Embed Size (px)
Citation preview

J
F
I
R
0d
ournal de radiologie (2011) 92, 308—322
ORMATION MÉDICALE CONTINUE : LE POINT SUR. . .
nsuffisance rénale et maladies kystiques du rein
enal failure and cystic kidney diseases
J.-M. Correas ∗, D. Joly, D. Chauveau,S. Richard, O. Hélénon
Service de radiologie, hôpital Necker, 149, rue de Sèvres, 75743 Paris cedex 15, France
MOTS CLÉSRein ;Génétique ;Kyste ;Insuffisance rénale
Résumé Les maladies kystiques du rein sont souvent découvertes soit lors du bilan d’uneinsuffisance rénale par le biais d’une échographie ou d’une enquête familiale, soit de faconfortuite lors d’un examen d’imagerie. Parmi les affections héréditaires, on distingue la poly-kystose rénale autosomique dominante ou récessive, la sclérose tubéreuse de Bourneville et lesmaladies kystiques de la médullaire. Dans sa forme dominante, la polykystose se caractérise parla présence de volumineux kystes rénaux qui apparaissent chez l’adulte jeune. L’insuffisancerénale se développe progressivement et devient sévère vers 50—60 ans. La présence de kystesatypiques (hémorragiques ou hyperdenses) est très fréquente au scanner ou en IRM. L’imageriejoue un rôle essentiel au cours des complications aiguës que sont l’hémorragie ou la surinfec-tion kystique. La polykystose autosomique récessive est le plus souvent découverte de faconprécoce en période néonatale, chez le nourrisson ou le jeune enfant. Elle associe une néphro-mégalie constituée de petits kystes et une atteinte hépatique (fibrose et dilatation des voiesbiliaires). Le diagnostic des formes tardives et d’évolution lente est plus complexe à distin-guer de la polykystose autosomique dominante. Ces maladies kystiques ne doivent pas êtreconfondues avec la multikystose rénale. Dans la sclérose tubéreuse de Bourneville, les kystesrénaux sont associés à des angiomyolipomes et parfois à la lymphangioléiomyomatose pulmo-naire. L’insuffisance rénale est inconstante. Les autres maladies kystiques héréditaires, commela maladie kystique de la médullaire et la néphronophtise, sont volontiers associées à une insuf-fisance rénale. Parmi les maladies kystiques non héréditaires, on distingue la dysplasie rénalemultikystique (par atrésie pyélo-infundibulaire complète ou par hydronéphrose), la maladiekystique rénale acquise (insuffisance rénale chronique, hémodialyse chronique) et les mala-
dies kystiques rénales diverses (la maladie plurikystique rénale, la maladie glomérulokystiquerénale, la maladie microkystique rénale).© 2011 Publie par Elsevier Masson SAS.∗ Auteur correspondant.Adresse e-mail : [email protected] (J.-M. Correas).
221-0363/$ — see front matter © 2011 Publie par Elsevier Masson SAS.oi:10.1016/j.jradio.2011.02.021

Insuffisance rénale et maladies kystiques du rein 309
KEYWORDSKidney;Genetics;Cyst;Renal failure
Abstract Cystic kidney diseases often are discovered at the time of initial work-up of renalfailure through ultrasound or family history, or incidentally at the time of an imaging test.Hereditary diseases include autosomal dominant or recessive polycystic kidney disease (PKD),tuberous sclerosis (TS) and medullary cystic kidney disease (MCKD). Autosomal dominant PKDis characterized by large renal cysts developing in young adults. Renal failure is progressiveand becomes severe around 50—60 years of age. Atypical cysts (hemorrhagic or hyperdense)are frequent on CT and MRI examinations. Imaging plays a valuable role in the managementof acute complications such as cyst hemorrhage or infection. Autosomal recessive PKD is oftendetected in neonates, infants or young adults. It is characterized by renal enlargement dueto the presence of small cysts and liver disease (fibrosis and biliary ductal dilatation). Latemanifestation or slow progression of autosomal recessive PKD may be more difficult to distin-guish from autosomal dominant PKD. These cystic kidney diseases should not be confused withnon-hereditary incidental multiple renal cysts. In tuberous sclerosis, renal cysts are associatedwith angiomyolipomas and sometimes pulmonary lymphangioleiomyomatosis. Renal failure isinconstant. Other hereditary cystic kidney diseases, including MCKD and nephronophtisis, areusually associated with renal failure. Non-hereditary cystic kidney diseases include multicysticrenal dysplasia (due to complete pelvi-ureteric atresia or hydronephrosis), acquired multicystickidney disease (chronic renal failure, chronic hemodialysis) and varied cystic kidney diseases(multicystic renal disease, glomerulocystic kidney disease, microcystic kidney disease).© 2011 Published by Elsevier Masson SAS.
L
OdaLtetctosthnprtsm
L(
QCasnhypertension artérielle, d’un retard de croissance ou de gros
Les maladies kystiques du rein regroupent un ensemblehétérogène d’affections qui ont pour seul point communla présence de kystes. Les mécanismes de la genèse deskystes, la disposition anatomique ou leur association àd’autres lésions rénales et extrarénales les distinguentdes kystes simples du rein. Cependant, le développementde la biologie moléculaire et de la génétique humainepermet progressivement l’émergence d’une meilleure noso-logie. Ces progrès ouvrent des possibilités, comme lediagnostic anténatal et le dépistage familial qui consti-tuent un progrès décisif, alors que le traitement desmaladies kystiques du rein demeure principalement symp-tomatique. Celles-ci représentent environ 25 % des causesd’insuffisance rénale terminale chez l’enfant et 10 % chezl’adulte [1].
Bien qu’il n’existe pas de classification simple etsatisfaisante des maladies kystiques rénales, on peut dis-tinguer, à côté des kystes simples compliqués ou nonet des kystes du sinus, sept catégories, en séparantles affections héréditaires de celles qui ne le sont pas[1].
Parmi les maladies kystiques héréditaires, on distinguela polykystose rénale autosomique dominante ou réces-sive, les maladies kystiques pouvant être associées à destumeurs du rein (maladie de von Hippel Lindau et sclérosetubéreuse de Bourneville) et les maladies kystiques de lamédullaire (maladie kystique de la médullaire et néphrono-phtise).
Parmi les maladies kystiques non héréditaires, ondistingue la dysplasie rénale multikystique (par atrésiepyélo-infundibulaire complète ou par hydronéphrose), lamaladie kystique rénale acquise (insuffisance rénale chro-nique, hémodialyse chronique) et les maladies kystiques
rénales diverses (la maladie plurikystique rénale, la maladieglomérulokystique rénale, la maladie microkystique rénaleet la maladie de Cacchi et Ricci).rsr
a polykystose rénale
n distingue, selon le mode de transmission génétique,eux formes différentes de polykystose rénale : la formeutosomique récessive et la forme autosomique dominante.a polykystose autosomique récessive (PKRAR) est carac-érisée par une dilatation des tubes collecteurs des reinst une atteinte hépatique constante par fibroadénoma-ose des voies biliaires, résultant en une fibrose hépatiqueongénitale combinant une hyperplasie des canaux hépa-iques avec une fibrose périportale. Elle atteint le nourrissonu l’enfant. L’expression clinique a une sévérité variable,ouvent dissociée entre le foie et le rein. La PKRAD est habi-uellement découverte chez l’enfant pour lequel l’atteinteépatique prédomine et plus rarement, en période néo-atale où l’atteinte rénale conditionne le pronostic. Laolykystose autosomique dominante (PKRAD) est caracté-isée par la formation de kystes développés à partir deous les segments du néphron ou du tube collecteur ; elle’associe à la présence de kystes hépatiques et éventuelle-ent d’anévrismes cérébraux.
a polykystose rénale autosomique récessivePKRAR)
uelques rappels anatomocliniques’est une maladie rare (0,25 à 1/10 000), à transmissionutosomique récessive liée à un gène localisé au chromo-ome 6p [2]. Elle est souvent découverte dans la périodeéonatale ou dans la petite enfance, à l’occasion d’une
eins, voire d’un gros foie ou d’une angiocholite. L’évolutione fait plus ou moins rapidement vers une insuffisanceénale. Le diagnostic différentiel avec les PKRAD à début
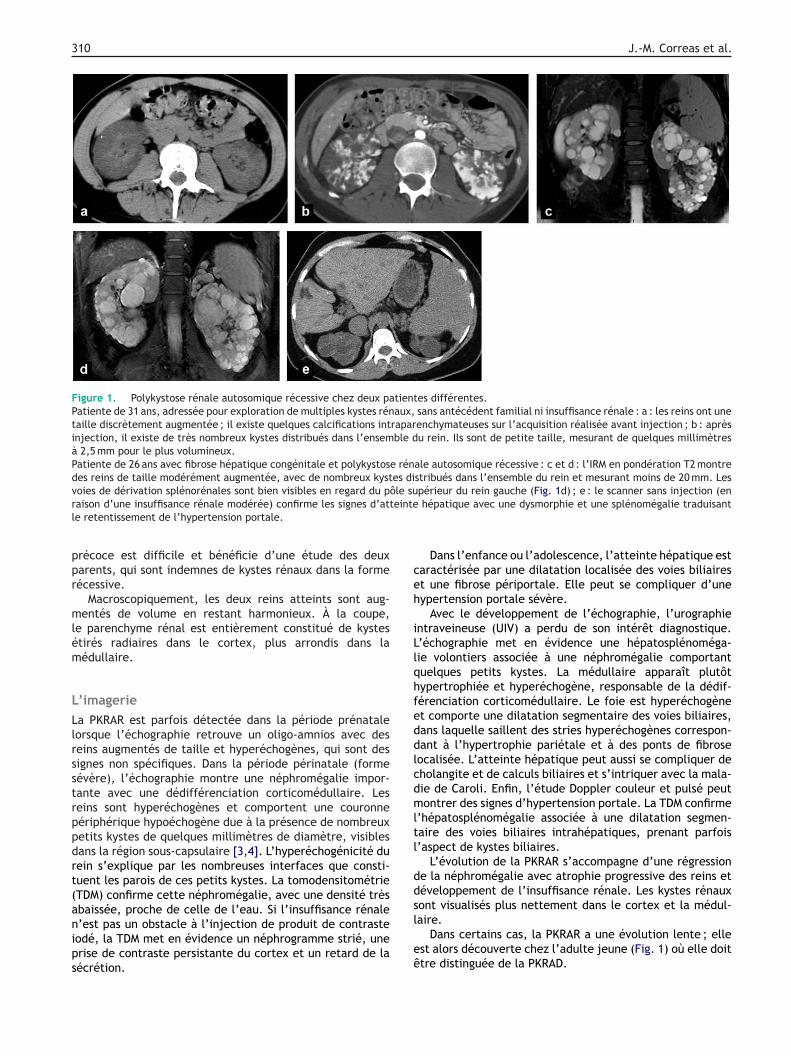
310 J.-M. Correas et al.
Figure 1. Polykystose rénale autosomique récessive chez deux patientes différentes.Patiente de 31 ans, adressée pour exploration de multiples kystes rénaux, sans antécédent familial ni insuffisance rénale : a : les reins ont unetaille discrètement augmentée ; il existe quelques calcifications intraparenchymateuses sur l’acquisition réalisée avant injection ; b : aprèsinjection, il existe de très nombreux kystes distribués dans l’ensemble du rein. Ils sont de petite taille, mesurant de quelques millimètresà 2,5 mm pour le plus volumineux.Patiente de 26 ans avec fibrose hépatique congénitale et polykystose rénale autosomique récessive : c et d : l’IRM en pondération T2 montredes reins de taille modérément augmentée, avec de nombreux kystes distribués dans l’ensemble du rein et mesurant moins de 20 mm. Lesvoies de dérivation splénorénales sont bien visibles en regard du pôle supérieur du rein gauche (Fig. 1d) ; e : le scanner sans injection (enr eintel
ppr
mlém
LLlrsstrppdrt(anips
ceh
iLlqhfeddlcdmltl
dds
aison d’une insuffisance rénale modérée) confirme les signes d’atte retentissement de l’hypertension portale.
récoce est difficile et bénéficie d’une étude des deuxarents, qui sont indemnes de kystes rénaux dans la formeécessive.
Macroscopiquement, les deux reins atteints sont aug-entés de volume en restant harmonieux. À la coupe,
e parenchyme rénal est entièrement constitué de kystestirés radiaires dans le cortex, plus arrondis dans laédullaire.
’imageriea PKRAR est parfois détectée dans la période prénataleorsque l’échographie retrouve un oligo-amnios avec deseins augmentés de taille et hyperéchogènes, qui sont designes non spécifiques. Dans la période périnatale (formeévère), l’échographie montre une néphromégalie impor-ante avec une dédifférenciation corticomédullaire. Leseins sont hyperéchogènes et comportent une couronneériphérique hypoéchogène due à la présence de nombreuxetits kystes de quelques millimètres de diamètre, visiblesans la région sous-capsulaire [3,4]. L’hyperéchogénicité duein s’explique par les nombreuses interfaces que consti-uent les parois de ces petits kystes. La tomodensitométrieTDM) confirme cette néphromégalie, avec une densité trèsbaissée, proche de celle de l’eau. Si l’insuffisance rénale
’est pas un obstacle à l’injection de produit de contrasteodé, la TDM met en évidence un néphrogramme strié, unerise de contraste persistante du cortex et un retard de laécrétion.l
eê
hépatique avec une dysmorphie et une splénomégalie traduisant
Dans l’enfance ou l’adolescence, l’atteinte hépatique estaractérisée par une dilatation localisée des voies biliairest une fibrose périportale. Elle peut se compliquer d’uneypertension portale sévère.
Avec le développement de l’échographie, l’urographientraveineuse (UIV) a perdu de son intérêt diagnostique.’échographie met en évidence une hépatosplénoméga-ie volontiers associée à une néphromégalie comportantuelques petits kystes. La médullaire apparaît plutôtypertrophiée et hyperéchogène, responsable de la dédif-érenciation corticomédullaire. Le foie est hyperéchogènet comporte une dilatation segmentaire des voies biliaires,ans laquelle saillent des stries hyperéchogènes correspon-ant à l’hypertrophie pariétale et à des ponts de fibroseocalisée. L’atteinte hépatique peut aussi se compliquer deholangite et de calculs biliaires et s’intriquer avec la mala-ie de Caroli. Enfin, l’étude Doppler couleur et pulsé peutontrer des signes d’hypertension portale. La TDM confirme
’hépatosplénomégalie associée à une dilatation segmen-aire des voies biliaires intrahépatiques, prenant parfois’aspect de kystes biliaires.
L’évolution de la PKRAR s’accompagne d’une régressione la néphromégalie avec atrophie progressive des reins etéveloppement de l’insuffisance rénale. Les kystes rénauxont visualisés plus nettement dans le cortex et la médul-
aire.Dans certains cas, la PKRAR a une évolution lente ; ellest alors découverte chez l’adulte jeune (Fig. 1) où elle doittre distinguée de la PKRAD.

Insuffisance rénale et maladies kystiques du rein 311
Figure 2. Polykystose rénale autosomique dominante débutante chez un patient de 35 ans, avec fonction rénale conservée. Les reins sontde grande taille ; il existe de nombreux kystes distribués dans l’ensemble du parenchyme, de taille variable mais dépassant pour les plusvolumineux 30 mm.
Figure 3. Polykystose rénale autosomique dominante de type PKD1 chez un patient de 51 ans, avec insuffisance rénale modérée : a : lescanner sans injection montre la néphromégalie, les nombreux kystes hypodenses, des calcifications intraparenchymateuses et un calcul
tiongauch
eddeta
LLkho
rrlàtt(
dans un calice du tiers moyen du rein gauche ; b : l’IRM en pondéraatypiques de signal très augmenté ou de signal intermédiaire (rein
La polykystose rénale autosomique dominante(PKRAD)
Quelques rappels anatomocliniquesLa PKRAD se caractérise le plus souvent chez l’adulte jeunepar la présence de multiples kystes rénaux associés à deskystes hépatiques et plus rarement à des anomalies car-diovasculaires (anévrismes intracérébraux, valvulopathies).Il s’agit d’une maladie héréditaire autosomique dominantefréquente (prévalence 1/1000) à expression variable, liéedans la très grande majorité des cas au gène PKD1 (chromo-some 16 dans environ 85 % des cas) et dans les autres cas augène PKD2 (chromosome 4) [5,6]. La fréquence des PKRADdue à une néomutation est faible (< 5 %) et l’interrogatoirerévèle une histoire familiale de maladie kystique rénale dansla majorité des cas.
L’insuffisance rénale terminale est la complication la plussévère et touche environ 80 % des patients avant 70 ans[7]. La PKRAD représente la quatrième cause d’insuffisancerénale chronique et environ 10 % des patients en hémodia-lyse chronique [8]. La fréquence du cancer du rein à cellulesclaires n’est pas augmentée par rapport à la population
générale [9,10].Macroscopiquement, les reins gardent leurs contours etils sont très volumineux, mesurant jusqu’à 30 cm de hauteuret pesant jusqu’à 6 kg. À la coupe, tout le parenchyme rénal
clkl
T1 confirme la présence d’une néphromégalie et révèle des kystese) ; c : en pondération T2, l’aspect est typique de PKAD.
st remplacé par des kystes de quelques millimètres à 5 cme diamètre. Ces kystes uniloculaires, à paroi fine, arron-is, sont répartis dans la corticale et la médullaire. Ils sontmplis d’un liquide clair, filant, parfois hémorragique, géla-ineux ou surinfecté. Les tiges pyélocalicielles sont étirées,llongées, mais elles restent harmonieuses.
’imagerie de l’atteinte rénalea séméiologie radiologique rénale est liée à la présence desystes et à leurs complications (hémorragie intrakystique,ématurie, infection kystique, calculs d’oxalate de calciumu d’acide urique, colique néphrétique).
L’aspect des kystes non compliqués est sans particula-ité (Fig. 2). Néanmoins, leur nombre et leur taille sontesponsables d’une néphromégalie parfois majeure lorsquea maladie est évoluée (hauteur rénale parfois supérieure20 cm). Les kystes sont principalement développés à par-
ir du cortex rénal et sont le plus souvent bilatéraux et deaille variable, bien que l’atteinte puisse être asymétriqueFig. 3 et 4) [11].
Sur les clichés d’abdomen sans préparation, inutiles dans
ette indication, plusieurs syndromes de masse déformentes contours des reins qui sont augmentés de taille. Lesystes peuvent effacer les contours du psoas ou même refou-er les structures digestives lorsqu’ils sont très volumineux.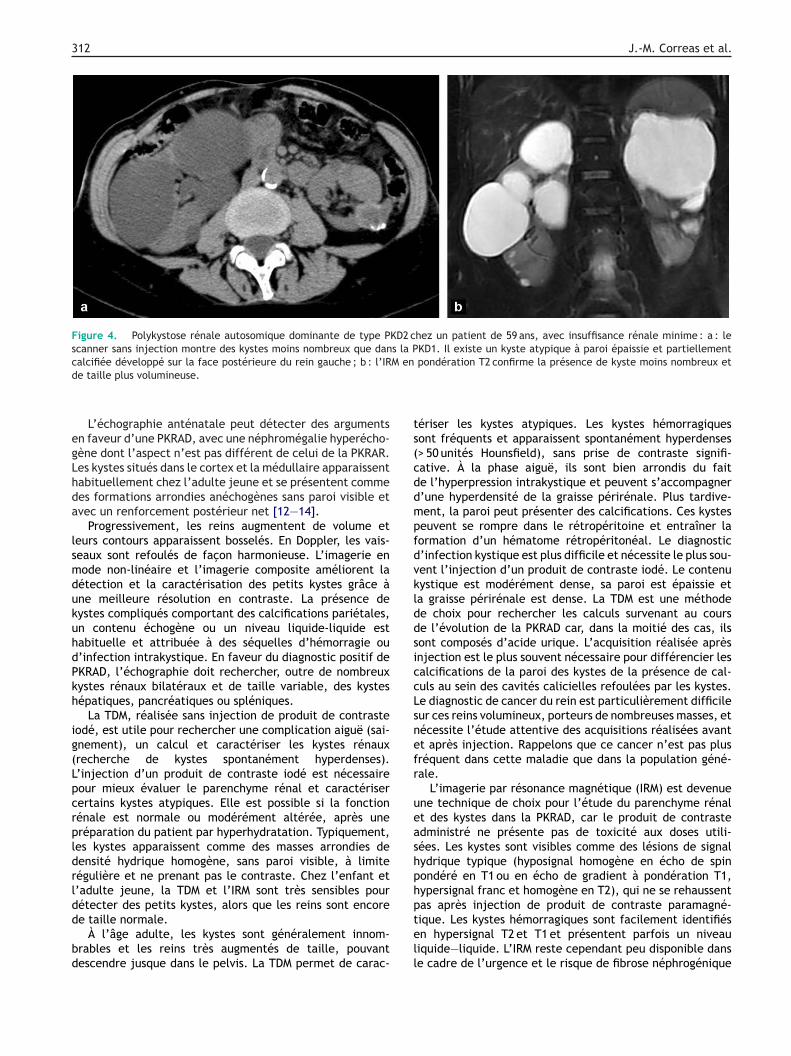
312 J.-M. Correas et al.
Figure 4. Polykystose rénale autosomique dominante de type PKD2 chez un patient de 59 ans, avec insuffisance rénale minime : a : lescanner sans injection montre des kystes moins nombreux que dans la PKD1. Il existe un kyste atypique à paroi épaissie et partiellementcalcifiée développé sur la face postérieure du rein gauche ; b : l’IRM en pondération T2 confirme la présence de kyste moins nombreux etd
egLhda
lsmdukuhdPkh
ig(Lpcrpldrldd
bd
ts(cddmpfdvklddsiccLsnefr
ueashphp
e taille plus volumineuse.
L’échographie anténatale peut détecter des argumentsn faveur d’une PKRAD, avec une néphromégalie hyperécho-ène dont l’aspect n’est pas différent de celui de la PKRAR.es kystes situés dans le cortex et la médullaire apparaissentabituellement chez l’adulte jeune et se présentent commees formations arrondies anéchogènes sans paroi visible etvec un renforcement postérieur net [12—14].
Progressivement, les reins augmentent de volume eteurs contours apparaissent bosselés. En Doppler, les vais-eaux sont refoulés de facon harmonieuse. L’imagerie enode non-linéaire et l’imagerie composite améliorent laétection et la caractérisation des petits kystes grâce àne meilleure résolution en contraste. La présence deystes compliqués comportant des calcifications pariétales,n contenu échogène ou un niveau liquide-liquide estabituelle et attribuée à des séquelles d’hémorragie ou’infection intrakystique. En faveur du diagnostic positif deKRAD, l’échographie doit rechercher, outre de nombreuxystes rénaux bilatéraux et de taille variable, des kystesépatiques, pancréatiques ou spléniques.
La TDM, réalisée sans injection de produit de contrasteodé, est utile pour rechercher une complication aiguë (sai-nement), un calcul et caractériser les kystes rénauxrecherche de kystes spontanément hyperdenses).’injection d’un produit de contraste iodé est nécessaireour mieux évaluer le parenchyme rénal et caractériserertains kystes atypiques. Elle est possible si la fonctionénale est normale ou modérément altérée, après uneréparation du patient par hyperhydratation. Typiquement,es kystes apparaissent comme des masses arrondies deensité hydrique homogène, sans paroi visible, à limiteégulière et ne prenant pas le contraste. Chez l’enfant et’adulte jeune, la TDM et l’IRM sont très sensibles pourétecter des petits kystes, alors que les reins sont encore
e taille normale.À l’âge adulte, les kystes sont généralement innom-rables et les reins très augmentés de taille, pouvantescendre jusque dans le pelvis. La TDM permet de carac-
tell
ériser les kystes atypiques. Les kystes hémorragiquesont fréquents et apparaissent spontanément hyperdenses> 50 unités Hounsfield), sans prise de contraste signifi-ative. À la phase aiguë, ils sont bien arrondis du faite l’hyperpression intrakystique et peuvent s’accompagner’une hyperdensité de la graisse périrénale. Plus tardive-ent, la paroi peut présenter des calcifications. Ces kysteseuvent se rompre dans le rétropéritoine et entraîner laormation d’un hématome rétropéritonéal. Le diagnostic’infection kystique est plus difficile et nécessite le plus sou-ent l’injection d’un produit de contraste iodé. Le contenuystique est modérément dense, sa paroi est épaissie eta graisse périrénale est dense. La TDM est une méthodee choix pour rechercher les calculs survenant au course l’évolution de la PKRAD car, dans la moitié des cas, ilsont composés d’acide urique. L’acquisition réalisée aprèsnjection est le plus souvent nécessaire pour différencier lesalcifications de la paroi des kystes de la présence de cal-uls au sein des cavités calicielles refoulées par les kystes.e diagnostic de cancer du rein est particulièrement difficileur ces reins volumineux, porteurs de nombreuses masses, etécessite l’étude attentive des acquisitions réalisées avantt après injection. Rappelons que ce cancer n’est pas plusréquent dans cette maladie que dans la population géné-ale.
L’imagerie par résonance magnétique (IRM) est devenuene technique de choix pour l’étude du parenchyme rénalt des kystes dans la PKRAD, car le produit de contrastedministré ne présente pas de toxicité aux doses utili-ées. Les kystes sont visibles comme des lésions de signalydrique typique (hyposignal homogène en écho de spinondéré en T1 ou en écho de gradient à pondération T1,ypersignal franc et homogène en T2), qui ne se rehaussentas après injection de produit de contraste paramagné-
ique. Les kystes hémorragiques sont facilement identifiésn hypersignal T2 et T1 et présentent parfois un niveauiquide—liquide. L’IRM reste cependant peu disponible danse cadre de l’urgence et le risque de fibrose néphrogénique
LLtldddndpcdsdprnsdaêdlT
LDldtpbcprcladirannr[
LrLnt42tà•
Insuffisance rénale et maladies kystiques du rein
systémique liée à l’injection de chélates de Gadolinium doitfaire discuter la nécessité de réaliser une injection de pro-duit de contraste.
Le diagnostic des complications rénalesL’échographie est un examen essentiel qui participe audiagnostic précoce des complications, dont la correctionpermet d’éviter l’accélération de la détérioration de lafonction rénale. Par rapport à la TDM, elle ne nécessite pasd’injection de produit de contraste iodé potentiellementnéphrotoxique.
En cas de douleurs lombaires, c’est le premier exa-men prescrit avec le cliché d’abdomen sans préparation.La présence de calcifications du parenchyme ou des paroiskystiques est assez banale. Il est souvent difficile de dif-férencier les calcifications parenchymateuses des calculslorsque la maladie est déjà évoluée. Les diagnostics sui-vants doivent être discutés : l’hémorragie intrakystique,l’infection kystique, la pyélonéphrite aiguë et la coliquenéphrétique.
Les kystes remaniésLa présence d’un kyste atypique hypoéchogène hétérogèneà paroi épaissie, voire calcifiée, est compatible avec lesdiagnostics de saignement ou d’infection intrakystique. Àla phase aiguë, on peut même identifier un aspect de sédi-ment déclive non spécifique, correspondant à des produitsde dégradation de la fibrine en cas de saignement ancien,ou de pus en cas d’infection. L’hémorragie intrakystiquepeut s’accompagner d’un hématome sous-capsulaire, voirepérirénal ; il témoigne alors d’une rupture du kyste. Lacommunication avec la voie excrétrice peut entraîner unehématurie macroscopique avec un caillotage des cavitéspyélocalicielles et un syndrome obstructif aigu. La TDM encoupes fines sans injection de produit de contraste permetgénéralement de faire le bilan de ce saignement. Les kysteshémorragiques sont spontanément hyperdenses à la phaseaiguë. Avec le temps, la densité de ces lésions diminue etdes calcifications des parois ou du contenu peuvent appa-raître. L’IRM a comme avantage d’étudier le parenchyme etles kystes atypiques avec injection d’un produit de contrastenon néphrotoxique.
Les infections kystiquesLe diagnostic d’infection kystique est surtout clinique etrepose sur l’association d’une douleur lombaire et d’unefièvre. L’échographie peut mettre en évidence un kysteatypique dense, avec de facon très inconstante un niveauliquidien. La TDM confirme l’existence d’un kyste densedont la paroi est volontiers épaissie et prend le contraste.L’épaississement du fascia de Gérota ou une densification dela graisse sont des signes inconstants et non spécifiques. Laprésence de bulles gazeuses, en l’absence de manœuvresurologiques rétrogrades, est rare mais spécifique du diag-nostic. Parfois, l’infection s’étend à l’espace périrénal. Letraitement, outre les antibiotiques, peut en cas d’échecrequérir un drainage écho- ou scanoguidé. Le PET-scan peut
permettre d’identifier un ou plusieurs kystes qui sont lesiège de remaniements inflammatoires et orienter la priseen charge thérapeutique en cas de résistance au traitementantibiotique.•
313
es douleurs lombairesa douleur lombaire, ou plutôt une sensation de pesan-eur, peut également être liée à la taille des kystesorsqu’ils sont très volumineux. Il s’agit d’un diagnostic’exclusion devant un bilan négatif (absence de fièvre,’hématurie, d’infection urinaire, de kyste compliqué oue dilatation pyélocalicielle à l’échographie ou au scan-er sans injection, voire parfois injecté). La présence’images anéchogènes dans le sinus du rein ne témoigneas nécessairement d’une dilatation des cavités pyélocali-ielles. En effet, le diagnostic échographique de dilatationes cavités pyélocalicielles est souvent difficile en pré-ence d’une PKRAD avancée. Il faut rechercher la présencee structures calicielles tubulaires ramifiées. Les kystesarapyéliques ou rénaux confluents dans le sinus peuventéaliser un aspect trompeur. Le diagnostic de coliqueéphrétique est alors posé essentiellement sur la TDMans injection de produit de contraste qui recherche uneilatation des cavités pyélocalicielles et de l’uretère enmont de l’obstacle lithiasique hyperdense. L’UIV peuttre utile lorsque la fonction rénale est peu dégra-ée. Elle est, en fait, de plus en plus remplacée par’uro-TDM, où des clichés de l’abdomen font suite à laDM.
e diagnostic des manifestations extrarénaleses kystes sont observés dans le foie (57 % des patients),
e pancréas (5 %), la rate (5 %) et plus exceptionnellementans l’épididyme, les vésicules séminales, les ovaires, lahyroïde ou le colon (diverticulose). L’atteinte hépatiqueeut entraîner une compression de l’estomac, des voiesiliaires intrahépatiques, des veines hépatiques, de la veineave inférieure, voire de l’oreillette et du ventricule droits,ouvant aboutir à une insuffisance cardiaque par défaut deetour veineux ou par défaut de remplissage des cavitésardiaques. L’angio-IRM est une technique de choix poure diagnostic de ces complications vasculaires. Les autrestteintes incluent le prolapsus mitral ou plus rarement celui’une autre valve cardiaque. La fréquence des anévrismesntracérébraux est plus élevée que dans la population géné-ale. Son dépistage par une angio-IRM chez les patientssymptomatiques apparaît légitime uniquement en cas deotion de rupture anévrismale dans la famille ou de signeeurologique évocateur. Un traitement prophylactique chi-urgical ou par voie endovasculaire peut être alors proposé15].
e diagnostic de la PKRAD chez les patients àisquea présence de plusieurs kystes rénaux chez un adulte jeunee fait pas le diagnostic de PKRAD. En effet, on peut détec-er au moins un kyste rénal chez 2 % des sujets âgés de 30 à9 ans, chez 11 % des sujets âgés de 50 à 70 ans, et chez2 % chez les sujets âgés de plus de 70 ans. Selon les cri-ères de Ravine, les arguments en faveur d’une PKRAD liéePKD1 sont les suivants [14] :le contexte familial de néphropathie à transmission auto-
somique dominante ;le nombre des kystes, avec avant 30 ans, la présence d’aumoins deux kystes dans l’un ou les deux reins, entre 30 et59 ans, la présence d’au moins deux kystes dans chaque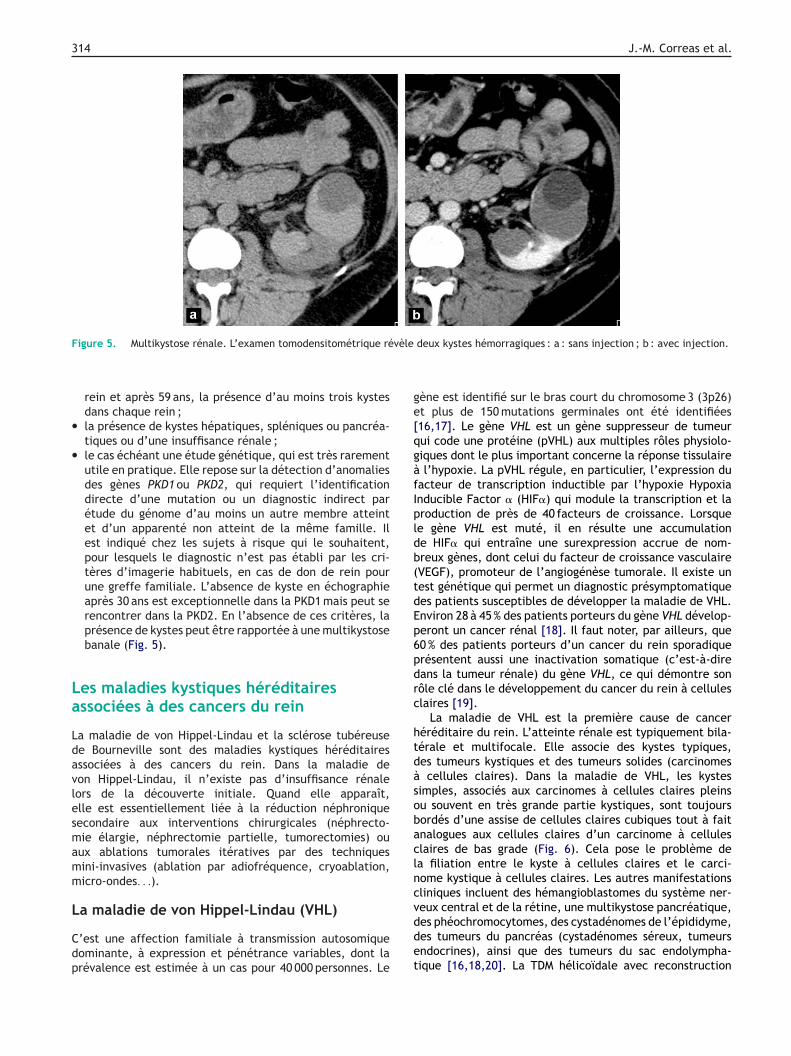
314 J.-M. Correas et al.
F vèle
•
•
La
Ldavlesmamm
L
Cdp
ge[qgàfIpldb(tdEp6pdrc
htdàsobaclncv
igure 5. Multikystose rénale. L’examen tomodensitométrique ré
rein et après 59 ans, la présence d’au moins trois kystesdans chaque rein ;la présence de kystes hépatiques, spléniques ou pancréa-tiques ou d’une insuffisance rénale ;le cas échéant une étude génétique, qui est très rarementutile en pratique. Elle repose sur la détection d’anomaliesdes gènes PKD1 ou PKD2, qui requiert l’identificationdirecte d’une mutation ou un diagnostic indirect parétude du génome d’au moins un autre membre atteintet d’un apparenté non atteint de la même famille. Ilest indiqué chez les sujets à risque qui le souhaitent,pour lesquels le diagnostic n’est pas établi par les cri-tères d’imagerie habituels, en cas de don de rein pourune greffe familiale. L’absence de kyste en échographieaprès 30 ans est exceptionnelle dans la PKD1 mais peut serencontrer dans la PKD2. En l’absence de ces critères, laprésence de kystes peut être rapportée à une multikystosebanale (Fig. 5).
es maladies kystiques héréditairesssociées à des cancers du rein
a maladie de von Hippel-Lindau et la sclérose tubéreusee Bourneville sont des maladies kystiques héréditairesssociées à des cancers du rein. Dans la maladie deon Hippel-Lindau, il n’existe pas d’insuffisance rénaleors de la découverte initiale. Quand elle apparaît,lle est essentiellement liée à la réduction néphroniqueecondaire aux interventions chirurgicales (néphrecto-ie élargie, néphrectomie partielle, tumorectomies) ou
ux ablations tumorales itératives par des techniquesini-invasives (ablation par adiofréquence, cryoablation,icro-ondes. . .).
a maladie de von Hippel-Lindau (VHL)
’est une affection familiale à transmission autosomiqueominante, à expression et pénétrance variables, dont larévalence est estimée à un cas pour 40 000 personnes. Le
ddet
deux kystes hémorragiques : a : sans injection ; b : avec injection.
ène est identifié sur le bras court du chromosome 3 (3p26)t plus de 150 mutations germinales ont été identifiées16,17]. Le gène VHL est un gène suppresseur de tumeurui code une protéine (pVHL) aux multiples rôles physiolo-iques dont le plus important concerne la réponse tissulairel’hypoxie. La pVHL régule, en particulier, l’expression du
acteur de transcription inductible par l’hypoxie Hypoxianducible Factor � (HIF�) qui module la transcription et laroduction de près de 40 facteurs de croissance. Lorsquee gène VHL est muté, il en résulte une accumulatione HIF� qui entraîne une surexpression accrue de nom-reux gènes, dont celui du facteur de croissance vasculaireVEGF), promoteur de l’angiogénèse tumorale. Il existe unest génétique qui permet un diagnostic présymptomatiquees patients susceptibles de développer la maladie de VHL.nviron 28 à 45 % des patients porteurs du gène VHL dévelop-eront un cancer rénal [18]. Il faut noter, par ailleurs, que0 % des patients porteurs d’un cancer du rein sporadiquerésentent aussi une inactivation somatique (c’est-à-direans la tumeur rénale) du gène VHL, ce qui démontre sonôle clé dans le développement du cancer du rein à celluleslaires [19].
La maladie de VHL est la première cause de canceréréditaire du rein. L’atteinte rénale est typiquement bila-érale et multifocale. Elle associe des kystes typiques,es tumeurs kystiques et des tumeurs solides (carcinomes
cellules claires). Dans la maladie de VHL, les kystesimples, associés aux carcinomes à cellules claires pleinsu souvent en très grande partie kystiques, sont toujoursordés d’une assise de cellules claires cubiques tout à faitnalogues aux cellules claires d’un carcinome à celluleslaires de bas grade (Fig. 6). Cela pose le problème dea filiation entre le kyste à cellules claires et le carci-ome kystique à cellules claires. Les autres manifestationsliniques incluent des hémangioblastomes du système ner-eux central et de la rétine, une multikystose pancréatique,
es phéochromocytomes, des cystadénomes de l’épididyme,es tumeurs du pancréas (cystadénomes séreux, tumeursndocrines), ainsi que des tumeurs du sac endolympha-ique [16,18,20]. La TDM hélicoïdale avec reconstruction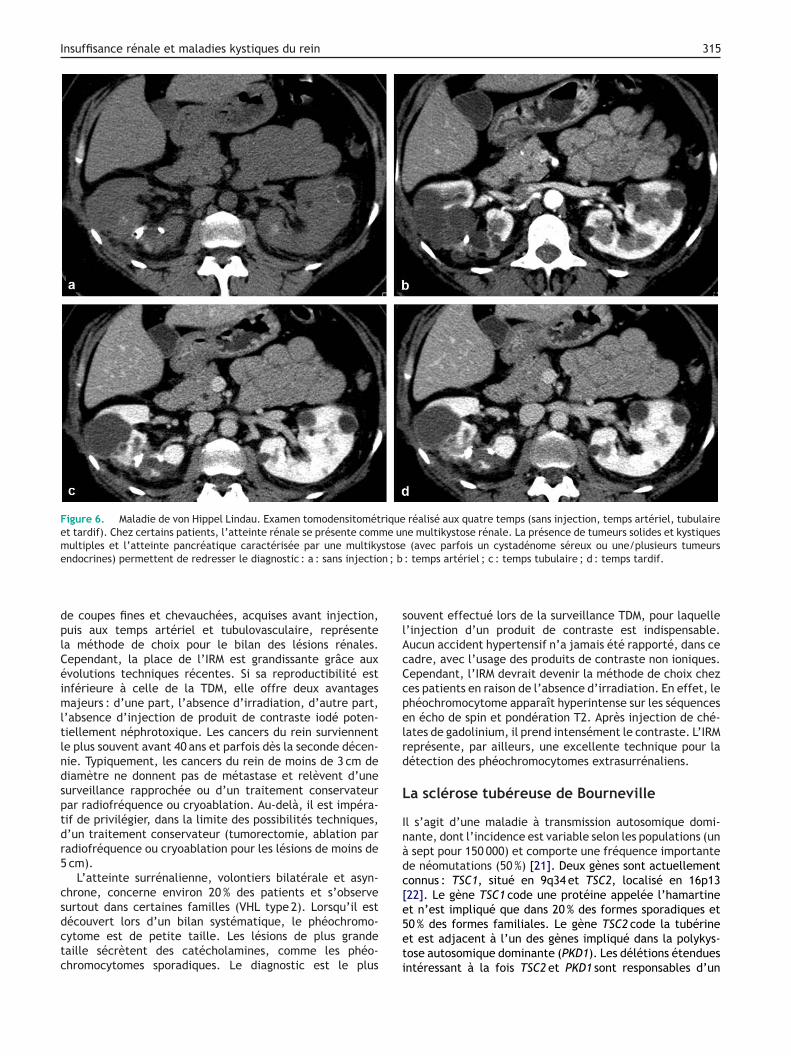
Insuffisance rénale et maladies kystiques du rein 315
Figure 6. Maladie de von Hippel Lindau. Examen tomodensitométrique réalisé aux quatre temps (sans injection, temps artériel, tubulaireet tardif). Chez certains patients, l’atteinte rénale se présente comme une multikystose rénale. La présence de tumeurs solides et kystiquesmultiples et l’atteinte pancréatique caractérisée par une multikystose (avec parfois un cystadénome séreux ou une/plusieurs tumeurs
n ; b
slAcCcpelrd
L
Inàdc[e
endocrines) permettent de redresser le diagnostic : a : sans injectio
de coupes fines et chevauchées, acquises avant injection,puis aux temps artériel et tubulovasculaire, représentela méthode de choix pour le bilan des lésions rénales.Cependant, la place de l’IRM est grandissante grâce auxévolutions techniques récentes. Si sa reproductibilité estinférieure à celle de la TDM, elle offre deux avantagesmajeurs : d’une part, l’absence d’irradiation, d’autre part,l’absence d’injection de produit de contraste iodé poten-tiellement néphrotoxique. Les cancers du rein surviennentle plus souvent avant 40 ans et parfois dès la seconde décen-nie. Typiquement, les cancers du rein de moins de 3 cm dediamètre ne donnent pas de métastase et relèvent d’unesurveillance rapprochée ou d’un traitement conservateurpar radiofréquence ou cryoablation. Au-delà, il est impéra-tif de privilégier, dans la limite des possibilités techniques,d’un traitement conservateur (tumorectomie, ablation parradiofréquence ou cryoablation pour les lésions de moins de5 cm).
L’atteinte surrénalienne, volontiers bilatérale et asyn-chrone, concerne environ 20 % des patients et s’observesurtout dans certaines familles (VHL type 2). Lorsqu’il est
découvert lors d’un bilan systématique, le phéochromo-cytome est de petite taille. Les lésions de plus grandetaille sécrètent des catécholamines, comme les phéo-chromocytomes sporadiques. Le diagnostic est le plus5eti
: temps artériel ; c : temps tubulaire ; d : temps tardif.
ouvent effectué lors de la surveillance TDM, pour laquelle’injection d’un produit de contraste est indispensable.ucun accident hypertensif n’a jamais été rapporté, dans ceadre, avec l’usage des produits de contraste non ioniques.ependant, l’IRM devrait devenir la méthode de choix chezes patients en raison de l’absence d’irradiation. En effet, lehéochromocytome apparaît hyperintense sur les séquencesn écho de spin et pondération T2. Après injection de ché-ates de gadolinium, il prend intensément le contraste. L’IRMeprésente, par ailleurs, une excellente technique pour laétection des phéochromocytomes extrasurrénaliens.
a sclérose tubéreuse de Bourneville
l s’agit d’une maladie à transmission autosomique domi-ante, dont l’incidence est variable selon les populations (unsept pour 150 000) et comporte une fréquence importantee néomutations (50 %) [21]. Deux gènes sont actuellementonnus : TSC1, situé en 9q34 et TSC2, localisé en 16p1322]. Le gène TSC1 code une protéine appelée l’hamartinet n’est impliqué que dans 20 % des formes sporadiques et
0 % des formes familiales. Le gène TSC2 code la tubérinet est adjacent à l’un des gènes impliqué dans la polykys-ose autosomique dominante (PKD1). Les délétions étenduesntéressant à la fois TSC2 et PKD1 sont responsables d’un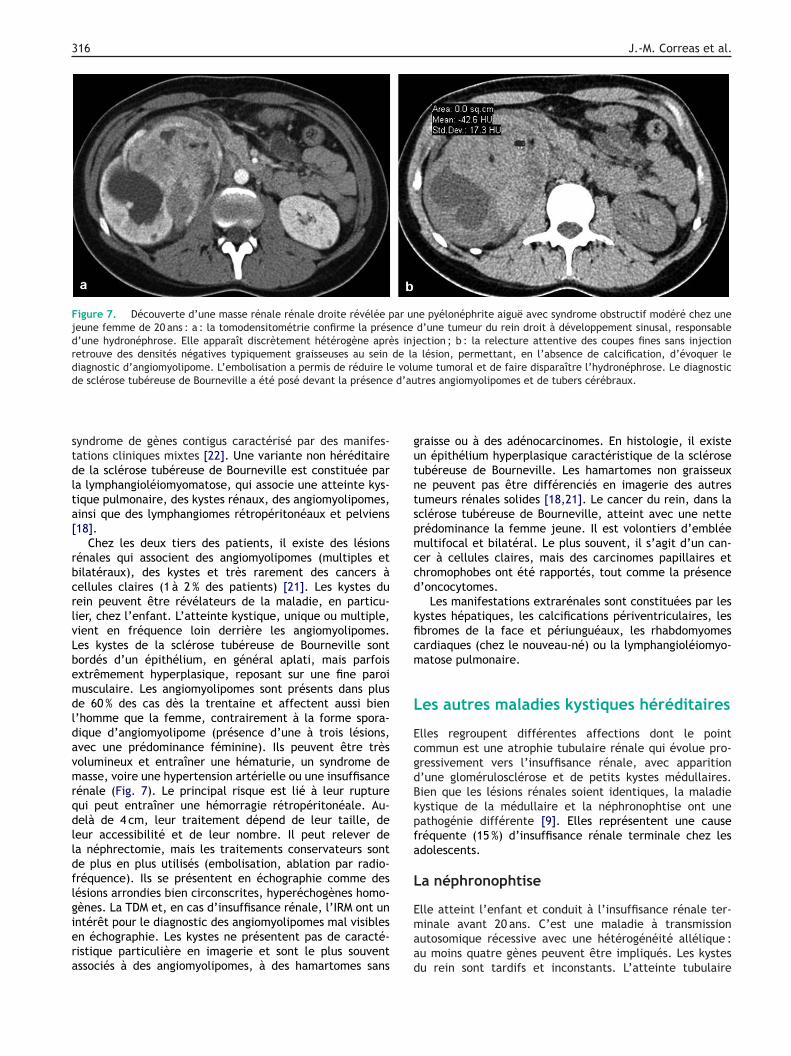
316 J.-M. Correas et al.
Figure 7. Découverte d’une masse rénale rénale droite révélée par une pyélonéphrite aiguë avec syndrome obstructif modéré chez unejeune femme de 20 ans : a : la tomodensitométrie confirme la présence d’une tumeur du rein droit à développement sinusal, responsabled’une hydronéphrose. Elle apparaît discrètement hétérogène après injection ; b : la relecture attentive des coupes fines sans injectionretrouve des densités négatives typiquement graisseuses au sein de la lésion, permettant, en l’absence de calcification, d’évoquer lediagnostic d’angiomyolipome. L’embolisation a permis de réduire le volume tumoral et de faire disparaître l’hydronéphrose. Le diagnosticde sclérose tubéreuse de Bourneville a été posé devant la présence d’autres angiomyolipomes et de tubers cérébraux.
stdlta[
rbcrlvLbemdldavmrqdlldflgiera
gutntspmccd
kficm
L
EcgdBkpfa
L
E
yndrome de gènes contigus caractérisé par des manifes-ations cliniques mixtes [22]. Une variante non héréditairee la sclérose tubéreuse de Bourneville est constituée para lymphangioléiomyomatose, qui associe une atteinte kys-ique pulmonaire, des kystes rénaux, des angiomyolipomes,insi que des lymphangiomes rétropéritonéaux et pelviens18].
Chez les deux tiers des patients, il existe des lésionsénales qui associent des angiomyolipomes (multiples etilatéraux), des kystes et très rarement des cancers àellules claires (1 à 2 % des patients) [21]. Les kystes duein peuvent être révélateurs de la maladie, en particu-ier, chez l’enfant. L’atteinte kystique, unique ou multiple,ient en fréquence loin derrière les angiomyolipomes.es kystes de la sclérose tubéreuse de Bourneville sontordés d’un épithélium, en général aplati, mais parfoisxtrêmement hyperplasique, reposant sur une fine paroiusculaire. Les angiomyolipomes sont présents dans pluse 60 % des cas dès la trentaine et affectent aussi bien’homme que la femme, contrairement à la forme spora-ique d’angiomyolipome (présence d’une à trois lésions,vec une prédominance féminine). Ils peuvent être trèsolumineux et entraîner une hématurie, un syndrome deasse, voire une hypertension artérielle ou une insuffisance
énale (Fig. 7). Le principal risque est lié à leur ruptureui peut entraîner une hémorragie rétropéritonéale. Au-elà de 4 cm, leur traitement dépend de leur taille, deeur accessibilité et de leur nombre. Il peut relever dea néphrectomie, mais les traitements conservateurs sonte plus en plus utilisés (embolisation, ablation par radio-réquence). Ils se présentent en échographie comme desésions arrondies bien circonscrites, hyperéchogènes homo-ènes. La TDM et, en cas d’insuffisance rénale, l’IRM ont un
ntérêt pour le diagnostic des angiomyolipomes mal visiblesn échographie. Les kystes ne présentent pas de caracté-istique particulière en imagerie et sont le plus souventssociés à des angiomyolipomes, à des hamartomes sansmaad
raisse ou à des adénocarcinomes. En histologie, il existen épithélium hyperplasique caractéristique de la scléroseubéreuse de Bourneville. Les hamartomes non graisseuxe peuvent pas être différenciés en imagerie des autresumeurs rénales solides [18,21]. Le cancer du rein, dans laclérose tubéreuse de Bourneville, atteint avec une netterédominance la femme jeune. Il est volontiers d’embléeultifocal et bilatéral. Le plus souvent, il s’agit d’un can-
er à cellules claires, mais des carcinomes papillaires ethromophobes ont été rapportés, tout comme la présence’oncocytomes.
Les manifestations extrarénales sont constituées par lesystes hépatiques, les calcifications périventriculaires, lesbromes de la face et périunguéaux, les rhabdomyomesardiaques (chez le nouveau-né) ou la lymphangioléiomyo-atose pulmonaire.
es autres maladies kystiques héréditaires
lles regroupent différentes affections dont le pointommun est une atrophie tubulaire rénale qui évolue pro-ressivement vers l’insuffisance rénale, avec apparition’une glomérulosclérose et de petits kystes médullaires.ien que les lésions rénales soient identiques, la maladieystique de la médullaire et la néphronophtise ont uneathogénie différente [9]. Elles représentent une causeréquente (15 %) d’insuffisance rénale terminale chez lesdolescents.
a néphronophtise
lle atteint l’enfant et conduit à l’insuffisance rénale ter-
inale avant 20 ans. C’est une maladie à transmissionutosomique récessive avec une hétérogénéité allélique :u moins quatre gènes peuvent être impliqués. Les kystesu rein sont tardifs et inconstants. L’atteinte tubulaire

Insuffisance rénale et maladies kystiques du rein 317
Figure 8. La maladie kystique de la médullaire—aspect en IRM : a et b : les acquisitions en écho de spin pondéré T2 (a) et en écho degradient pondéré T1 (b) après injection de chélates de gadolinium mettent en évidence de multiples kystes typiques, de taille centimétrique,presque exclusivement situés dans la médullaire rénale.
hp
mdcefi
ccrttepde(
led
llahpnskg
L
est responsable d’un syndrome polyuropolydypsique, d’uneénurésie secondaire, d’un retard de croissance, d’une fuiteobligatoire en sodium et d’une insuffisance rénale qui abou-tit à la dialyse.
Macroscopiquement, les reins sont de tailleréduite, jusqu’à la moitié de la normale, fermeset pâles. La corticale et la médullaire sont amin-cies, plusieurs petits kystes infracentimétriques àparoi fine peuvent s’observer à la jonction corti-comédullaire, mais ils n’existent que chez 75 % despatients.
Les atteintes extrarénales peuvent associer une rétinitepigmentaire, une dégénérescence tapétorétinienne bilaté-rale ou d’autres anomalies oculaires (colobome, nystagmus,myopie, strabisme, amblyopie, atrophie du nerf optique),voire une fibrose hépatique congénitale, une dysplasie dusquelette ou un retard mental.
La maladie kystique de la médullaire
Maladie à transmission autosomique dominante, elle secaractérise par la présence de kystes localisés à la médul-laire rénale (Fig. 8). Elle s’observe chez l’adulte jeune (25 à30 ans) et ne s’accompagne pas de manifestation extraré-nale. Elle aboutit plus tardivement à l’insuffisance rénaleterminale (25—40 ans).
Les affections non héréditaires
La dysplasie rénale multikystique (ou aplasie,dysplasie, dysgénésie rénale)
On regroupe sous cette dénomination les anomalies dudéveloppement du rein secondaires à un défaut de perméa-bilité pyélique ou urétérale, aboutissant à la formation de
multiples kystes de taille variable et à la destruction duparenchyme rénal. L’atteinte est le plus souvent unilatéraleet touche l’ensemble du rein. Cependant, certaines formeslocalisées ou bilatérales ont été rapportées et des formesL4dl
éréditaires commencent à être dénombrées, par exemplear mutation de HNF-1B.
En histologie, le rein est volumineux, transformé en uneasse multikystique, avec de multiples kystes périphériquese grande taille (4 à 5 cm de diamètre) et un noyau fibreuxentral. Il n’y a aucune connexion entre les kystes et la voiexcrétrice qui elle-même est atrésique au sein du noyaubreux central, avec un uretère atrésique ou absent.
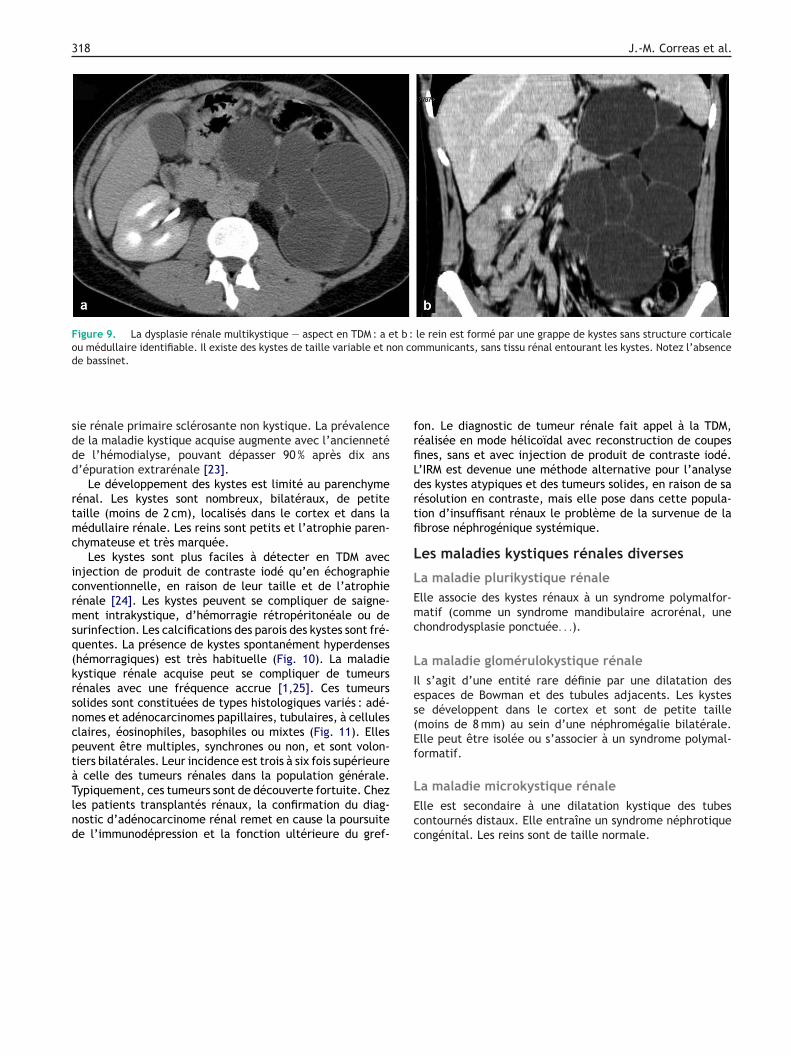
La dysplasie rénale multikystique est le plus souvent asso-iée à une atrésie pyélo-urétérale complète précoce auours de la vie fœtale. Le bassinet ne se développe pas et leein est formé par une grappe de kystes sans structure cor-icale ou médullaire identifiable. L’aspect échographique etomodensitométrique associe des kystes de taille variablet non communicants (les kystes les plus gros se situant enériphérie), l’absence de tissu rénal entourant les kystes oue bassinet et la présence de petites zones hyperéchogènesxcentrées correspondant à un reliquat mésenchymateuxFig. 9).
Lorsque les kystes ne sont pas visibles ou de petite taille,a maladie est découverte de facon fortuite à l’âge adulte,t l’on parlera plus volontiers d’aplasie, de dysplasie ou deysgénésie rénale.
Lorsque l’obstacle est sévère mais incomplet au cours dea néphrogénèse, le résultat histologique est identique, maise bassinet est dilaté et communique avec les kystes. Il s’agitlors d’une dysplasie rénale multikystique associée à uneydronéphrose. En échographie, il existe plusieurs kystesériphériques entourant une formation anéchogène volumi-euse médiane qui correspond au bassinet. Ces différentestructures kystiques peuvent communiquer. Les parois desystes peuvent présenter plusieurs calcifications. La tomo-raphie précise au mieux ces différents aspects.
a maladie kystique rénale acquise
a maladie kystique rénale acquise touche environ 40 à5 % des sujets en hémodialyse chronique et non porteurs’une maladie kystique initiale. Elle débute dès le stade de’insuffisance rénale et représente l’évolution d’une dyspla-
318 J.-M. Correas et al.
Figure 9. La dysplasie rénale multikystique — aspect en TDM : a et b : le rein est formé par une grappe de kystes sans structure corticaleou médullaire identifiable. Il existe des kystes de taille variable et non communicants, sans tissu rénal entourant les kystes. Notez l’absencede bassinet.
sddd
rtmc
icrmsq(krsncptàTlnd
frfiLdrtfi
L
LEmc
LIes(Ef
L
ie rénale primaire sclérosante non kystique. La prévalencee la maladie kystique acquise augmente avec l’anciennetée l’hémodialyse, pouvant dépasser 90 % après dix ans’épuration extrarénale [23].
Le développement des kystes est limité au parenchymeénal. Les kystes sont nombreux, bilatéraux, de petiteaille (moins de 2 cm), localisés dans le cortex et dans laédullaire rénale. Les reins sont petits et l’atrophie paren-
hymateuse et très marquée.Les kystes sont plus faciles à détecter en TDM avec
njection de produit de contraste iodé qu’en échographieonventionnelle, en raison de leur taille et de l’atrophieénale [24]. Les kystes peuvent se compliquer de saigne-ent intrakystique, d’hémorragie rétropéritonéale ou de
urinfection. Les calcifications des parois des kystes sont fré-uentes. La présence de kystes spontanément hyperdenseshémorragiques) est très habituelle (Fig. 10). La maladieystique rénale acquise peut se compliquer de tumeursénales avec une fréquence accrue [1,25]. Ces tumeursolides sont constituées de types histologiques variés : adé-omes et adénocarcinomes papillaires, tubulaires, à celluleslaires, éosinophiles, basophiles ou mixtes (Fig. 11). Elleseuvent être multiples, synchrones ou non, et sont volon-iers bilatérales. Leur incidence est trois à six fois supérieure
celle des tumeurs rénales dans la population générale.
ypiquement, ces tumeurs sont de découverte fortuite. Chezes patients transplantés rénaux, la confirmation du diag-ostic d’adénocarcinome rénal remet en cause la poursuitee l’immunodépression et la fonction ultérieure du gref-Ecc
on. Le diagnostic de tumeur rénale fait appel à la TDM,éalisée en mode hélicoïdal avec reconstruction de coupesnes, sans et avec injection de produit de contraste iodé.’IRM est devenue une méthode alternative pour l’analysees kystes atypiques et des tumeurs solides, en raison de saésolution en contraste, mais elle pose dans cette popula-ion d’insuffisant rénaux le problème de la survenue de labrose néphrogénique systémique.
es maladies kystiques rénales diverses
a maladie plurikystique rénalelle associe des kystes rénaux à un syndrome polymalfor-atif (comme un syndrome mandibulaire acrorénal, une
hondrodysplasie ponctuée. . .).
a maladie glomérulokystique rénalel s’agit d’une entité rare définie par une dilatation desspaces de Bowman et des tubules adjacents. Les kystese développent dans le cortex et sont de petite taillemoins de 8 mm) au sein d’une néphromégalie bilatérale.lle peut être isolée ou s’associer à un syndrome polymal-ormatif.
a maladie microkystique rénale
lle est secondaire à une dilatation kystique des tubesontournés distaux. Elle entraîne un syndrome néphrotiqueongénital. Les reins sont de taille normale.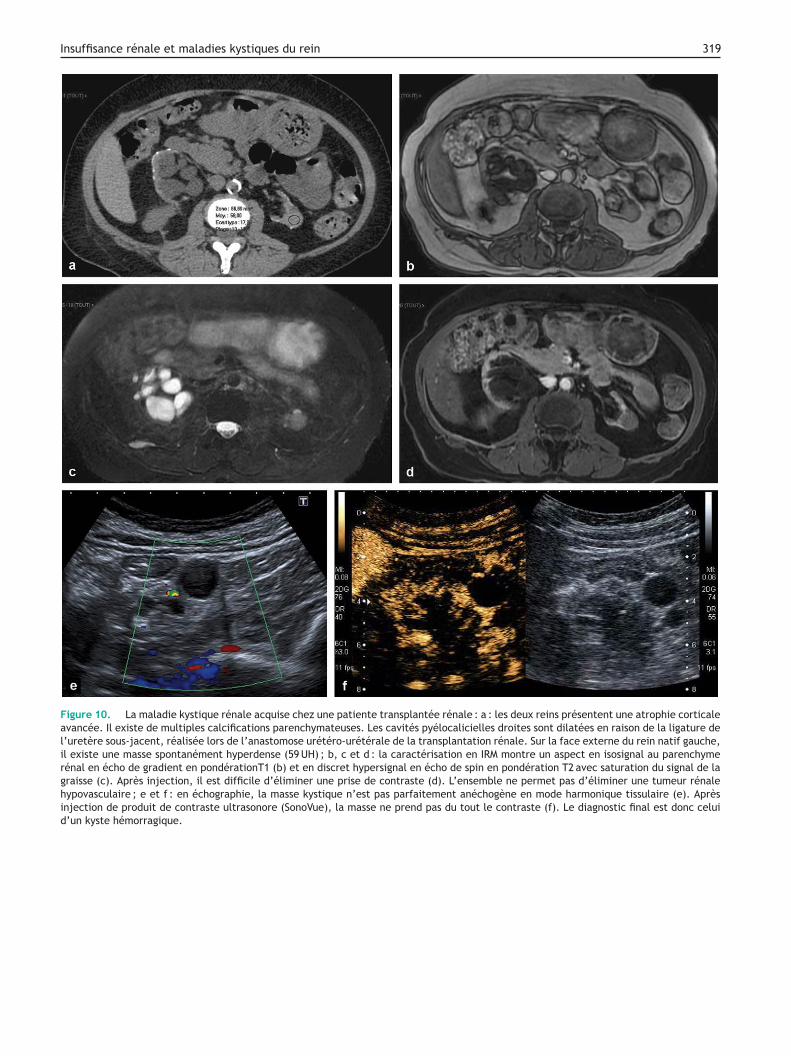
Insuffisance rénale et maladies kystiques du rein 319
Figure 10. La maladie kystique rénale acquise chez une patiente transplantée rénale : a : les deux reins présentent une atrophie corticaleavancée. Il existe de multiples calcifications parenchymateuses. Les cavités pyélocalicielles droites sont dilatées en raison de la ligature del’uretère sous-jacent, réalisée lors de l’anastomose urétéro-urétérale de la transplantation rénale. Sur la face externe du rein natif gauche,il existe une masse spontanément hyperdense (59 UH) ; b, c et d : la caractérisation en IRM montre un aspect en isosignal au parenchymerénal en écho de gradient en pondérationT1 (b) et en discret hypersignal en écho de spin en pondération T2 avec saturation du signal de lagraisse (c). Après injection, il est difficile d’éliminer une prise de contraste (d). L’ensemble ne permet pas d’éliminer une tumeur rénalehypovasculaire ; e et f : en échographie, la masse kystique n’est pas parfaitement anéchogène en mode harmonique tissulaire (e). Aprèsinjection de produit de contraste ultrasonore (SonoVue), la masse ne prend pas du tout le contraste (f). Le diagnostic final est donc celuid’un kyste hémorragique.
320 J.-M. Correas et al.
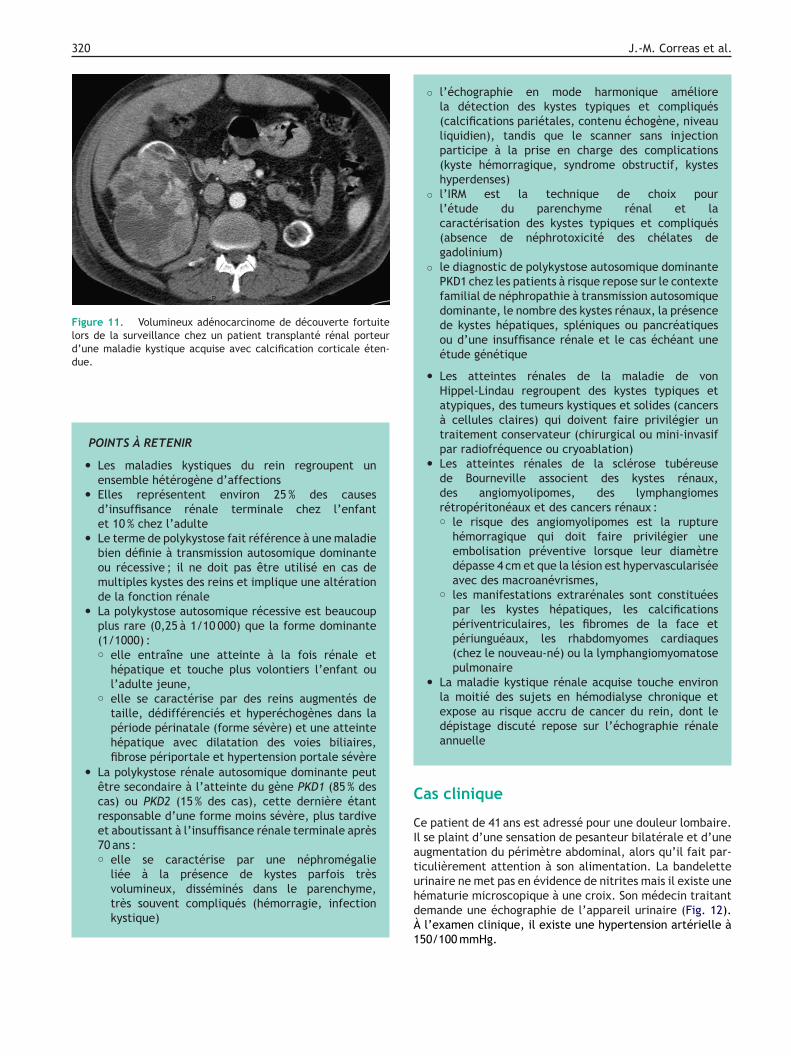
Figure 11. Volumineux adénocarcinome de découverte fortuitelors de la surveillance chez un patient transplanté rénal porteurd’une maladie kystique acquise avec calcification corticale éten-due.
◦ l’échographie en mode harmonique améliorela détection des kystes typiques et compliqués(calcifications pariétales, contenu échogène, niveauliquidien), tandis que le scanner sans injectionparticipe à la prise en charge des complications(kyste hémorragique, syndrome obstructif, kysteshyperdenses)
◦ l’IRM est la technique de choix pourl’étude du parenchyme rénal et lacaractérisation des kystes typiques et compliqués(absence de néphrotoxicité des chélates degadolinium)
◦ le diagnostic de polykystose autosomique dominantePKD1 chez les patients à risque repose sur le contextefamilial de néphropathie à transmission autosomiquedominante, le nombre des kystes rénaux, la présencede kystes hépatiques, spléniques ou pancréatiquesou d’une insuffisance rénale et le cas échéant uneétude génétique
• Les atteintes rénales de la maladie de vonHippel-Lindau regroupent des kystes typiques etatypiques, des tumeurs kystiques et solides (cancersà cellules claires) qui doivent faire privilégier untraitement conservateur (chirurgical ou mini-invasifpar radiofréquence ou cryoablation)
• Les atteintes rénales de la sclérose tubéreusede Bourneville associent des kystes rénaux,des angiomyolipomes, des lymphangiomesrétropéritonéaux et des cancers rénaux :◦ le risque des angiomyolipomes est la rupture
hémorragique qui doit faire privilégier uneembolisation préventive lorsque leur diamètredépasse 4 cm et que la lésion est hypervasculariséeavec des macroanévrismes,
◦ les manifestations extrarénales sont constituéespar les kystes hépatiques, les calcificationspériventriculaires, les fibromes de la face etpériunguéaux, les rhabdomyomes cardiaques(chez le nouveau-né) ou la lymphangiomyomatosepulmonaire
• La maladie kystique rénale acquise touche environla moitié des sujets en hémodialyse chronique etexpose au risque accru de cancer du rein, dont ledépistage discuté repose sur l’échographie rénale
C
CIatuhématurie microscopique à une croix. Son médecin traitantdemande une échographie de l’appareil urinaire (Fig. 12).À l’examen clinique, il existe une hypertension artérielle à150/100 mmHg.
POINTS À RETENIR
• Les maladies kystiques du rein regroupent unensemble hétérogène d’affections
• Elles représentent environ 25 % des causesd’insuffisance rénale terminale chez l’enfantet 10 % chez l’adulte
• Le terme de polykystose fait référence à une maladiebien définie à transmission autosomique dominanteou récessive ; il ne doit pas être utilisé en cas demultiples kystes des reins et implique une altérationde la fonction rénale
• La polykystose autosomique récessive est beaucoupplus rare (0,25 à 1/10 000) que la forme dominante(1/1000) :◦ elle entraîne une atteinte à la fois rénale et
hépatique et touche plus volontiers l’enfant oul’adulte jeune,
◦ elle se caractérise par des reins augmentés detaille, dédifférenciés et hyperéchogènes dans lapériode périnatale (forme sévère) et une atteintehépatique avec dilatation des voies biliaires,fibrose périportale et hypertension portale sévère
• La polykystose rénale autosomique dominante peutêtre secondaire à l’atteinte du gène PKD1 (85 % descas) ou PKD2 (15 % des cas), cette dernière étantresponsable d’une forme moins sévère, plus tardiveet aboutissant à l’insuffisance rénale terminale après70 ans :◦ elle se caractérise par une néphromégalie
liée à la présence de kystes parfois trèsvolumineux, disséminés dans le parenchyme,très souvent compliqués (hémorragie, infectionkystique)
annuelle
as clinique
e patient de 41 ans est adressé pour une douleur lombaire.l se plaint d’une sensation de pesanteur bilatérale et d’uneugmentation du périmètre abdominal, alors qu’il fait par-iculièrement attention à son alimentation. La bandeletterinaire ne met pas en évidence de nitrites mais il existe une
Insuffisance rénale et maladies kystiques du rein
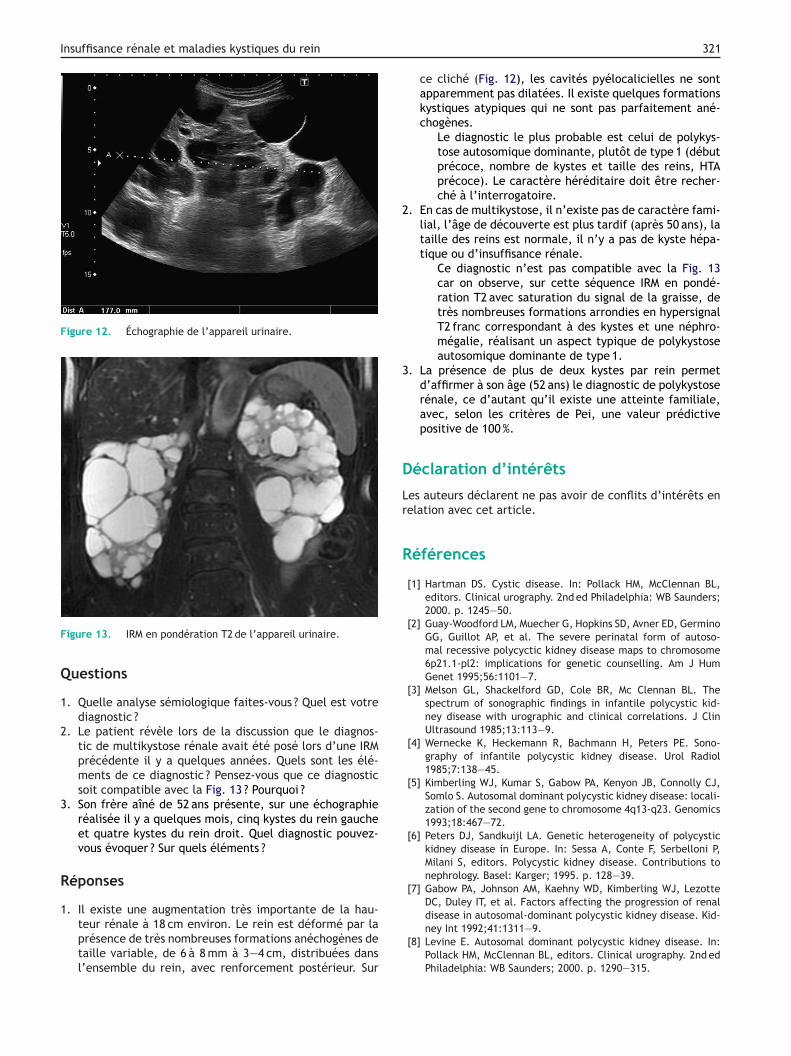
Figure 12. Échographie de l’appareil urinaire.
2
3
D
Lr
R
Figure 13. IRM en pondération T2 de l’appareil urinaire.
Questions
1. Quelle analyse sémiologique faites-vous ? Quel est votrediagnostic ?
2. Le patient révèle lors de la discussion que le diagnos-tic de multikystose rénale avait été posé lors d’une IRMprécédente il y a quelques années. Quels sont les élé-ments de ce diagnostic ? Pensez-vous que ce diagnosticsoit compatible avec la Fig. 13 ? Pourquoi ?
3. Son frère aîné de 52 ans présente, sur une échographieréalisée il y a quelques mois, cinq kystes du rein gaucheet quatre kystes du rein droit. Quel diagnostic pouvez-vous évoquer ? Sur quels éléments ?
Réponses
1. Il existe une augmentation très importante de la hau-teur rénale à 18 cm environ. Le rein est déformé par la
présence de très nombreuses formations anéchogènes detaille variable, de 6 à 8 mm à 3—4 cm, distribuées dansl’ensemble du rein, avec renforcement postérieur. Sur321
ce cliché (Fig. 12), les cavités pyélocalicielles ne sontapparemment pas dilatées. Il existe quelques formationskystiques atypiques qui ne sont pas parfaitement ané-chogènes.
Le diagnostic le plus probable est celui de polykys-tose autosomique dominante, plutôt de type 1 (débutprécoce, nombre de kystes et taille des reins, HTAprécoce). Le caractère héréditaire doit être recher-ché à l’interrogatoire.
. En cas de multikystose, il n’existe pas de caractère fami-lial, l’âge de découverte est plus tardif (après 50 ans), lataille des reins est normale, il n’y a pas de kyste hépa-tique ou d’insuffisance rénale.
Ce diagnostic n’est pas compatible avec la Fig. 13car on observe, sur cette séquence IRM en pondé-ration T2 avec saturation du signal de la graisse, detrès nombreuses formations arrondies en hypersignalT2 franc correspondant à des kystes et une néphro-mégalie, réalisant un aspect typique de polykystoseautosomique dominante de type 1.
. La présence de plus de deux kystes par rein permetd’affirmer à son âge (52 ans) le diagnostic de polykystoserénale, ce d’autant qu’il existe une atteinte familiale,avec, selon les critères de Pei, une valeur prédictivepositive de 100 %.
éclaration d’intérêts
es auteurs déclarent ne pas avoir de conflits d’intérêts enelation avec cet article.
éférences
[1] Hartman DS. Cystic disease. In: Pollack HM, McClennan BL,editors. Clinical urography. 2nd ed Philadelphia: WB Saunders;2000. p. 1245—50.
[2] Guay-Woodford LM, Muecher G, Hopkins SD, Avner ED, GerminoGG, Guillot AP, et al. The severe perinatal form of autoso-mal recessive polycyctic kidney disease maps to chromosome6p21.1-pl2: implications for genetic counselling. Am J HumGenet 1995;56:1101—7.
[3] Melson GL, Shackelford GD, Cole BR, Mc Clennan BL. Thespectrum of sonographic findings in infantile polycystic kid-ney disease with urographic and clinical correlations. J ClinUltrasound 1985;13:113—9.
[4] Wernecke K, Heckemann R, Bachmann H, Peters PE. Sono-graphy of infantile polycystic kidney disease. Urol Radiol1985;7:138—45.
[5] Kimberling WJ, Kumar S, Gabow PA, Kenyon JB, Connolly CJ,Somlo S. Autosomal dominant polycystic kidney disease: locali-zation of the second gene to chromosome 4q13-q23. Genomics1993;18:467—72.
[6] Peters DJ, Sandkuijl LA. Genetic heterogeneity of polycystickidney disease in Europe. In: Sessa A, Conte F, Serbelloni P,Milani S, editors. Polycystic kidney disease. Contributions tonephrology. Basel: Karger; 1995. p. 128—39.
[7] Gabow PA, Johnson AM, Kaehny WD, Kimberling WJ, LezotteDC, Duley IT, et al. Factors affecting the progression of renaldisease in autosomal-dominant polycystic kidney disease. Kid-
ney Int 1992;41:1311—9.[8] Levine E. Autosomal dominant polycystic kidney disease. In:Pollack HM, McClennan BL, editors. Clinical urography. 2nd edPhiladelphia: WB Saunders; 2000. p. 1290—315.

3
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[of acquired cystic kidney disease and renal tumors in long-term
22
[9] Chauveau D, Grimbert P, Grünfeld JP. Néphropathies hérédi-taires. Encycl Med Chir (Elsevier, Paris), Néphrologie-Urologie,18-050-A-10, 1997. 11 p.
10] Keith DS, Torres VE, King BF, Zincki H, Farrow GM. Renal cellcarcinoma in autosomal dominant polycystic kidney disease. JAm Soc Nephrol 1994;4:1661—9.
11] Delaney VB, Adler S, Bruns FJ, Licinia M, Segel DP, Fraley DS.Autosomal dominant polycystic kidney disease: presentation,complications, and prognosis. Am J Kidney Dis 1985;5:104—11.
12] Hélénon O, Souissi M, Rotkopf L, Denys A, Cornud F, Moreau JF.Kyste simple du rein. Radiodiagnostic-Urologie-Gynécologie,34119 B30. Paris, France: Éditions techniques. Encycl Med Chir;1992. 16 p.
13] Lawson TL, Mc Clennan BL, Shirkhoda A. Adult polycystic kidneydisease: ultrasonographic and computed tomographic appea-rance. J Clin Ultrasound 1978;6:297—302.
14] Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-SmithP, Danks DM. Evaluation of ultrasonographic diagnostic crite-ria for autosomal dominant polycystic kidney disease 1. Lancet1994;343:824—7.
15] Levey AS. Screening for occult intracranial aneurysms in poly-cystic kidney disease: interim guidelines. J Am Soc Nephrol1990;1:9—12.
16] Chauveau D, Duvic C, Chrétien Y, et al. Renal involvement invon Hippel-Lindau disease. Kidney Int 1996;50:944—51.
17] Ogawa O, Habuchi T, Kakehi Y, Koshiba M, Sugiyama T, YoshidaO. Allelic losses at chromosome 17p in human renal cell carci-
[
J.-M. Correas et al.
noma are inversely related to allelic losses at chromosome 3p.Cancer Res 1992;52:1881—5.
18] Choyke PL, Glenn GM, Walther MM, Zbar B, Linehan WM. Here-ditary renal cancers. Radiology 2003;226:33—46.
19] Richard S. Von Hippel-Lindau disease: recent advancesand therapeutic perspectives. Exp Rev Anticancer Ther2003;3:215—33.
20] Levine E, Collins DL, Horton WA, Schimke RN. CT scree-ning of the abdomen in Von Hippel Lindau disease. AJR1982;139:505—10.
21] Smirniotopoulos JG, Hartman DS. Renal cystic disease asso-ciated with tuberous sclerosis. In: Pollack HM, McClennan BL,editors. Clinical urography. 2nd ed Philadelphia: WB Saunders;2000. p. 1359—67.
22] Brook-Carter PT, Peral B, Ward CJ, et al. Deletion of theTSC2 and PKD1 genes associated with severe infantile polycys-tic kidney disease — a contiguous gene syndrome. Nat Genet1994;8:328—32.
23] Matson MA, Cohen EP. Acquired cystic kidney disease: occur-rence, prevalence, and renal cancers. Medicine (Baltimore)1990;69:217—26.
24] Levine E, Grantham JJ, Slusher SL, Greathouse JL, Krohn BP. CT
dialysis patients. AJR 1984;142:125—31.25] Scanlon MH, Karasick SR. Acquired renal cystic disease and
neoplasia: complications of chronic hemodialysis. Radiology1983;147:837—8.



















![Cancer du Rein [Réparé] - CHU de Reims · 2017. 6. 22. · • Rasage du mamelon au pubis Complications de la chirurgie • Hémorragie, hématome loge rénale ou de paroi • Urinome,](https://img.pdfslide.fr/doc/110x75/6110480b15aecb47f57b9021/cancer-du-rein-rpar-chu-de-reims-2017-6-22-a-rasage-du-mamelon-au.jpg)









