Embed Size (px)
Citation preview

THÉMATIQUE À TAPER
REVUE FRANCOPHONE DES LABORATOIRES - JANVIER 2008 - N°398 // 53
ANATOMIE ET CYTOLOGIE PATHOLOGIQUES
Histologie et pathologie moléculaire des tumeurs stromales gastrointestinales (GIST)Jean-François Emilea, b, c,*, Jean-Baptiste Bacheta, b, c, Séverine Tabone-Eglingerd, Sabrina Brahimia, b, c
a Service de pathologieHôpital universitaire Ambroise-Paré (AP-HP)9, av. Charles-de-Gaulle92104 Boulogne cedexb Faculté de médecine PIFO, UVSQ, Guyancourt.c INSERM U602 – Villejuif.d INSERM U590 – Lyon.
article reçu le 21 septembre, accepté le 7 octobre 2007.
© 2007 – Elsevier Masson SAS – Tous droits réservés.
RÉSUMÉ
Les tumeurs stromales gastrointestinales (GIST) sont les sarcomes les plus fréquents du tube digestif. Ces tumeurs peuvent se développer à partir de tous les segments du tractus digestif, depuis l’œsophage jusqu’à l’anus, ou exceptionnellement à partir du mésentère et du péritoine. Les circons-tances de découverte les plus fréquentes sont des douleurs abdominales, des saignements digestifs, des troubles du transit ou la présence d’une masse abdominale palpable. Toutefois ces tumeurs restent longtemps peu symptomatiques. De ce fait, 10 à 20 % sont de découverte fortuite et 15 à 50 % sont découvertes à un stade métastatique. Macroscopiquement, les GIST sont généralement des tumeurs bien limitées, à développement extra-pariétal, de consistance ferme, de couleur chair de poisson avec de fréquents remaniements hémorragiques. Les GIST se caractérisent his-tologiquement par une prolifération importante de cellules le plus souvent fusiformes ou épithélioïdes. Le diagnostic doit être confirmé par l’immuno-histochimie, avec une positivité de KIT dans 95 % des cas, ou par la biologie moléculaire, avec des mutations de KIT ou PDGFRA dans 85 % des cas. Toute GIST est a priori maligne, et le risque de récidive ou de métastase est actuellement évalué en fonction de la taille tumorale et l’activité mitotique. Des métastases se développent chez 30 à 50 % des patients atteints de GIST. Celles-ci sont totalement résistantes aux chimiothérapies convention-nelles, mais l’utilisation des thérapies ciblées avec des anti-oncogènes tels le Glivec® (Imatinib Mesylate), constitue l’un des progrès thérapeutiques majeurs de ces dernières années en cancérologie.
Tumeur – sarcome – tractus digestif – KIT – récepteurs tyrosine kinase.
SUMMARY
Histology and molecular pathology of
gastrointestinal stromal tumors (GIST)
Gastrointestinal stromal tumors (GIST) are the most frequent sarcoma of the digestive tract. These tumors can arise anywhere in the gastrointestinal tract, from oesophagus to anus, and rarely from peritoneal cavity or mesentery. The most frequent symptoms are abdominal pain, gastrointestinal bleeding, abdominal mass or bowel movement disorder. GIST usually remain asymptomatic for a long time, and 10 to 20 % are found incidentally at endoscopy or at time of surgery for others rea-sons. At diagnosis, metastases are found in 15 to 50 % of cases. Macroscopicaly, GIST are usually well limited, with extra-parietal development, firm in consistency, like flesh of fish in color and with frequent intra-tumoral hemorrage. Histologicaly, they are characterized by an important cellular proliferation constituted of epithelioid or spindle cell. In all cases, diagnosis must be confirmed by immunohistochemistry, with positivity of KIT in 95 % of cases, or by molecular biology, with «gain of function» mutation of KIT or PDGFRA in 85 % of cases. All GIST have a malignant po-tential. Mitotic activity and tumor size are the two principal prognostic factors to assess the risk of relapse or metastastatic evolution. Metastases occured in 30 to 50 % of patients with GIST. GIST are highly resistant to conventional chemotherapy and results obtained with new targeted therapies like Glivec® are one of the major progress in on-cology during the last ten years.
Tumor – sarcoma – digestive tract – KIT – tyrosine kinase receptors.
1. Introduction
Les tumeurs stromales gastrointestinales (GIST) ont été décrites dès 1983 [17], mais ce n’est que depuis la décou-verte du phénotype particulier des cellules tumorales, avec l’expression fréquente du CD34 [26] et celle quasi constante de KIT [15], il y a une dizaine d’années, que ces
tumeurs sont réellement individualisées. En pratique, ce n’est que depuis le début du millénaire que les GIST sont diagnostiquées en routine, et clairement distinguées des autres tumeurs mésenchymateuses digestives telles les léiomyomes, léiomyosarcomes ou schwannomes.La localisation des GIST à la musculeuse, tout le long du tractus digestif, et leur expression quasi constante de KIT suggèrent fortement que ces tumeurs dérivent de progé-

54 // REVUE FRANCOPHONE DES LABORATOIRES - JANVIER 2008 - N°398
niteurs des cellules interstitielles de Cajal. Ces cellules, décrites comme étant responsables du péristaltisme digestif et qui expriment KIT à l’état normal, sont localisées entre les cellules musculaires, souvent concentrées autour des plexus d’Auerbach. L’hyperplasie majeure des cellules interstitielles de Cajal et la fréquence importante de GIST chez les patients ayant une mutation constitutionnelle de KIT [4] sont des arguments supplémentaires en faveur de cette hypothèse phylogénique.
2. Le diagnostic de GIST
Le diagnostic de GIST est de plus en plus fréquemment évoqué par les radiologues, devant une tumeur hypervas-cularisée développée à partir de la paroi de l’estomac ou du grêle et sans métastase ganglionnaire. Toutefois, seule l’analyse histologique permet de confirmer le diagnostic de GIST. Les GIST sont des tumeurs bien limitées mais
sans encapsulation, qui se développent préférentiellement sur le versant séreux de la paroi digestive (figure 1). Lors de la macroscopie, la mesure du diamètre maximal de la tumeur primitive est un paramètre majeur pour l’évaluation du potentiel évolutif (tableau I). Il est important de bien échantillonner la tumeur, pour le diagnostic différentiel avec d’autres sarcomes (exemple : liposarcome), et parce qu’il peut exister des variations dans l’index de prolifération. À l’histologie, la densité cellulaire est généralement forte et homogène, et des remaniements nécrotiques, œdémateux et/ou hémorragiques sont d’autant plus fréquents que les tumeurs sont de grande taille. Les cellules sont fusiformes dans 70 % des cas, avec une architecture le plus souvent fasciculée (figure 2a), évoquant une prolifération muscu-laire lisse. Plus rarement, les cellules fusiformes se dis-posent en palissade (figure 2b) ou en « bulbe d’oignon ». Les cellules fusiformes ont un noyau ovalaire court, et un cytoplasme éosinophile qui présente fréquemment des pseudo-vacuolisations évocatrices du diagnostic. Le stroma tumoral est généralement très peu abondant, constitué de capillaires sanguins, et peut, dans les localisations intes-tinales, contenir des fibres skénoïdes très spécifiques du diagnostic (figure 2c). Dans environ 20 % des cas, les cellules sont épithélioïdes (figure 2d). Ce type histologi-que est plus fréquent dans les localisations gastriques. Les autres variantes histologiques sont plus rares, avec des variantes mixtes (fusiformes et épithélioïdes), ou des aspects devant faire discuter d’autres diagnostics (formes myxoïdes, pléomorphes, carcinoïdes…). Pour l’évaluation du risque de reprise évolutive, le compte mitotique se fait sur 50 champs au fort grossissement (tableau I) ; ce qui n’est généralement possible que sur les pièces opératoires.Même lorsque la clinique et l’histologie sont évocatrices, le diagnostic de GIST doit être confirmé par une étude immunohistochimique, qui permet notamment de mettre en évidence une expression de KIT par les cellules tumo-rales dans 95 % des cas. L’expression de KIT est, dans la grande majorité des cas, forte et diffuse à l’ensemble des cellules tumorales. Le marquage peut être membra-naire, cytoplasmique diffus et/ou cytoplasmique « golgien » (figure 3). Dans quelques cas, le marquage est hétérogène au sein de la tumeur et seuls de petits territoires tumoraux sont positifs, ce qui peut poser des problèmes de faux négatifs sur des biopsies ou des micro-biopsies. Toutefois, la présence quasi constante de témoins positifs sur les coupes histologiques, tels les mastocytes intra-muqueux ou intra-tumoraux et les cellules interstitielles de Cajal de la musculeuse, permet d’éliminer facilement un problème technique. À l’inverse, pour éviter les faux positifs, il est recommandé d’utiliser les anticorps dans des conditions assez stringentes ; ainsi l’anticorps polyclonal commercialisé par Dako doit être utilisé soit à la dilution finale de 1/300 après restauration antigénique dans du tampon citrate à pH6, soit au 1/50 sans restauration antigénique. Enfin, il faut se souvenir que plusieurs autres tumeurs peuvent exprimer KIT [29] dans la majorité (séminome, carcinome adénoïde kystique) ou dans une proportion variable de cas (mélanome, carcinomes bronchique, etc…). Environ 5 % des GIST sont négatives pour KIT ; le diagnostic nécessite alors la mise en évidence de mutations des gènes KIT ou PDGFRA au sein de l’ADN tumoral [1]. Les GIST les
Tableau I – Consensus international pour l’évaluation
du risque de récidive ou métastase d’une GIST localisée
(d’après [Fletcher 02]).
Diamètre tumoral maximal Nombre de mitoses/50 hpf*
Très faible risque moins de 2 cm et moins de 5 mitoses
Faible risque 2 à 5 cm et moins de 5 mitoses
Risque intermédiaire
moins de 5 cmou
5 à 10 cm
et 6 à 10 mitoses
et moins de 5 mitoses
Haut risque
5 à 10 cmou
plus de 10 cmou
indifférent
et 6 à 10 mitoses
indifférent
plus de 10 mitoses
* hpf : champs au grossissement x 400.
Figure 1
Macroscopie d’une volumineuse GIST développée sur la face externe de la paroi
gastrique. Il s’agit d’une gastrectomie atypique car un curage ganglionnaire n’est
pas nécessaire ; la paroi gastrique reséquée est donc de petite taille (à droite).
La tumeur (à gauche) est arrondie, bien limitée en périphérie, avec quelques
remaniements œdémateux et hémorragiques.
REVUE FRANCOPHONE DES LABORATOIRES - JANVIER 2008 - N°398 // 55
ANATOMIE ET CYTOLOGIE PATHOLOGIQUES
plus fréquemment négatives pour KIT sont des variantes épithélioïdes de localisation gastrique, avec une mutation du PDGFRA [16].D’autres marqueurs peuvent être utiles pour le diagnostic de GIST. Le CD34 est positif dans 70 % des GIST gastri-ques et 50 % des GIST extra-gastriques. Il est préférable de considérer la tumeur comme positive seulement lors-que plus de 20 % des cellules tumorales sont marquées. Environ 70 % des GIST sont positifs pour la h-Caldes-mone [6]. D’autres marqueurs, tels DOG-1 [30] ou PKC theta [13] ont été décrits comme spécifiques des GIST. Dans environ 5 % des GIST, des cellules tumorales sont positives pour la protéine S100, mais une positivité diffuse doit faire envisager un autre diagnostic, tel une tumeur nerveuse périphérique ou un mélanome. La desmine est un marqueur important pour le diagnostic différentiel avec un léiomyosarcome, car contrairement à ceux-ci les GIST ne sont qu’exceptionnellement positifs.Les diagnostics différentiels de GIST sont nombreux avec notamment des léiomyomes et léiomyosarcomes, des schwannomes et neurofibromes, des liposarcomes dédif-férenciés ou d’autres sarcomes d’origine abdominale ou pelvienne. Ces tumeurs sont habituellement KIT négatives, ce qui implique d’être particulièrement vigilant au risque de faux positifs. Les métastases de mélanomes, notamment sur le grêle, peuvent quant à elles être positives pour KIT et poser un problème difficile de diagnostic différentiel. Les tumeurs desmoïdes abdominales constituent également un diagnostic différentiel important, à distinguer sur des critères histologiques.En pratique courante, le diagnostic de GIST, de plus en plus souvent évoqué par le radiologue ou le chirurgien, est confirmé facilement par l’histologie et l’immunohistochimie avec un panel de 3 ou 4 anticorps (KIT, CD34, PS100, +/- h-caldesmone). Toutefois certains cas nécessitent le recours à la biologie moléculaire pour mettre en évidence une mutation spécifique de KIT ou PDGFRA [1].
3. Epidémiologie des GIST
Les GIST surviennent généralement chez l’adulte, avec un âge moyen et médian au diagnostic de 60 ans, et la fréquence est globalement identique entre les sexes. L’in-cidence est actuellement évaluée par des études rétros-pectives entre 10 et 20 nouveaux cas/an/million d’habitant [19, 20], et ne semble pas varier en fonction des origines
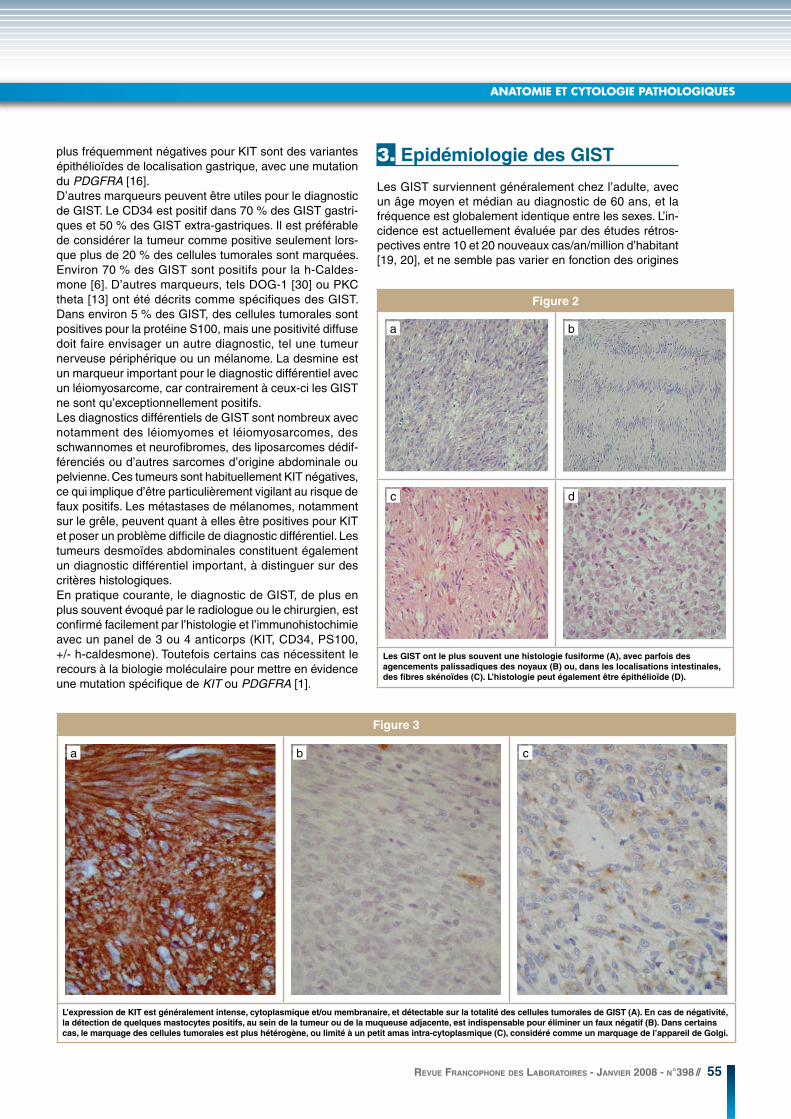
Figure 2
Les GIST ont le plus souvent une histologie fusiforme (A), avec parfois des
agencements palissadiques des noyaux (B) ou, dans les localisations intestinales,
des fibres skénoïdes (C). L’histologie peut également être épithélioïde (D).
a b
c d
Figure 3
L’expression de KIT est généralement intense, cytoplasmique et/ou membranaire, et détectable sur la totalité des cellules tumorales de GIST (A). En cas de négativité,
la détection de quelques mastocytes positifs, au sein de la tumeur ou de la muqueuse adjacente, est indispensable pour éliminer un faux négatif (B). Dans certains
cas, le marquage des cellules tumorales est plus hétérogène, ou limité à un petit amas intra-cytoplasmique (C), considéré comme un marquage de l’appareil de Golgi.
a b c

56 // REVUE FRANCOPHONE DES LABORATOIRES - JANVIER 2008 - N°398
géographiques et ethniques des patients. Toutefois, les données épidémiologiques sont encore très limitées pour ces tumeurs dont le diagnostic était encore rarement fait il y a 10 ans. Des études de registre, telles MolecGIST (http://www.gist-france.org) sont encore nécessaires pour préciser ces données.Les GIST se développent à partir de l’estomac dans 60 à 70 % des cas et de l’intestin grêle dans 20 à 30 % des cas, mais tous les autres segments digestifs, œsophage (5 %), côlon, appendice, rectum, ainsi que le mésentère et la cavité péritonéale peuvent être atteints. Dans l’estomac, les adé-nocarcinomes sont de loin les cancers les plus fréquents, mais environ 95 % des tumeurs mésenchymateuses sont des GIST, et les diagnostics différentiels de schwannome et léiomyosarcome sont donc rares. Dans le grêle, environ 1/4 des tumeurs malignes sont des GIST.Les GIST constituent 20 à 30 % des sarcomes des tissus mous [18]. Leur survenue est sporadique dans la grande majorité des cas, mais il existe quelques prédispositions familiales, telles la neurofibromatose de type I [28] et d’ex-ceptionnelles formes familiales décrites par Carney et Stratakis [22] ou liées à une mutation constitutionnelle de KIT [21] ou PDGFRA [5].
4. Les mutations de KIT
KIT est un récepteur membranaire de la famille des tyro-sines kinases, qui est la principale famille d’oncogènes en pathologie humaine [3]. La fixation à KIT de son ligand spécifique, le stem cell factor (SCF), induit son activation et la transmission de signaux intracellulaires responsables de la prolifération et de la survie des cellules. Dans 70 à 80 % des GIST, mais également parfois dans d’autres tumeurs telles les mastocytoses ou les leucémies, une mutation du gène KIT est responsable de l’activation spon-tanée du récepteur KIT, y compris en l’absence de SCF ; il s’agit d’une mutation dite « gain de fonction » [15]. Il est intéressant de noter que KIT est activé au sein des GIST, même en l’absence de mutations « gain de fonction » [25]. Dans 5 à 10 % des GIST, une mutation « gain de fonction » est détectée sur un gène très proche de KIT, le PDGFRA [13]. Les mutations de ces deux gènes sont mutuellement exclusives. Elles sont de nature très variable dans leur localisation et leur type (délétion, insertion, substitution). Les plus fréquentes sont localisées sur les exons 11 et 9 de KIT et 18 de PDGFRA (tableau II).La détection des mutations peut se faire sur des blocs de tumeurs fixées en formol et incluses en paraffine. Après contrôle histologique, l’ADN tumoral est extrait puis amplifié par PCR. Le séquençage peut être précédé d’une étape de screening (figure 4) par LAPP [9] ou DHPLC [13]. La recherche de mutation est indispensa-ble pour le diagnostic de GIST KIT négative [1], ou les GIST d’histologie inhabituelle. Elle est souvent requise par les oncologues pour les patients atteints de GIST sévère. En effet, la présence de mutations de l’exon 11 de KIT est associée à une forte fréquence de réponse au traitement par le Glivec® [14], alors que lorsqu’il existe une mutation de l’exon 9 de KIT, un doublement de la dose est préférable [27]. Pour les autres patients, la recherche systématique de mutations serait également utile pour mieux connaître les formes clinico-biologiques, leur pronostic et leur sensibilité aux thérapies ciblées. C’est l’objectif de l’étude MolecGIST, qui propose de rechercher les mutations chez tous les patients dont le diagnostic à été fait en France entre juin 2006 et juin 2008 (http://www.gist-france.org).
5. Évolution et traitement
Toutes les GIST sont potentiellement malignes, et le risque de récidive après résection peut être évalué selon la taille et l’index mitotique (tableau I). Il est probable que d’autres paramètres, tels la localisation gastrique, la présence de nécrose et le type de mutation, aient une valeur pronostique [10], mais ceci mérite d’être évalué sur de grandes séries prospectives de patients. La valeur pronostique des muta-tions, publiée initialement [24], est actuellement débattue [11]. Les métastases de GIST se localisent au foie dans 2/3 des cas ou dans le péritoine dans 1/4 des cas. Les métastases ganglionnaires sont rares, ce qui justifie de ne pas faire de curage lorsque le diagnostic est suspecté. Les métastases pulmonaires sont également rares, et leur survenue peut justifier de rediscuter le diagnostic.
Tableau II – Les mutations de KIT et PDGFRA dans les GIST
(d’après [Corless 04]).
Gène Exon Fréquence Type de mutation Sensibilité au Glivec®
KIT 9 10 % Unique (insertion) Oui (mieux à 800 mg/j*)
KIT 11 66 % Très variable Oui
KIT 13 1 % Unique (substitution) Oui
KIT 17 < 1 % variable variable
PDGFRA 12 < 1 % variable Oui
PDGFRA 14 < 1 % variable Oui
PDGFRA 18 6 % variableDépend du type
de mutation
* la posologie usuelle est de 400 mg/j per os.
Figure 4
L’ADN tumoral peut être extrait sous contrôle histologique à partir des blocs fixés ou congelés, puis les mutations de KIT et PDGFRA sont identifiées par séquençage des produits d’amplification. Une étape de screening par LAPP (A) permet ici de détecter un pic correspondant à l’allèle sauvage (flèche rouge) et un allèle muté (flèche bleu). Dans le cas présent, le pic muté est nettement plus élevé, attestant d’une mutation homozygote, qui est ensuite identifiée par séquençage (B).
a
b

REVUE FRANCOPHONE DES LABORATOIRES - JANVIER 2008 - N°398 // 57
ANATOMIE ET CYTOLOGIE PATHOLOGIQUES
Le seul traitement curatif des GIST est la résection chirurgi-cale des tumeurs localisées. Les GIST métastatiques sont actuellement incurables. Elles sont totalement résistantes aux chimiothérapies usuelles [2] et à la radiothérapie. Le développement de thérapies ciblées avec des inhibiteurs des récepteurs tyrosine kinase, tels le Glivec® (Imatinib Mesylate) a heureusement fortement amélioré la survie des patients atteints de GIST métastatiques. En effet, alors que dans les séries historiques la survie globale de ces patients était de l’ordre de 18 à 24 mois, le Glivec® permet d’obtenir une réponse tumorale (réponse objective ou stabilité de plus de 6 mois) chez 85 % à 90 % d’entre eux, avec des survies sans progression de 24 mois et des survies globa-les supérieures à 36 mois. [23]. La réponse tumorale est nettement corrélée à la présence de mutations de KIT [14], avec des réponses plus fréquentes chez les patients ayant des mutations de l’exon 11 de KIT. De plus, les patients ayant une mutation de l’exon 9 de KIT ont une survie aug-mentée lorsqu’ils sont traités à double dose (800 ou lieu de 400 mg/jour) [27]. De ce fait, les oncologues demandent de plus en plus fréquemment la recherche de mutations chez les patients atteints de formes métastatiques.
Un traitement adjuvant, après résection complète d’une GIST, est également probablement utile [8]. Un essai cli-nique visant à confirmer ceci est actuellement en cours en Europe sur des patients ayant un risque intermédiaire ou élevé de récidive. Ici encore, la recherche de mutation pourrait être extrêmement utile pour la prise en charge des patients.
6. Conclusion
Les GIST sont un des meilleurs exemples de l’intérêt de la recherche actuelle en cancérologie. En effet, en l’espace de 3 ans, une tumeur pratiquement inconnue a pu être individualisée et facilement diagnostiquée en raison de l’identification d’un oncogène (KIT), puis traitée efficace-ment avec un inhibiteur spécifique de cet oncogène (Gli-vec®). Encore 3 ans plus tard, l’étude de grandes cohortes de patients a permis de démontrer l’association entre le type de mutation et la réponse à ce traitement, et permet maintenant d’envisager des traitements « à la carte » pour chaque patient.
Références
[1] Blay J.Y., Bonvalot S., Casali P., Choi H., Debiec-Richter M., Dei Tos A.P., Emile J.F., Gronchi A., Hogendoorn P.C., Joensuu H., Le Cesne A., McClure J., Maurel J., Nupponen N., Ray-Coquard I., Reichardt P., Sciot R., Stroobants S., van Glabbeke M., van Oosterom A., Demetri G.D., GIST consensus meeting panellists, Consensus meeting for the management of gastrointestinal stromal tumors, Report of the GIST Consensus Conference of 20-21 March 2004, under the auspices of ESMO, Ann. Oncol. 16(4) (2005) 566-578.
[2] Blay J.Y., Landi B., Bonvalot S., Monges G., Ray-Coquard I., Duffaud F., Bui N.B., Bugat R., Chayvialle J.A., Rougier P., Bouche O., Bonichon F., Lassau N., Vanel D., Nordlinger B., Stoeckle E., Meeus P., Coindre J.M., Scoazec J.Y., Emile J.F., Ranchere D., Le Cesne A., Recommendations for the management of GIST patients, Bull. Cancer. 92(10) (2005) 907-918.
[3] Blume-Jensen P., Hunter T., Oncogenic kinase signalling, Nature 411(6835) (2001) 355-365.
[4] Chen H., Hirota S., Isozaki K., Sun H., Ohashi A., Kinoshita K., O’Brien P., Kapusta L., Dardick I., Obayashi T., Okazaki T., Shinomura Y., Matsuzawa Y., Kitamura Y., Polyclonal nature of diffuse proliferation of interstitial cells of Cajal in patients with familial and multiple gastrointes-tinal stromal tumours, Gut 51(6) (2002) 793-796.
[5] Chompret A., Kannengiesser C., Barrois M., Terrier P., Dahan P., Tursz T., Lenoir G.M., Bressac-De Paillerets B., PDGFRA germline mutation in a family with multiple cases of gastrointestinal stromal tumor, Gastroenterology 126(1) (2004) 318-321.
[6] Coindre J.M., Emile J.F., Monges G., Ranchere-Vince D., Scoazec J.Y., Gastrointestinal stromal tumors: definition, histological, immunohis-tochemical, and molecular features, and diagnostic strategy, Ann. Pathol. 25(5) (2005) 358-385.
[7] Corless C.L, Fletcher J.A., Heinrich M.C., Biology of gastrointestinal stromal tumors, J. Clin. Oncol. 22(18) (2004) 3813-3825.
[8] DeMatteo R., Owzar K., Maki R., Pisters P., Blackstein M., Antonescu C., Blanke C., Demetri G., von Mehren M., Ballman K. and the American College of Surgeons Oncology Group (ACOSOG) Intergroup
Adjuvant GIST Study Team, Adjuvant imatinib mesylate increases recur-rence free survival (RFS) in patients with completely resected local-ized primary gastrointestinal stromal tumor (GIST): North American Intergroup Phase III trial ACOSOG Z9001, ASCO Ann. Meet. Proc. No: 10079 (communication orale).[9] Emile J.F., Lemoine A., Bienfait N., Terrier P., Azoulay D., Debuire B., Length analysis of polymerase chain reaction products: a sensitive and reliable technique for the detection of mutations in KIT exon 11 in gas-trointestinal stromal tumors, Diagn. Mol. Pathol. 11(2) (2002) 107-112.[10] Emile J.F., Theou N., Tabone S., Cortez A., Terrier P., Chaumette M.T., Julie C., Bertheau P., Lavergne-Slove A., Donadieu J., Barrier A., Le Cesne A., Debuire B., Lemoine A., Groupe d’étude des GIST, Clinicopathologic, phenotypic, and genotypic characteristics of gastroin-testinal mesenchymal tumors, Clin. Gastroenterol. Hepatol. 2(7) (2004) 597-605.[11] Emile J.F., Tabone-Eglinger S., Theou-Anton N., Lemoine A., Prognostic value of KIT exon 11 deletions in GISTs, Gastroenterology 131(3) (2006) 976-977.[12] Fletcher C.D., Berman J.J., Corless C., Gorstein F., Lasota J., Longley B.J., Miettinen M., O’Leary T.J., Remotti H., Rubin B.P., Shmookler B., Sobin L.H., Weiss S.W., Diagnosis of gastrointestinal stro-mal tumors : a consensus approach, Hum. Pathol. 33(5) (2002) 459-465.[13] Heinrich M.C., Corless C.L., Duensing A., McGreevey L., Chen C.J., Joseph N., Singer S., Griffith D.J., Haley A., Town A., Demetri G.D., Fletcher C.D., Fletcher J.A., PDGFRA activating mutations in gastroin-testinal stromal tumors, Science 299(5607) (2003) 708-710.[14] Heinrich M.C., Corless C.L., Demetri G.D., Blanke C.D., von Mehren M., Joensuu H., McGreevey L.S., Chen C.J., Van den Abbeele A.D., Druker B.J., Kiese B., Eisenberg B., Roberts P.J., Singer S., Fletcher C.D., Silberman S., Dimitrijevic S., Fletcher J.A., Kinase muta-tions and imatinib response in patients with metastatic gastrointestinal stromal tumor, J. Clin. Oncol. 21(23) (2003) 4342-4349.[15] Hirota S., Isozaki K., Moriyama Y., Hashimoto K., Nishida T., Ishiguro S., Kawano K., Hanada M., Kurata A., Takeda M., Muhammad Tunio G., Matsuzawa Y., Kanakura Y., Shinomura Y., Kitamura Y., Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors, Science 279(5350) (1998) 577-580.

58 // REVUE FRANCOPHONE DES LABORATOIRES - JANVIER 2008 - N°398
[16] Lasota J., Dansonka-Mieszkowska A., Stachura T., Schneider-Stock R., Kallajoki M., Steigen S.E., Sarlomo-Rikala M., Boltze C., Kordek R., Roessner A., Stachura J., Miettinen M., Gastrointestinal stro-mal tumors with internal tandem duplications in 3’ end of KIT juxtamem-brane domain occur predominantly in stomach and generally seem to have a favorable course, Mod. Pathol. 16(12) (2003) 1257-1264.[17] Mazur M.T., Clark H.B., Gastric stromal tumors, Reappraisal of his-togenesis, Am. J. Surg. Pathol. 7(6) (1983) 507-519.[18] Miettinen M., Majidi M., Lasota J., Pathology and diagnostic criteria of gastrointestinal stromal tumors (GISTs): a review, Eur. J. Cancer 38 Suppl 5 (2002) S39-51.[19] Monges G., Coindre J.M., Scoazec J.Y., Bouvier A., Blay J.Y., Loria-Kanza Y., Mathieu-Boue A., Bisot-Locard S., Incidence of gastrointesti-nal stromal tumors (GISTs) in France: results of the PROGIST survey conducted among pathologists, J. Clin. Oncol. ASCO 2007, Ann. Meet. Proc., Part I, Vol. 25,18S, 2007, 10047.[20] Nilsson B., Bumming P, Meis-Kindblom J.M., Oden A., Dortok A., Gustavsson B., Sablinska K., Kindblom L.G., Gastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era-a population-based study in western Sweden, Cancer 103(4) (2005) 821-829.[21] Nishida T., Hirota S., Taniguchi M., Hashimoto K., Isozaki K., Nakamura H., Kanakura Y., Tanaka T., Takabayashi A., Matsuda H., Kitamura Y., Familial gastrointestinal stromal tumours with germline mutation of the KIT gene, Nat. Genet. 19(4) (1998) 323-324.[22] Pasini B., McWhinney S.R., Bei T., Matyakhina L., Stergiopoulos S., Muchow M., Boikos S.A., Ferrando B., Pacak K., Assie G., Baudin E., Chompret A., Ellison J.W., Briere J.J., Rustin P., Gimenez-Roqueplo A.P., Eng C., Carney J.A., Stratakis C.A., Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB,
SDHC, and SDHD, Eur. J. Hum. Genet. (2007 Aug. 1) [Epub ahead of print].[23] Rubin B.P., Heinrich M.C., Corless C.L., Gastrointestinal stromal tumour, Lancet 369(9574) (2007) 1731-1741.[24] Taniguchi M., Nishida T., Hirota S., Isozaki K., Ito T., Nomura T., Matsuda H., Kitamura Y., Effect of c-kit mutation on prognosis of gas-trointestinal stromal tumors, Cancer Res. 59(17) (1999) 4297-4300.[25] Theou-Anton N., Tabone S., Brouty-Boye D., Saffroy R., Ronnstrand L., Lemoine A., Emile J.F., Co expression of SCF and KIT in gastrointestinal stromal tumours (GISTs) suggests an autocrine/para-crine mechanism, Br. J. Cancer 94(8) (2006) 1180-1185.[26] van de Rijn M., Hendrickson M.R., Rouse R.V., CD34 expression by gastrointestinal tract stromal tumors, Hum. Pathol. 25(8) (1994) 766-771.[27] Van Glabbeke M., Owzar K., Rankin C., Simes J., Crowley J., GIST Meta-analysis Group (MetaGIST) Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors (GIST): a meta-analyis based on 1 640 patients, J. Clin. Oncol. ASCO 2007, Ann. Meet. Proc., Part I, Vol. 25, 18S, 2007, 1 000.[28] Yantiss R.K., Rosenberg A.E., Sarran L., Besmer P., Antonescu C.R., Multiple gastrointestinal stromal tumors in type I neurofibromatosis: a pathologic and molecular study, Mod. Pathol. 18(4) (2005) 475-484.[29] Went P.T., Dirnhofer S., Bundi M., Mirlacher M., Schraml P., Mangialaio S., Dimitrijevic S., Kononen J., Lugli A., Simon R., Sauter G., Prevalence of KIT expression in human tumors, J. Clin. Oncol. 22(22) (2004) 4514-4522.[30] West R.B., Corless C.L., Chen X., Rubin B.P., Subramanian S., Montgomery K., Zhu S., Ball C.A., Nielsen T.O., Patel R., Goldblum J.R., Brown P.O., Heinrich M.C., van de Rijn M., The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status, Am. J. Pathol. 165(1) (2004) 107-113.








![Chapitre : 12 Tumeurs stromales gastro instestinales (GIST) · 12.1.2. Biopsies Les biopsies [1,2] endoscopiques sont généralement négatives. L’indication d’une ponction-biopsie](https://img.pdfslide.fr/doc/110x75/5f5a496ef96f0525a65bca7a/chapitre-12-tumeurs-stromales-gastro-instestinales-gist-1212-biopsies-les.jpg)