2000.
[62] P. Peveri, A. Walz, B. Dewald, and M. Baggiolini, “A novelneutrophil-activating factor produced by human mononu-clear phagocytes,” The Journal of Experimental Medicine,vol. 167, no. 5, pp. 1547–1559, 1988.
[63] B. Moser, C. Schumacher, V. von Tscharner, I. Clark-Lewis,and M. Baggiolini, “Neutrophil-activating peptide 2 andgro/melanoma growth-stimulatory activity interact withneutrophil-activating peptide 1/interleukin 8 receptors onhuman neutrophils,” The Journal of Biological Chemistry,vol. 266, no. 16, pp. 10666–10671, 1991.
[64] P. V. Afonso, M. Janka-Junttila, Y. J. Lee et al., “LTB4 is asignal-relay molecule during neutrophil chemotaxis,” Devel-opmental Cell, vol. 22, no. 5, pp. 1079–1091, 2012.
[65] E. S. Van Amersfoort, T. J. Van Berkel, and J. Kuiper, “Recep-tors, mediators, and mechanisms involved in bacterial sepsisand septic shock,” Clinical Microbiology Reviews, vol. 16,no. 3, pp. 379–414, 2003.
[66] O. Chertov, D. F. Michiel, L. Xu et al., “Identification ofdefensin-1, defensin-2, and CAP37/azurocidin as T-cell che-moattractant proteins released from interleukin-8-stimulatedneutrophils,” The Journal of Biological Chemistry, vol. 271,no. 6, pp. 2935–2940, 1996.
[67] S. Ikeda, H. Takahashi, Y. Suga et al., “Therapeutic depletionof myeloid lineage leukocytes in patients with generalizedpustular psoriasis indicates a major role for neutrophilsin the immunopathogenesis of psoriasis,” Journal of theAmerican Academy of Dermatology, vol. 68, no. 4,pp. 609–617, 2013.
[68] P. C. Res, G. Piskin, O. J. de Boer et al., “Overrepresentationof IL-17A and IL-22 producing CD8 T cells in lesional skinsuggests their involvement in the pathogenesis of psoriasis,”PLoS One, vol. 5, no. 11, article e14108, 2010.
11Mediators of Inflammation

[69] R. R. Keijsers, A. G. Hendriks, P. E. van Erp et al., “In vivoinduction of cutaneous inflammation results in the accumu-lation of extracellular trap-forming neutrophils expressingRORγt and IL-17,” The Journal of Investigative Dermatology,vol. 134, no. 5, pp. 1276–1284, 2014.
[70] K. Reich, K. A. Papp, R. T. Matheson et al., “Evidencethat a neutrophil-keratinocyte crosstalk is an early targetof IL-17A inhibition in psoriasis,” Experimental Dermatology,vol. 24, no. 7, pp. 529–535, 2015.
[71] A. M. Lin, C. J. Rubin, R. Khandpur et al., “Mast cells andneutrophils release IL-17 through extracellular trap forma-tion in psoriasis,” Journal of Immunology, vol. 187, no. 1,pp. 490–500, 2011.
[72] H. Sumida, K. Yanagida, Y. Kita et al., “Interplay betweenCXCR2 and BLT1 facilitates neutrophil infiltration andresultant keratinocyte activation in a murine model ofimiquimod-induced psoriasis,” Journal of Immunology,vol. 192, no. 9, pp. 4361–4369, 2014.
[73] L. Li, L. Huang, A. L. Vergis et al., “IL-17 produced byneutrophils regulates IFN-γ-mediated neutrophil migrationin mouse kidney ischemia-reperfusion injury,” The Journalof Clinical Investigation, vol. 120, no. 1, pp. 331–342, 2010.
[74] S. C. Liang, X. Y. Tan, D. P. Luxenberg et al., “Interleukin(IL)-22 and IL-17 are coexpressed by Th17 cells and cooper-atively enhance expression of antimicrobial peptides,” TheJournal of Experimental Medicine, vol. 203, no. 10,pp. 2271–2279, 2006.
[75] K. E. Nograles, L. C. Zaba, E. Guttman-Yassky et al.,“Th17 cytokines interleukin (IL)-17 and IL-22 modulate dis-tinct inflammatory and keratinocyte-response pathways,”The British Journal of Dermatology, vol. 159, no. 5,pp. 1092–1102, 2008.
[76] M. Peric, S. Koglin, S. M. Kim et al., “IL-17A enhances vita-min D3-induced expression of cathelicidin antimicrobialpeptide in human keratinocytes,” Journal of Immunology,vol. 181, no. 12, pp. 8504–8512, 2008.
[77] K. El Malki, S. H. Karbach, J. Huppert et al., “An alternativepathway of imiquimod-induced psoriasis-like skin inflamma-tion in the absence of interleukin-17 receptor a signaling,”The Journal of Investigative Dermatology, vol. 133, no. 2,pp. 441–451, 2013.
[78] A. L. Croxford, S. Karbach, F. C. Kurschus et al., “IL-6 regu-lates neutrophil microabscess formation in IL-17A-drivenpsoriasiform lesions,” The Journal of Investigative Dermatol-ogy, vol. 134, no. 3, pp. 728–735, 2014.
[79] G. S. Garcia-Romo, S. Caielli, B. Vega et al., “Netting neutro-phils are major inducers of type I IFN production in pediatricsystemic lupus erythematosus,” Science TranslationalMedicine, vol. 3, no. 73, p. 73ra20, 2011.
[80] R. Lande, E. Botti, C. Jandus et al., “The antimicrobial peptideLL37 is a T-cell autoantigen in psoriasis,” Nature Communi-cations, vol. 5, p. 5621, 2014.
[81] N. A. Cicco, A. Lindemann, J. Content et al., “Inducible pro-duction of interleukin-6 by human polymorphonuclear neu-trophils: role of granulocyte-macrophage colony-stimulatingfactor and tumor necrosis factor-alpha,” Blood, vol. 75,no. 10, pp. 2049–2052, 1990.
[82] D. B. Dubravec, D. R. Spriggs, J. A. Mannick, and M. L.Rodrick, “Circulating human peripheral blood granulocytessynthesize and secrete tumor necrosis factor alpha,” Proceed-ings of the National Academy of Sciences, vol. 87, no. 17,pp. 6758–6761, 1990.
[83] E. J. Gosselin, K. Wardwell, W. F. Rigby, and P. M. Guyre,“Induction of MHC class II on human polymorphonuclearneutrophils by granulocyte/macrophage colony-stimulatingfactor, IFN-gamma, and IL-3,” Journal of Immunology,vol. 151, no. 3, pp. 1482–1490, 1993.
[84] A. Matsukawa and M. Yoshinaga, “Neutrophils as a source ofcytokines in inflammation,” Histology and Histopathology,vol. 14, no. 2, pp. 511–516, 1999.
[85] E. Glowacka, P. Lewkowicz, H. Rotsztejn, and A. Zalewska,“IL-8, IL-12 and IL-10 cytokines generation by neutrophils,fibroblasts and neutrophils- fibroblasts interaction in psoria-sis,” Advances in Medical Sciences, vol. 55, no. 2, pp. 254–260,2010.
[86] M. Uribe-Herranz, L. H. Lian, K. M. Hooper, K. A. Milora,and L. E. Jensen, “IL-1R1 signaling facilitates Munro’s micro-abscess formation in psoriasiform imiquimod-induced skininflammation,” The Journal of Investigative Dermatology,vol. 133, no. 6, pp. 1541–1549, 2013.
[87] M. Rahmoun, J. P. Moles, N. Pedretti et al., “Cytokine-induced CEACAM1 expression on keratinocytes is character-istic for psoriatic skin and contributes to a prolonged lifespanof neutrophils,” The Journal of Investigative Dermatology,vol. 129, no. 3, pp. 671–681, 2009.
[88] X. Xue, P. Soroosh, A. De Leon-Tabaldo et al., “Pharmaco-logic modulation of RORγt translates to efficacy inpreclinical and translational models of psoriasis andinflammatory arthritis,” Scientific Reports, vol. 6, article37977, 2016.
[89] I. Khmaladze, T. Kelkka, S. Guerard et al., “Mannan inducesROS-regulated, IL-17A-dependent psoriasis arthritis-likedisease in mice,” Proceedings of the National Academy ofSciences, vol. 111, no. 35, pp. E3669–E3678, 2014.
[90] H. Watanabe, M. Kawaguchi, S. Fujishima et al., “Functionalcharacterization of IL-17F as a selective neutrophil attractantin psoriasis,” The Journal of Investigative Dermatology,vol. 129, no. 3, pp. 650–656, 2009.
[91] K. Yamanaka, Y. Umezawa, A. Yamagiwa et al., “Biologictherapy improves psoriasis by decreasing the activity ofmonocytes and neutrophils,” The Journal of Dermatology,vol. 41, no. 8, pp. 679–685, 2014.
[92] B. J. Nickoloff, S. L. Kunkel, M. Burdick, and R. M. Strieter,“Severe combined immunodeficiency mouse and human pso-riatic skin chimeras. Validation of a new animal model,” TheAmerican Journal of Pathology, vol. 146, no. 3, pp. 580–588,1995.
[93] A. Gilhar, M. David, Y. Ullmann, T. Berkutski, and R. S.Kalish, “T-lymphocyte dependence of psoriatic pathologyin human psoriatic skin grafted to SCID mice,” TheJournal of Investigative Dermatology, vol. 109, no. 3,pp. 283–288, 1997.
[94] S. Guerrero-Aspizua, M. Garcia, R. Murillas et al., “Develop-ment of a bioengineered skin-humanized mouse model forpsoriasis: dissecting epidermal-lymphocyte interacting path-ways,” The American Journal of Pathology, vol. 177, no. 6,pp. 3112–3124, 2010.
[95] M. Del Rio, F. Larcher, F. Serrano et al., “A preclinical modelfor the analysis of genetically modified human skin in vivo,”Human Gene Therapy, vol. 13, no. 8, pp. 959–968, 2002.
[96] S. Bracke, M. Carretero, S. Guerrero-Aspizua et al., “Targetedsilencing of DEFB4 in a bioengineered skin-humanizedmouse model for psoriasis: development of siRNA
12 Mediators of Inflammation

SECosome-based novel therapies,” Experimental Dermatol-ogy, vol. 23, no. 3, pp. 199–201, 2014.
[97] J. Jean, M. Lapointe, J. Soucy, and R. Pouliot, “Developmentof an in vitro psoriatic skin model by tissue engineering,”Journal of Dermatological Science, vol. 53, no. 1, pp. 19–25,2009.
[98] G. Tjabringa, M. Bergers, D. van Rens, R. de Boer, E. Lamme,and J. Schalkwijk, “Development and validation of humanpsoriatic skin equivalents,” The American Journal of Pathol-ogy, vol. 173, no. 3, pp. 815–823, 2008.
[99] C. Pouliot-Berube, K. Zaniolo, S. L. Guerin, and R. Pouliot,“Tissue-engineered human psoriatic skin supplementedwith cytokines as an in vitro model to study plaque psori-asis,” Regenerative Medicine, vol. 11, no. 6, pp. 545–557,2016.
[100] M. Carretero, S. Guerrero-Aspizua, N. Illera et al., “Differen-tial features between chronic skin inflammatory diseasesrevealed in skin-humanized psoriasis and atopic dermatitismouse models,” The Journal of Investigative Dermatology,vol. 136, no. 1, pp. 136–145, 2016.
[101] A. Gazel, M. Rosdy, B. Bertino, C. Tornier, F. Sahuc, andM. Blumenberg, “A characteristic subset of psoriasis-associated genes is induced by oncostatin-M in reconstitutedepidermis,” The Journal of Investigative Dermatology,vol. 126, no. 12, pp. 2647–2657, 2006.
[102] C. Dezutter-Dambuyant, A. Black, N. Bechetoille et al.,“Evolutive skin reconstructions: from the dermal collagen–glycosaminoglycan– chitosane substrate to an immuno-competent reconstructed skin,” Bio-medical Materials andEngineering, vol. 16, Supplement 4, pp. S85–S94, 2006.
[103] D. Y. Chau, C. Johnson, S. MacNeil, J. W. Haycock, andA. M. Ghaemmaghami, “The development of a 3D immu-nocompetent model of human skin,” Biofabrication, vol. 5,no. 3, article 035011, 2013.
[104] N. Bechetoille, H. Vachon, A. Gaydon et al., “A new orga-notypic model containing dermal-type macrophages,”Experimental Dermatology, vol. 20, no. 12, pp. 1035–1037, 2011.
[105] E. H. van den Bogaard, G. S. Tjabringa, I. Joosten et al.,“Crosstalk between keratinocytes and T cells in a 3Dmicroenvironment: a model to study inflammatory skindiseases,” The Journal of Investigative Dermatology, vol. 134,no. 3, pp. 719–727, 2014.
[106] H. Pageon, H. Zucchi, F. Rousset, S. Girardeau-Hubert,E. Tancrede, and D. Asselineau, “Glycation stimulates cuta-neous monocyte differentiation in reconstructed skinin vitro,” Mechanisms of Ageing and Development, vol. 162,pp. 18–26, 2017.
[107] D. G. Nguyen, J. Funk, J. B. Robbins et al., “Bioprinted 3Dprimary liver tissues allow assessment of organ-level responseto clinical drug induced toxicity in vitro,” PLoS One, vol. 11,no. 7, article e0158674, 2016.
[108] C. Maier, A. Ramming, C. Bergmann et al., “Inhibition ofphosphodiesterase 4 (PDE4) reduces dermal fibrosis byinterfering with the release of interleukin-6 from M2 macro-phages,” Annals of the Rheumatic Diseases, vol. 76, 2017.
[109] H. Bessar, I. Venditti, L. Benassi et al., “Functionalized goldnanoparticles for topical delivery of methotrexate for thepossible treatment of psoriasis,” Colloids and Surfaces. B,Biointerfaces, vol. 141, pp. 141–147, 2016.
[110] T. Ramezanli, B. E. Kilfoyle, Z. Zhang, and B. B. Michniak-Kohn, “Polymeric nanospheres for topical delivery of vitaminD3,” International Journal of Pharmaceutics, vol. 516, no. 1-2,pp. 196–203, 2017.
[111] C. L. Barker, M. T. McHale, A. K. Gillies et al., “Thedevelopment and characterization of an in vitro model ofpsoriasis,” Journal of Investigative Dermatology, vol. 123,no. 5, pp. 892–901, 2004.
13Mediators of Inflammation
Submit your manuscripts athttps://www.hindawi.com
Stem CellsInternational
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
MEDIATORSINFLAMMATION
of
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Behavioural Neurology
EndocrinologyInternational Journal of
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Disease Markers
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
BioMed Research International
OncologyJournal of
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Oxidative Medicine and Cellular Longevity
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
PPAR Research
The Scientific World JournalHindawi Publishing Corporation http://www.hindawi.com Volume 2014
Immunology ResearchHindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Journal of
ObesityJournal of
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Computational and Mathematical Methods in Medicine
OphthalmologyJournal of
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Diabetes ResearchJournal of
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Research and TreatmentAIDS
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Gastroenterology Research and Practice
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Parkinson’s Disease
Evidence-Based Complementary and Alternative Medicine
Volume 2014Hindawi Publishing Corporationhttp://www.hindawi.com





























