Embed Size (px)
Citation preview

Annales de chirurgie plastique esthétique 51 (2006) 339–346
ava i lab le at www.sc ienced i rect .com
journa l homepage: www.e l sev ier.com/locate/annp la
Hémangiomes congénitaux et autre tumeursvasculaires infantiles rares
Congenital haemangiomas and other rare infantilevascular tumours
O. Enjolras*, A. Picard, V. Soupre
Consultations des angiomes, service du Professeur-Marie-Paule-Vazquez, hôpital d’enfants Armand-Trousseau, Inserm U714,université Paris-VI, Assistance-publique–Hôpitaux de Paris, rue du Docteur-Arnold-Netter, 75571 Paris cedex 12, France
02do
MOTS CLÉSHémangiome ;Congénital ;Angiome en touffes ;Hémangioendothéliomekaposiforme ;Syndrome de Kasabach-Merritt
* Auteur correspondant.Adresse e-mail : secretaria
94-1260/$ - see front mattei:10.1016/j.anplas.2006.07.
t.enjolr
r © 200006
Résumé Un certain nombre de tumeurs infantiles bien moins fréquentes que l’hémangiomeinfantile ont longtemps été confondues avec lui. Elles sont aujourd’hui clairement individuali-sées, sur des bases cliniques et histologiques, et leurs différences sont confortées par l’emploide marqueurs endothéliaux phénotypiques de découverte récente comme GLUT1. GLUT1 estprésent dans 100 % des hémangiomes infantiles et le marquage est toujours négatif dans lesautres tumeurs vasculaires infantiles. Les hémangiomes congénitaux constituent un groupe detumeurs dont l’analyse est encore en évolution car le groupe est hétérogène. Leur pronostic aété affiné, et leur prise en charge thérapeutique améliorée. Tous sont pleinement développésin utero et contrairement à l’hémangiome infantile ils ne subissent pas de prolifération post-natale. Certains cependant (RICH — rapidly involuting congenital haemangioma) régressentdans la première année. D’autres (NICH — non involuting congenital haemangioma) persistentindéfiniment. L’angiome en touffes et l’hémangioendothéliome kaposiforme sont bien carac-térisés histologiquement et, en outre, considérés aujourd’hui comme faisant partie d’unmême spectre tumoral, impliquant le tissu endothélial lymphatique. Ces deux tumeurs sontreconnues comme base du syndrome de thrombopénie sévère, à risque vital, qu’est le syn-drome de Kasabach-Merritt. Elles existent aussi isolément, avec une évolution souvent chro-nique qui pose des problèmes esthétiques, et parfois un développement invalidant.© 2006 Elsevier Masson SAS. Tous droits réservés.
KEYWORDSHaemangioma;Congenital;Tufted angioma;
Abstract A number of infantile tumours, far less frequent than infantile haemangiomas, werelong assimilated to them. Today they are clearly individualised, based on distinctive clinicaland pathologic features, and this difference has been supported by the discovery of newimmunophenotypic markers such as GLUT1. GLUT1 stains 100% of infantile haemangiomas andnone of the other infantile vascular tumours. Congenital haemangiomas represent a group of
[email protected] (O. Enjolras).
6 Elsevier Masson SAS. Tous droits réservés.

O. Enjolras et al.340
Kaposiformhaemangioendothelio-ma;Kasabach-Merrittsyndrome
vascular tumours still under evaluation as they have slightly heterogeneous presentation.Their prognosis is better appraised and their therapeutic management has improved. Theyare all fully grown in utero and they do not experience postnatal proliferation like haemangio-mas do. Some of them (RICH — Rapidly Involuting Congenital Haemangioma) undergo sponta-neous involution during the first year. Others (NICH — Non Involuting Congenital Haeman-gioma) persist lifelong. Tufted angioma and kaposiform haemangioendothelioma arehistopathologically well characterized; in addition they are now considered as part of a samespectrum of vascular tumours, with the contribution of lymphatic endothelial cells in theirproliferation. Both are clearly the tumours able to create platelet trapping, thrombocytopeniaand the life-threatening Kasabach-Merritt syndrome. However they may occur as isolatedtumours, without thrombocytopenia but with cosmetic, and sometimes function-impairing,consequences.© 2006 Elsevier Masson SAS. Tous droits réservés.
Introduction
La classification des angiomes a beaucoup évolué depuis1976, date du premier Workshop on Vascular Anomalies. Cegroupe de travail international n’a cessé de se réunirdepuis, tous les deux ans. Il comporte aujourd’hui une par-ticipation multidisciplinaire issue de tous les continents. En1992, fut créée l’International Society for the Study of Vas-cular Anomalies (ISSVA) qui regroupe les membres du Works-hop. L’ISSVA continue à assurer tous les deux ans cette réu-nion avec un but principal : l’amélioration desthérapeutiques.
La classification adoptée par l’ISSVA lors de son Works-hop de Rome en 1996 prend en compte l’essentiel des don-nées cliniques, radiologiques, histologiques et biologiques.Elle repose sur les travaux biologiques fondateurs de Mulli-ken et Glowacki au début des années 1980. Le suffixe« ome » impliquant une notion de prolifération, il est désor-mais réservé aux tumeurs et le terme générique « angiome »ne recouvre plus l’ensemble de ces lésions. On parle désor-mais d’« anomalies vasculaires » qui sont classées en deuxgroupes : les tumeurs vasculaires et les malformationsvasculaires [1].
Les malformations sont faites de vaisseaux mal forméssans accélération du turnover cellulaire. Selon leur hémo-dynamique, elles sont à flux lent (et selon le type de vais-seau altéré de façon prédominante, elles sont capillaires,veineuses ou lymphatiques) ou à flux rapide (ce sont lesmalformations artérioveineuses avec fistules artérioveineu-ses).
Les tumeurs se développent par hyperplasie cellulaire.Parmi elles, l’hémangiome infantile est une tumeur bénignetrès commune. D’autres tumeurs infantiles longtempsconfondues avec l’hémangiome infantile en sont aujourd’-hui clairement différenciées.
Une seule tumeur, l’hémangiome infantile, est fré-quente touchant de 7 à 10 % des enfants à l’âge d’un anet jusqu’à 30 % des prématurés de moins de 1500 g. Elleévolue par phases : prolifération postnatale puis régression.L’adolescent et l’adulte n’en conservent que les éventuel-les séquelles. Cette prolifération tumorale bénigne de cel-lules endothéliales formant des capillaires est habituelle-ment sans gravité [2,3]. Le phénotype des cellulesendothéliales proliférant au cours du développement d’unhémangiome se rapproche de celui des microvaisseaux pla-
centaires [4,5] du fait de l’expression de marqueurs parti-culiers que sont GLUT1, la mérosine, l’antigène Lewis Y etle Fc gamma-récepteur II. Cette découverte a été uneconfirmation immunophénotypique de ce que laissaientdéjà supposer la clinique et l’histologie depuis quelquesannées [6–8] : toutes les tumeurs vasculaires infantiles nesont pas des hémangiomes.
Il existe donc d’autres tumeurs vasculaires chez le nour-risson, plus rares, également susceptibles de régressionpour certaines, persistantes et parfois se compliquant pourd’autres. On en distingue actuellement principalementtrois groupes : les hémangiomes congénitaux, l’angiomeen touffes et l’hémangioendothéliome kaposiforme.Aucune d’entre elles ne montre de marquage pour GLUT1,marqueur aujourd’hui indispensable au diagnostic histolo-gique, dès qu’une biopsie soulève des doutes quand audiagnostic d’hémangiome infantile [9,10]. D’autres tumeursplus rares portent l’étiquette d’hémangioendothéliome (cf.texte Anatomie Pathologique, M. Wassef).
Aspects cliniques, diagnostic, traitement
Hémangiomes congénitaux
On en distingue actuellement deux types au pronostic biendifférent. Ils sont désignés par deux acronymes en langueanglaise : le RICH (rapidly involuting congenital heman-gioma) et le NICH (non involuting congenital hemangioma).
RICHC’est une lésion bien différente des prodromes d’héman-giome (faux angiome plan, macule blanche « anémique »).Cette tumeur, pleinement développée in utero, ne subit pasde poussée postnatale, et régresse en quelques mois aprèsla naissance (de 6 à 18 mois) [9,11]. C’est une tumeur trom-peuse, inquiétante, dont le diagnostic prénatal par écho-graphie est de plus en plus fréquent. Elle siège essentielle-ment sur les membres près d’une articulation, ou en régioncéphalique près de l’oreille (front, tempe, joue ou scalp).C’est tantôt une infiltration massive violacée avec un haloplus clair rosé ou blanc bleuté, ou plus souvent une tumeursaillante de plusieurs centimètres de diamètre, à fin haloblanc, ou globalement pâle et cireuse. Elle est marquéede télangiectasies grossières à disposition radiée ou est par-semée au centre de quelques nodules rouges lisses

Hémangiomes congénitaux et autre tumeurs vasculaires infantiles rares 341
(Figs. 1, 2). Elle est parfois ferme au palper ou de consis-tance irrégulière et parfois multilobée (Fig. 3) et toujoursbien différente des aspects d’hémangiomes mixtes ou sous-cutanés développés après la naissance. Une zone centralenécrotique ou cicatricielle linéaire peut être observée(Fig. 2) justifiant, en urgence postnatale, un examenécho-doppler et IRM afin de vérifier s’il existe de largesvaisseaux rapides susceptibles de se rompre et de créerune hémorragie grave. La tumeur que nous appelons RICHa également été rapportée sous le nom de « non-progressive congenital hemangioma » en 2001 [10].
Le diagnostic peut être aidé par l’écho-doppler couplé àl’IRM [12,13]. Dans quelques cas, une biopsie en milieuchirurgical pédiatrique (ces biopsies sont sources desaignement marqué) peut être indispensable pour écarterd’autres diagnostics (rhabdomyosarcome embryonnaire,forme congénitale d’angiome en touffes ou d’hémangioen-dothéliome kaposiforme, myofibromatose infantile, derma-tofibrosarcome congénital de Darier-ferrand, etc.). Lesmarqueurs phénotypiques nouveaux (tel GLUT-1, toujourspositif dans les lésions d’hémangiome infantile, et négatifdans ce cas) semblent bien indiquer que le RICH est unetumeur différente de l’hémangiome infantile à croissancepostnatale [9] ; mais il peut aussi s’agir d’une simple diffé-rence de maturité cellulaire endothéliale ; en effet, il a étémontré que les cellules GLUT1 positives de l’hémangiomeinfantile perdent ce marquage en culture cellulaire.
L’hémangiome congénital de type RICH ne nécessite sou-vent pas de traitement, car il a ce potentiel de régression
Figure 1 RICH : forme saillante et télangiectasique, avechalo blanc bleuté, situé en regard du genou. a : à la naissance ;b : après régression spontanée.
rapide, laissant une zone de lipoatrophie, parfois rosée ettélangiectasique, ou un excès cutané esthétiquementacceptable (Fig. 1 a, b, Fig. 4 a, b). Certains cas cependantsont excisés en période néonatale, en particulier ceux dontle siège et la base d’implantation s’y prêtent sans problèmede fermeture de la plaie chirurgicale (Fig. 3). Ailleurs, c’estune exérèse de principe, s’il existe un risque hémorragique(la zone de nécrose centrale linéaire peut, si elle est situéeen regard de larges vaisseaux, représenter un risqued’hémorragie).
Figure 2 RICH : télangiectasies et cicatrice centrale linéaire.
Figure 3 RICH : masse trilobée palpable.
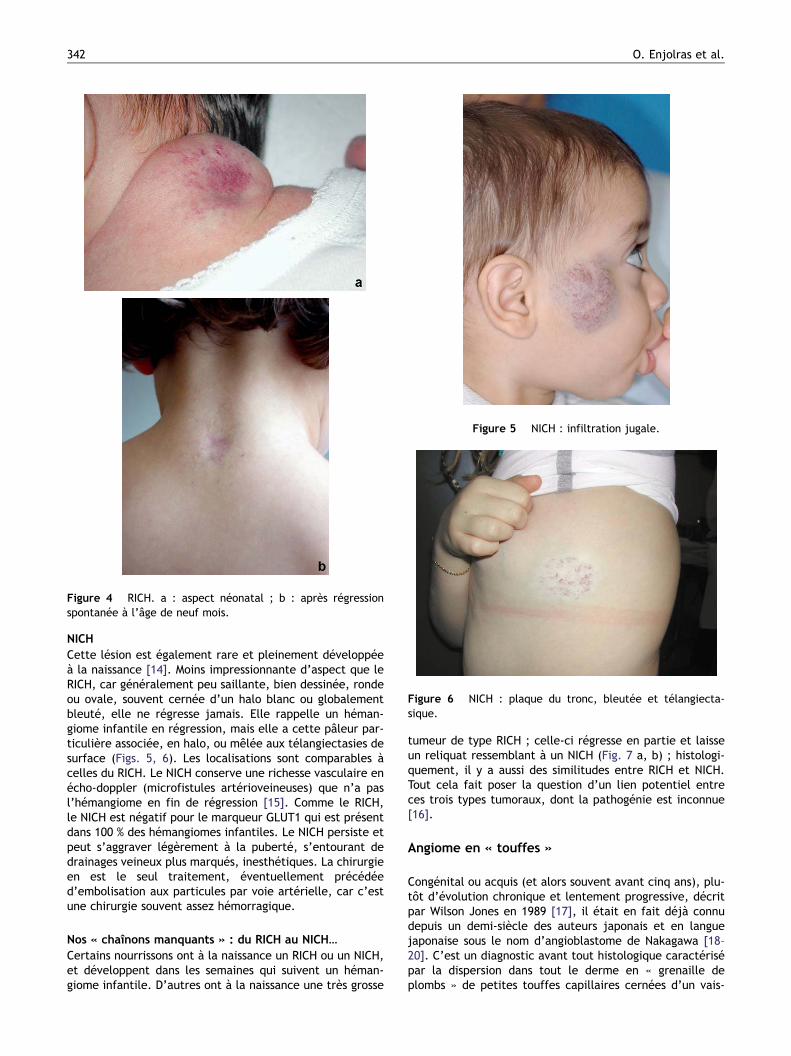
Figure 4 RICH. a : aspect néonatal ; b : après régressionspontanée à l’âge de neuf mois.
Figure 5 NICH : infiltration jugale.
Figure 6 NICH : plaque du tronc, bleutée et télangiecta-sique.
O. Enjolras et al.342
NICHCette lésion est également rare et pleinement développéeà la naissance [14]. Moins impressionnante d’aspect que leRICH, car généralement peu saillante, bien dessinée, rondeou ovale, souvent cernée d’un halo blanc ou globalementbleuté, elle ne régresse jamais. Elle rappelle un héman-giome infantile en régression, mais elle a cette pâleur par-ticulière associée, en halo, ou mêlée aux télangiectasies desurface (Figs. 5, 6). Les localisations sont comparables àcelles du RICH. Le NICH conserve une richesse vasculaire enécho-doppler (microfistules artérioveineuses) que n’a pasl’hémangiome en fin de régression [15]. Comme le RICH,le NICH est négatif pour le marqueur GLUT1 qui est présentdans 100 % des hémangiomes infantiles. Le NICH persiste etpeut s’aggraver légèrement à la puberté, s’entourant dedrainages veineux plus marqués, inesthétiques. La chirurgieen est le seul traitement, éventuellement précédéed’embolisation aux particules par voie artérielle, car c’estune chirurgie souvent assez hémorragique.
Nos « chaînons manquants » : du RICH au NICH…Certains nourrissons ont à la naissance un RICH ou un NICH,et développent dans les semaines qui suivent un héman-giome infantile. D’autres ont à la naissance une très grosse
tumeur de type RICH ; celle-ci régresse en partie et laisseun reliquat ressemblant à un NICH (Fig. 7 a, b) ; histologi-quement, il y a aussi des similitudes entre RICH et NICH.Tout cela fait poser la question d’un lien potentiel entreces trois types tumoraux, dont la pathogénie est inconnue[16].
Angiome en « touffes »
Congénital ou acquis (et alors souvent avant cinq ans), plu-tôt d’évolution chronique et lentement progressive, décritpar Wilson Jones en 1989 [17], il était en fait déjà connudepuis un demi-siècle des auteurs japonais et en languejaponaise sous le nom d’angioblastome de Nakagawa [18–20]. C’est un diagnostic avant tout histologique caractérisépar la dispersion dans tout le derme en « grenaille deplombs » de petites touffes capillaires cernées d’un vais-
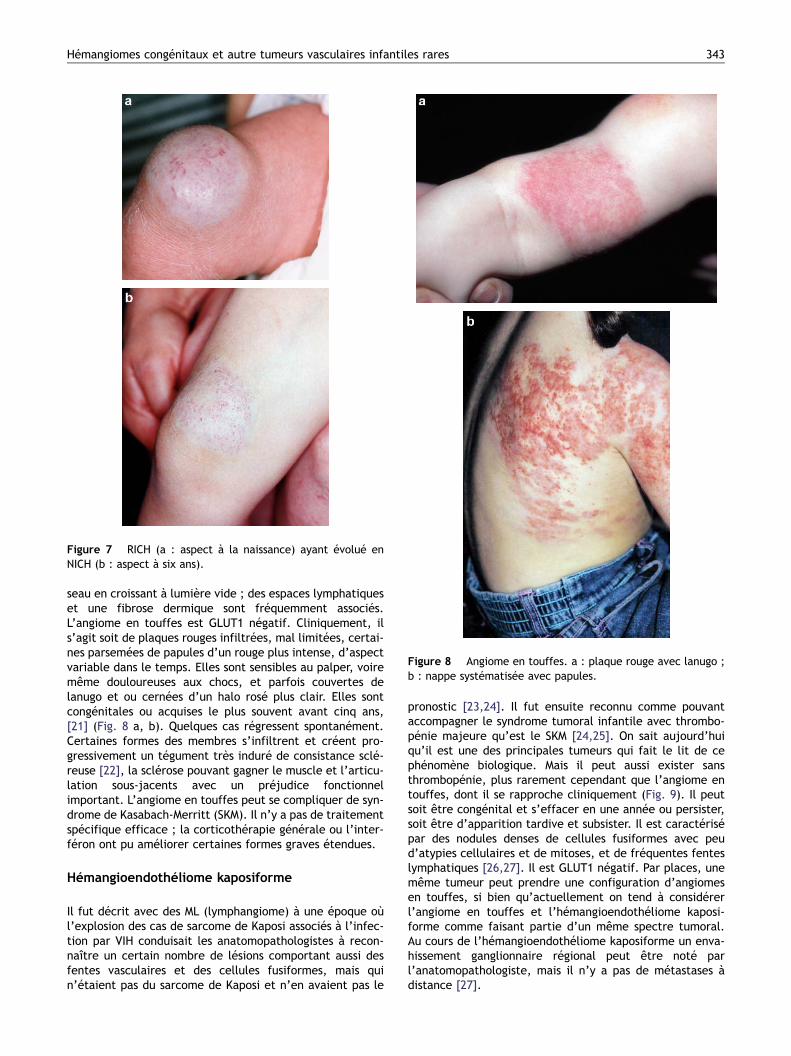
Figure 7 RICH (a : aspect à la naissance) ayant évolué enNICH (b : aspect à six ans).
Figure 8 Angiome en touffes. a : plaque rouge avec lanugo ;b : nappe systématisée avec papules.
Hémangiomes congénitaux et autre tumeurs vasculaires infantiles rares 343
seau en croissant à lumière vide ; des espaces lymphatiqueset une fibrose dermique sont fréquemment associés.L’angiome en touffes est GLUT1 négatif. Cliniquement, ils’agit soit de plaques rouges infiltrées, mal limitées, certai-nes parsemées de papules d’un rouge plus intense, d’aspectvariable dans le temps. Elles sont sensibles au palper, voiremême douloureuses aux chocs, et parfois couvertes delanugo et ou cernées d’un halo rosé plus clair. Elles sontcongénitales ou acquises le plus souvent avant cinq ans,[21] (Fig. 8 a, b). Quelques cas régressent spontanément.Certaines formes des membres s’infiltrent et créent pro-gressivement un tégument très induré de consistance sclé-reuse [22], la sclérose pouvant gagner le muscle et l’articu-lation sous-jacents avec un préjudice fonctionnelimportant. L’angiome en touffes peut se compliquer de syn-drome de Kasabach-Merritt (SKM). Il n’y a pas de traitementspécifique efficace ; la corticothérapie générale ou l’inter-féron ont pu améliorer certaines formes graves étendues.
Hémangioendothéliome kaposiforme
Il fut décrit avec des ML (lymphangiome) à une époque oùl’explosion des cas de sarcome de Kaposi associés à l’infec-tion par VIH conduisait les anatomopathologistes à recon-naître un certain nombre de lésions comportant aussi desfentes vasculaires et des cellules fusiformes, mais quin’étaient pas du sarcome de Kaposi et n’en avaient pas le
pronostic [23,24]. Il fut ensuite reconnu comme pouvantaccompagner le syndrome tumoral infantile avec thrombo-pénie majeure qu’est le SKM [24,25]. On sait aujourd’huiqu’il est une des principales tumeurs qui fait le lit de cephénomène biologique. Mais il peut aussi exister sansthrombopénie, plus rarement cependant que l’angiome entouffes, dont il se rapproche cliniquement (Fig. 9). Il peutsoit être congénital et s’effacer en une année ou persister,soit être d’apparition tardive et subsister. Il est caractérisépar des nodules denses de cellules fusiformes avec peud’atypies cellulaires et de mitoses, et de fréquentes fenteslymphatiques [26,27]. Il est GLUT1 négatif. Par places, unemême tumeur peut prendre une configuration d’angiomesen touffes, si bien qu’actuellement on tend à considérerl’angiome en touffes et l’hémangioendothéliome kaposi-forme comme faisant partie d’un même spectre tumoral.Au cours de l’hémangioendothéliome kaposiforme un enva-hissement ganglionnaire régional peut être noté parl’anatomopathologiste, mais il n’y a pas de métastases àdistance [27].
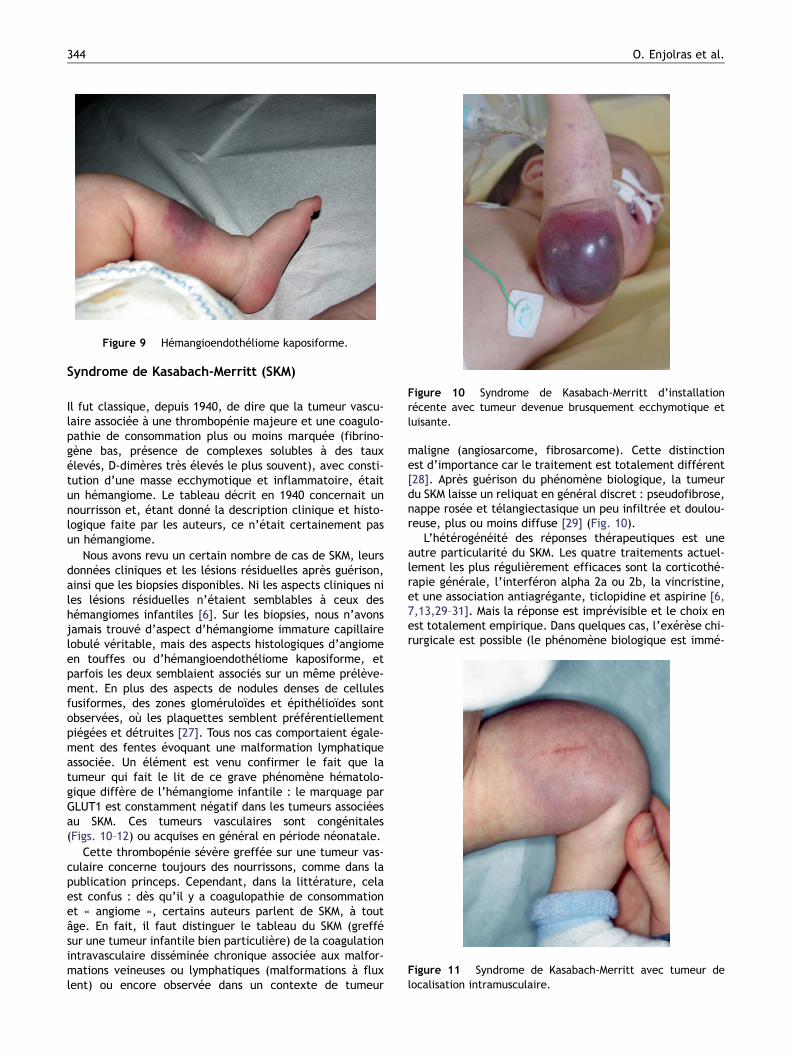
Figure 9 Hémangioendothéliome kaposiforme.
Figure 10 Syndrome de Kasabach-Merritt d’installationrécente avec tumeur devenue brusquement ecchymotique etluisante.
Figure 11 Syndrome de Kasabach-Merritt avec tumeur delocalisation intramusculaire.
O. Enjolras et al.344
Syndrome de Kasabach-Merritt (SKM)
Il fut classique, depuis 1940, de dire que la tumeur vascu-laire associée à une thrombopénie majeure et une coagulo-pathie de consommation plus ou moins marquée (fibrino-gène bas, présence de complexes solubles à des tauxélevés, D-dimères très élevés le plus souvent), avec consti-tution d’une masse ecchymotique et inflammatoire, étaitun hémangiome. Le tableau décrit en 1940 concernait unnourrisson et, étant donné la description clinique et histo-logique faite par les auteurs, ce n’était certainement pasun hémangiome.
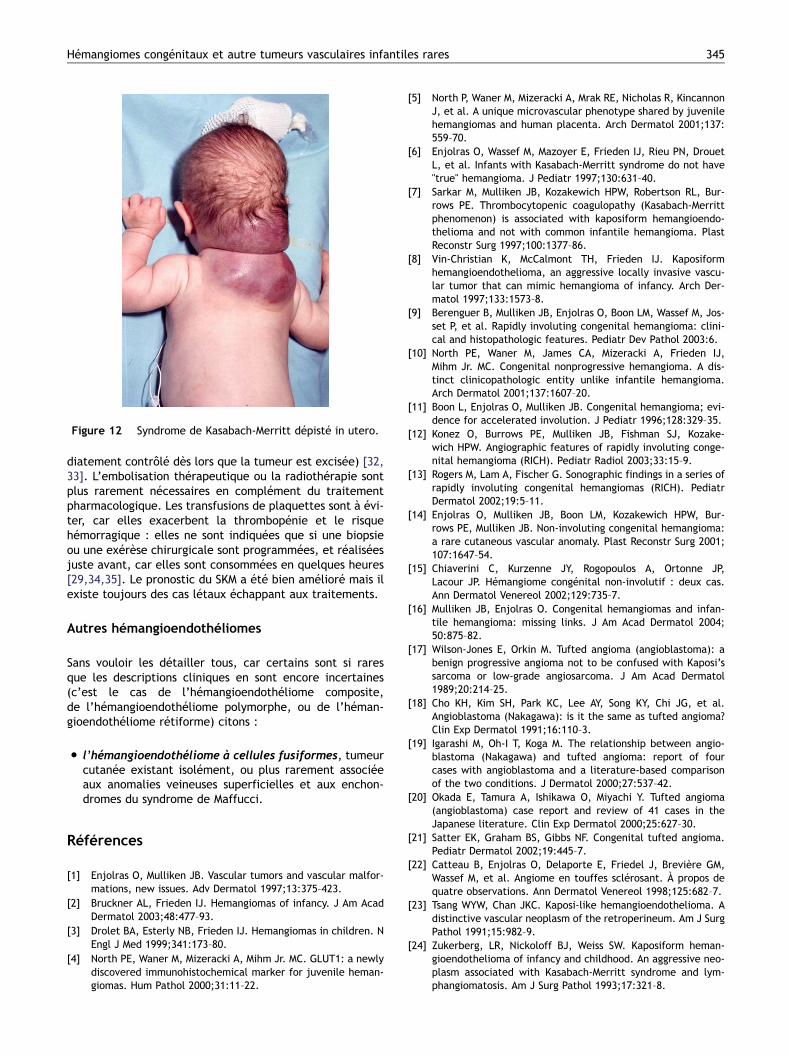
Nous avons revu un certain nombre de cas de SKM, leursdonnées cliniques et les lésions résiduelles après guérison,ainsi que les biopsies disponibles. Ni les aspects cliniques niles lésions résiduelles n’étaient semblables à ceux deshémangiomes infantiles [6]. Sur les biopsies, nous n’avonsjamais trouvé d’aspect d’hémangiome immature capillairelobulé véritable, mais des aspects histologiques d’angiomeen touffes ou d’hémangioendothéliome kaposiforme, etparfois les deux semblaient associés sur un même prélève-ment. En plus des aspects de nodules denses de cellulesfusiformes, des zones gloméruloïdes et épithélioïdes sontobservées, où les plaquettes semblent préférentiellementpiégées et détruites [27]. Tous nos cas comportaient égale-ment des fentes évoquant une malformation lymphatiqueassociée. Un élément est venu confirmer le fait que latumeur qui fait le lit de ce grave phénomène hématolo-gique diffère de l’hémangiome infantile : le marquage parGLUT1 est constamment négatif dans les tumeurs associéesau SKM. Ces tumeurs vasculaires sont congénitales(Figs. 10–12) ou acquises en général en période néonatale.
Cette thrombopénie sévère greffée sur une tumeur vas-culaire concerne toujours des nourrissons, comme dans lapublication princeps. Cependant, dans la littérature, celaest confus : dès qu’il y a coagulopathie de consommationet « angiome », certains auteurs parlent de SKM, à toutâge. En fait, il faut distinguer le tableau du SKM (greffésur une tumeur infantile bien particulière) de la coagulationintravasculaire disséminée chronique associée aux malfor-mations veineuses ou lymphatiques (malformations à fluxlent) ou encore observée dans un contexte de tumeur
maligne (angiosarcome, fibrosarcome). Cette distinctionest d’importance car le traitement est totalement différent[28]. Après guérison du phénomène biologique, la tumeurdu SKM laisse un reliquat en général discret : pseudofibrose,nappe rosée et télangiectasique un peu infiltrée et doulou-reuse, plus ou moins diffuse [29] (Fig. 10).
L’hétérogénéité des réponses thérapeutiques est uneautre particularité du SKM. Les quatre traitements actuel-lement les plus régulièrement efficaces sont la corticothé-rapie générale, l’interféron alpha 2a ou 2b, la vincristine,et une association antiagrégante, ticlopidine et aspirine [6,7,13,29–31]. Mais la réponse est imprévisible et le choix enest totalement empirique. Dans quelques cas, l’exérèse chi-rurgicale est possible (le phénomène biologique est immé-
[5]
[6]
[7]
[8]
[9]
[10
[11
[12
[13
[14
[15
[16
[17
[18
[19
[20
Figure 12 Syndrome de Kasabach-Merritt dépisté in utero.
Hémangiomes congénitaux et autre tumeurs vasculaires infantiles rares 345
diatement contrôlé dès lors que la tumeur est excisée) [32,33]. L’embolisation thérapeutique ou la radiothérapie sontplus rarement nécessaires en complément du traitementpharmacologique. Les transfusions de plaquettes sont à évi-ter, car elles exacerbent la thrombopénie et le risquehémorragique : elles ne sont indiquées que si une biopsieou une exérèse chirurgicale sont programmées, et réaliséesjuste avant, car elles sont consommées en quelques heures[29,34,35]. Le pronostic du SKM a été bien amélioré mais ilexiste toujours des cas létaux échappant aux traitements.
Autres hémangioendothéliomes
Sans vouloir les détailler tous, car certains sont si raresque les descriptions cliniques en sont encore incertaines(c’est le cas de l’hémangioendothéliome composite,de l’hémangioendothéliome polymorphe, ou de l’héman-gioendothéliome rétiforme) citons :
● l’hémangioendothéliome à cellules fusiformes, tumeurcutanée existant isolément, ou plus rarement associéeaux anomalies veineuses superficielles et aux enchon-dromes du syndrome de Maffucci.
[21
[22
[23
[24
Références
[1] Enjolras O, Mulliken JB. Vascular tumors and vascular malfor-mations, new issues. Adv Dermatol 1997;13:375–423.
[2] Bruckner AL, Frieden IJ. Hemangiomas of infancy. J Am AcadDermatol 2003;48:477–93.
[3] Drolet BA, Esterly NB, Frieden IJ. Hemangiomas in children. NEngl J Med 1999;341:173–80.
[4] North PE, Waner M, Mizeracki A, Mihm Jr. MC. GLUT1: a newlydiscovered immunohistochemical marker for juvenile heman-giomas. Hum Pathol 2000;31:11–22.
North P, Waner M, Mizeracki A, Mrak RE, Nicholas R, KincannonJ, et al. A unique microvascular phenotype shared by juvenilehemangiomas and human placenta. Arch Dermatol 2001;137:559–70.Enjolras O, Wassef M, Mazoyer E, Frieden IJ, Rieu PN, DrouetL, et al. Infants with Kasabach-Merritt syndrome do not have"true" hemangioma. J Pediatr 1997;130:631–40.Sarkar M, Mulliken JB, Kozakewich HPW, Robertson RL, Bur-rows PE. Thrombocytopenic coagulopathy (Kasabach-Merrittphenomenon) is associated with kaposiform hemangioendo-thelioma and not with common infantile hemangioma. PlastReconstr Surg 1997;100:1377–86.Vin-Christian K, McCalmont TH, Frieden IJ. Kaposiformhemangioendothelioma, an aggressive locally invasive vascu-lar tumor that can mimic hemangioma of infancy. Arch Der-matol 1997;133:1573–8.Berenguer B, Mulliken JB, Enjolras O, Boon LM, Wassef M, Jos-set P, et al. Rapidly involuting congenital hemangioma: clini-cal and histopathologic features. Pediatr Dev Pathol 2003:6.
] North PE, Waner M, James CA, Mizeracki A, Frieden IJ,Mihm Jr. MC. Congenital nonprogressive hemangioma. A dis-tinct clinicopathologic entity unlike infantile hemangioma.Arch Dermatol 2001;137:1607–20.
] Boon L, Enjolras O, Mulliken JB. Congenital hemangioma; evi-dence for accelerated involution. J Pediatr 1996;128:329–35.
] Konez O, Burrows PE, Mulliken JB, Fishman SJ, Kozake-wich HPW. Angiographic features of rapidly involuting conge-nital hemangioma (RICH). Pediatr Radiol 2003;33:15–9.
] Rogers M, Lam A, Fischer G. Sonographic findings in a series ofrapidly involuting congenital hemangiomas (RICH). PediatrDermatol 2002;19:5–11.
] Enjolras O, Mulliken JB, Boon LM, Kozakewich HPW, Bur-rows PE, Mulliken JB. Non-involuting congenital hemangioma:a rare cutaneous vascular anomaly. Plast Reconstr Surg 2001;107:1647–54.
] Chiaverini C, Kurzenne JY, Rogopoulos A, Ortonne JP,Lacour JP. Hémangiome congénital non-involutif : deux cas.Ann Dermatol Venereol 2002;129:735–7.
] Mulliken JB, Enjolras O. Congenital hemangiomas and infan-tile hemangioma: missing links. J Am Acad Dermatol 2004;50:875–82.
] Wilson-Jones E, Orkin M. Tufted angioma (angioblastoma): abenign progressive angioma not to be confused with Kaposi’ssarcoma or low-grade angiosarcoma. J Am Acad Dermatol1989;20:214–25.
] Cho KH, Kim SH, Park KC, Lee AY, Song KY, Chi JG, et al.Angioblastoma (Nakagawa): is it the same as tufted angioma?Clin Exp Dermatol 1991;16:110–3.
] Igarashi M, Oh-I T, Koga M. The relationship between angio-blastoma (Nakagawa) and tufted angioma: report of fourcases with angioblastoma and a literature-based comparisonof the two conditions. J Dermatol 2000;27:537–42.
] Okada E, Tamura A, Ishikawa O, Miyachi Y. Tufted angioma(angioblastoma) case report and review of 41 cases in theJapanese literature. Clin Exp Dermatol 2000;25:627–30.
] Satter EK, Graham BS, Gibbs NF. Congenital tufted angioma.Pediatr Dermatol 2002;19:445–7.
] Catteau B, Enjolras O, Delaporte E, Friedel J, Brevière GM,Wassef M, et al. Angiome en touffes sclérosant. À propos dequatre observations. Ann Dermatol Venereol 1998;125:682–7.
] Tsang WYW, Chan JKC. Kaposi-like hemangioendothelioma. Adistinctive vascular neoplasm of the retroperineum. Am J SurgPathol 1991;15:982–9.
] Zukerberg, LR, Nickoloff BJ, Weiss SW. Kaposiform heman-gioendothelioma of infancy and childhood. An aggressive neo-plasm associated with Kasabach-Merritt syndrome and lym-phangiomatosis. Am J Surg Pathol 1993;17:321–8.

[25
[26
[27
[28
[29
[30
[31
[32
[33
[34
[35
O. Enjolras et al.346
] Fukunaga M, Ushigome S, Ishikawa E. Kaposiform hemangioen-dothelioma associated with Kasabach-Merritt syndrome. His-topathology 1996;28:281–4.
] Debelenko, Perz-Atayde AR, Mulliken JB, Liang MG, Archi-bald TH, Kozakewich HP. D2-40 immunohistochemical analysisof pediatric vascular tumors reveals positivity in kaposiformhemangioendothelioma. Mod Pathol 2005 (May online).
] Lyons LL, North PE, Mac-Moune Lai F, Stoler MH, Folpe AL,Weiss SW. Kaposiform hemangioendothelioma. A study of 33cases emphasizing its pathologic, immunophenotypic, andbiologic uniqueness from juvenile hemangioma. Am J SurgPathol 2004;28:559–68.
] Mazoyer E, Enjolras O, Laurian C, Houdart E, Drouet L. Coagu-lation abnormalities associated with extensive venous malfor-mations of the limbs: differentiation from Kasabach-Merrittsyndrome. Clin Lab Haematol 2002;24:243–51.
] Enjolras O, Mulliken JB, Wassef M, Frieden IJ, Rieu PN, Bur-rows PE, et al. Residual lesions after Kasabach-Merritt pheno-menon in 41 patients. J Am Acad Dermatol 2000;42:275–9.
] Enjolras O. Syndrome de Kasabach-Merritt. Dermatologie Pra-tique 2003;269:10–1.
] Haisley-Royster C, Enjolras O, Frieden IJ, Garzon MD, OranjeA, Gonzalez F, et al. Kasabach-Merritt phenomeon: a retro-spective study of treatment with vincristine. J Pediatr Hema-tol Oncol 2002;24:459–62.
] Drolet BA, Scott LA, Esterly NB, Gosain AK. Early surgicalintervention in a patient with Kasabach-Merritt phenomenon.J Pediatr 2001;138:756–8.
] Velin P, Dupont D, Golkar A, Valla JS. Syndrome de Kasabach-Merritt neonatal guéri par exérèse chirurgicale complète del’angiome. Arch Pediatr 1998;5:295–7.
] Mulliken JB, Anupindi S, Ezekowitz RAB, Mihm Jr. MC. Case 13-2004: a newborn girl with a large cutaneous lesion, thrombo-cytopenia, and anemia. N Eng J Med 2004;350(17):1764–75.
] Enjolras O, Wassef M, Dosquet C, Drouet L, Fortier G, Josset P,et al. Syndrome de Kasabach-Merritt sur angiome en touffescongénital. Ann Dermatol Venereol 1998;125:257–60.





























