Embed Size (px)
Citation preview

Microporous and Mesoporous Materials 195 (2014) 102–111
Contents lists available at ScienceDirect
Microporous and Mesoporous Materials
journal homepage: www.elsevier .com/locate /micromeso
Investigation of the surfactant type effect on characteristicsand bioactivity of new mesoporous bioactive glass in the ternarysystem SiO2–CaO–P2O5: Structural, textural and reactivity studies
http://dx.doi.org/10.1016/j.micromeso.2014.03.0351387-1811/� 2014 Elsevier Inc. All rights reserved.
⇑ Corresponding authors. Address: Chimie du Solide et Matériaux, UMR 6226CNRS, Université de Rennes 1, Institut des Sciences Chimiques de Rennes, Campusde Beaulieu, Avenue du Général Leclerc, 35042 Rennes Cedex, France. Tel.: +3375376022; fax: +33 02 23 23 66 59 (N. Letaïef).
E-mail addresses: [email protected] (N. Letaïef), [email protected](A. Lucas-Girot).
Nouha Letaïef a,b,⇑, Anita Lucas-Girot a,⇑, Hassane Oudadesse a, Rachida Dorbez-Sridi b, Philippe Boullay c
a Chimie du Solide et Matériaux, UMR 6226 CNRS, Université de Rennes 1, Institut des Sciences Chimiques de Rennes, Campus de Beaulieu, Avenue du Général Leclerc, 35042 RennesCedex, Franceb Laboratoire de Physico-Chimie des Matériaux, Département de Physique, Faculté des Sciences de Monastir, Avenue de l’environnement, 5019 Monastir, Tunisiac Laboratoire de Cristallographie et Sciences des Matériaux, CRISMAT-UMR6508 CNRS/ENSICAEN, 6 Bd du Maréchal Juin, 14050 Caen Cedex 4, France
a r t i c l e i n f o a b s t r a c t
Article history:Received 19 July 2013Received in revised form 12 March 2014Accepted 24 March 2014Available online 4 April 2014
Keywords:Mesoporous materialsBioactivityApatiteCTABP123
In this study, a mesoporous bioactive glass 92S6 (92% SiO2, 6% CaO, and 2% P2O5), was successfullyprepared using two different surfactants. The template was further removed by calcination to generateunorganized or well-ordered pores. The bioactive glass was characterized by wide angle X-ray diffraction(WAXRD) analysis, small angle X-ray diffraction (SAXRD) analysis, Fourier Transform Infrared Spectros-copy (FTIR), Transmission Electron Microscopy (TEM) and Scanning Electron Microscopy (SEM). Fromthe isotherm desorption branch, the surface area was determined using the Brunauer–Emmett–Teller(BET) method, while pore volume and pore size distribution were determined by the Barrett–Joyner–Halenda (BJH) method. The in vitro bioactivity tests were also conducted in simulated body fluid (SBF).Finally, the samples were analyzed to quantify the apatite formation ability when soaked in SBF solution.The evolutions of silicon (Si), phosphorus (P) and calcium (Ca) concentrations in SBF were evaluated byinductively coupled plasma optical emission spectrometry (ICP-OES).
The SAXRD and TEM studies evidence the influence of the structure-directing agent (ionic surfactantCTAB or non-ionic P123) in the generation of unorganized or well ordered pores in the sol–gel synthesisof a bioactive glass in the ternary system SiO2–CaO–P2O5.
As observed from small-angle XRD patterns and TEM images, the presence of non-ionic surfactant andsubsequent calcination lead to the formation of highly ordered mesoporous glass.
The better textural properties observed in the ‘‘ordered mesoporous glasses’’ compared to those of‘‘non-ordered mesoporous glasses’’ lead to a faster in vitro bioactivity kinetics.
� 2014 Elsevier Inc. All rights reserved.
1. Introduction
Among biomaterials used in orthopedic surgery and dentalgrafting some of them, known as bioactive, are able to chemicallyattach to bone [1]. When implanted into the human body, thesebioactive materials can promote bone regeneration by inducing aspecific biological response at the interface of the material, whichresults in a formation of a new bone. They could spontaneouslybond to living bone without forming fibrous tissue around them
[2–4]. Their bone-bonding ability has been attributed to the depo-sition and growth of a hydroxylapatite (HA) layer, very close incomposition to that of bone mineral. This layer at the glass surfaceis observed when materials are soaked in physiological body fluidor implanted in vivo [5]. The interfacial reactions leading to HAformation involve surface leaching with calcium-proton exchange,formation of silanols groups, formation of a hydrated silica gel filmby polycondensation of silanols, development of an amorphouscalcium phosphate layer into the silica-rich layer and crystalliza-tion of HA [6].
The ability of biomaterials to enhance bone formation to facili-tate implant integration and tissue repair is related not only totheir chemical composition and structure (e.g. Si/Ca/P molar ratio),but also to their textural properties, such as porosity, pore volume,pore size and pore structure [7–10]. Alcaide et al. [1] reported thatmesoporous (pore size ranging from 2 to 50 nm) glasses may

N. Letaïef et al. / Microporous and Mesoporous Materials 195 (2014) 102–111 103
induce rapid crystallization of HA at their surface. Vallet-Regí et al.have demonstrated that the formation of HA is greatly acceleratedwhen the specific area and pore volume of the bioactive glassincrease [11].
In this sense, since the discovery of silica-based MCM (MobilComposition of Matter) 41 in 1992 [12], the first mesoporous solidthat showed a regularly ordered pore arrangement and a very nar-row pore-size distribution, the research interest focused on thesynthesis of new mesoporous biomaterials based on the MCM-41synthesis concept [13]. Most of them contain both SiO2, CaO andP2O5 as main components [14–19].
Mesoporous materials are used technically as adsorbents of bio-logical metabolites, sensors [20] and catalyst supports [21] owingto their textural properties: surface areas and porosity. They arealso suggested to promote cell adhesion, and resorbability at a con-trolled rate, to match to that of bone repair [22]. Moreover, themesoporous biomaterials with high specific surface areas and largepore volumes have been addressed to become ideal for encapsula-tion of pharmaceutical drugs, proteins and other biogenic mole-cules [14,23–24] such as for the delivery of anti-tumor agentsand antibiotics for osteomyelitis treatment [25].
It was also demonstrated that the pores diameter between 2and 6 nm allow the loading of different drug molecules [26].
Numerous papers have been published on the synthesis andcharacterization of mesoporous bioactive glasses with composi-tions based on ternary composition, SiO2–CaO–P2O5 systems, byemploying a non-ionic block copolymers surfactant, having a shortor a long PEO (poly(ethylene oxide)) chain length, as the structure-directing agent which lead to ordered mesoporous templatedglasses [14,17,18,27]. There are also reports on the presence ofthe cationic surfactant CTAB as a co-surfactant with the triblockcopolymer in the reaction [28]. There are also few reports on theuse of copolymer blends of P123 [29] or even nonionic oligomericsurfactants [30]. The comparative study of the surfactant’s natureeffects is the originality of this work.
In the present work, a glass, based on the molar composition 92SiO2
�6 CaO�2 P2O5 (mol%) has been synthesized and characterized.Our method is based on the synthesis of MCM-41 [31].
The purpose of this present study was first to investigate the effectof the surfactant type on the properties of the SiO2–CaO–P2O5:structural characteristics, morphology, textural properties andthen to investigate the effect of the surfactant type on the glassbioactivity.
2. Experimental procedure
2.1. Materials
Tetraethyl orthosilicate (TEOS) (99%; Aldrich, France) was usedas a silica source, Triethyl phosphate (TEP) (99.8%; Aldrich, France)was used as source of phosphorous, calcium carbonate CaCO3
(>98.5%; Merck, France) or calcium nitrate tetrahydrate Ca(NO3)2-
�4H2O (P99%, Fluka, Germany) were used as source of calcium.The surfactant n-hexadecyltrimethylammonium bromide CTAB(98%; Alfa Aesar, Germany) and the surfactant pluronic P123(EO20-PO70-EO20) (Sigma–Aldrich, Germany) were used as a struc-ture-directing agent. Water was used as solvent. All materials wereused as received without further purification.
Table 1Ion concentrations of the simulated body fluid and human blood plasma (mM).
Types Ion concentrations (mM)
Na+ K+ Mg2+ Ca2+ Cl� HCO32� HPO4
2� SO42�
SBF 142.0 5.0 1.5 2.5 147.8 4.2 1.0 0.5Blood plasma 142.0 5.0 1.5 2.5 103.0 27.0 1.0 0.5
2.2. Synthesis
2.2.1. Preparation of 92S6 using cationic surfactant (CTAB)Samples were prepared by the sol–gel process. The synthesis
was carried out according to the modified Stöber’s methoddescribed for synthesis of monodisperse silica spheres MCM-41
[31]. In the first step, the solution was prepared as follows: 2.5 gof CTAB was dissolved in 50 ml of deionized water and mixed with75 ml of absolute ethanol, at room temperature. 3.6 g of TEOS isfurther added to this solution and kept under strong stirring forone hour. Under stirring, 0.16 g of TEP and acidic CaCO3 solutionwere added. The final pH was 4.5. Then, the mixture was stirredfor 24 h and finally 18.53 ml of aqueous ammonia solution (25%wt., Fluka, France) was added to this clear solution and stirredfor 15 min. At the end of the addition of aqueous ammonia solu-tion, the gel formation immediately began. The white precipitatewas isolated by filtration and washed several times with waterand ethanol until neutral pH was reached. Then the sample wasdried at 90 �C overnight. The surfactant was further removed bycalcinations, treating the dried gel at 650 �C for 6 h. The resultingglass was crushed and sieved to select grain size less than63 lm. The glass was given the code BI (Bioactive glass synthesizedwith an Ionic surfactant).
2.2.2. Preparation of 92S6 using non-ionic surfactant (P123)Mesoporous 92S6 was synthesized by a two-step acid-catalyzed
self-assembly process combined with hydrothermal treatment inan inorganic–organic system [32]. Glass composition was the sameas the glass synthesized with CTAB. The P123 amphiphilic blockcopolymer: poly(ethylene oxide)-block-poly(propylene oxide)-block-poly(ethylene oxide) (PEO-PPO-PEO) was used as organicstructure-directing agent. Our glass was prepared by dissolving,under stirring, at 40 �C, 6 g of P123 in 120 ml of 2 M HNO3 and30 ml of distilled water solution, until the solution became clear.Then 3.6 g of TEOS, 0.16 g of TEP and 0.53 g of Ca(NO3)2�4H2O (toobtain a molar ratio Si/Ca/P = 92/6/2) were then added to the clearsolution. The final pH was 0.3. The mixture was stirred at 40 �C for12 h, and then hydrothermalized at 100 �C for 48 h. Without anyfiltering and washing, the resulting precipitate was directly driedat 100 �C for 20 h in air. The as-synthesized powders were calcinedat 650 �C in air for 6 h in order to remove the organic structure-directing agent completely. The resulting glass was crushed andsieved to select grain size less than 63 lm. The glass was giventhe code BNI for simplicity (Bioactive glass synthesized with aNon-Ionic surfactant).
2.3. Preparation of SBF
The simulated body fluid solution (SBF) was prepared by dis-solving reagent-grade NaCl; KCl; NaHCO3; MgCl2�6H2O; CaCl2 andKH2PO4�3H2O into distilled water and buffered at pH 7.4 withtris(hydroxymethyl)aminomethane (TRIS) and HCl 6 N at 37 �Caccording to Kokubo’s protocol [33]. The SBF solution had an ioniccomposition similar to that of human blood plasma as shown inTable 1 [34].
2.4. Structural characterization
The wide angle X-ray diffraction (WAXRD) patterns of the sam-ples were recorded on a Bruker AXS diffractometer with mono-chromatized CuKa radiation (k = 1.5406 Å), operating at 40 kV,40 mA, with 0.02� step size and a counting time of 300 ms per step,over a range of 5� < 2h < 80�, at room temperature. The Fourier
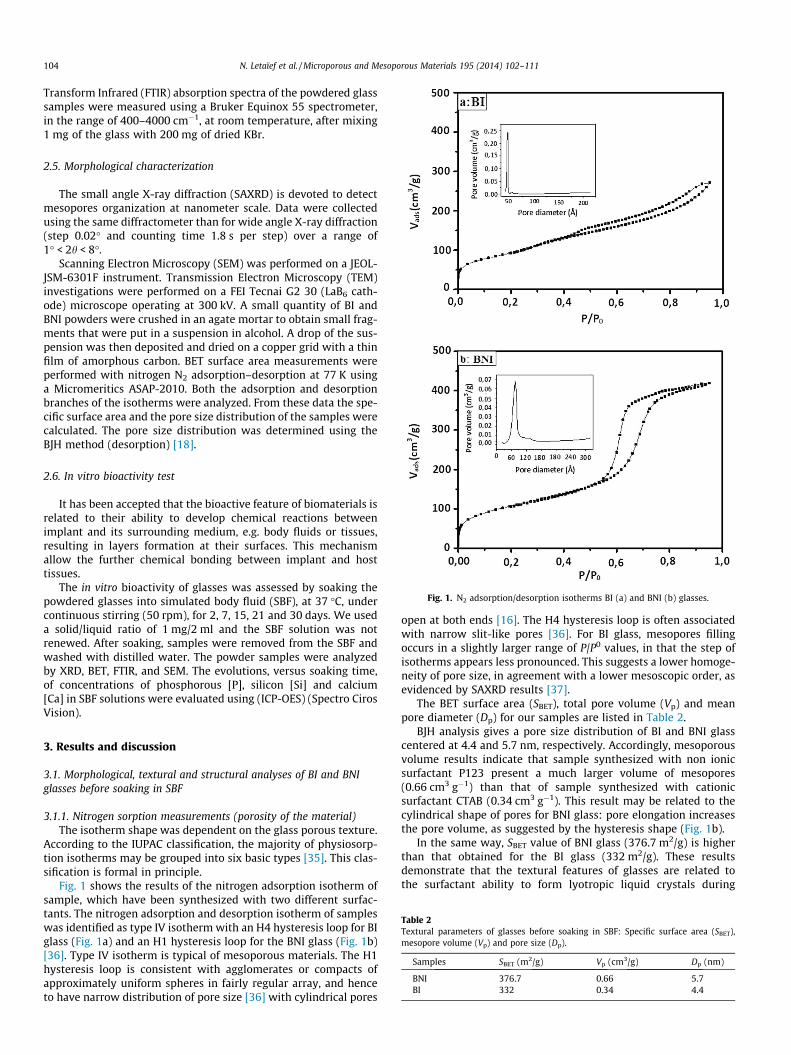
Fig. 1. N2 adsorption/desorption isotherms BI (a) and BNI (b) glasses.
104 N. Letaïef et al. / Microporous and Mesoporous Materials 195 (2014) 102–111
Transform Infrared (FTIR) absorption spectra of the powdered glasssamples were measured using a Bruker Equinox 55 spectrometer,in the range of 400–4000 cm�1, at room temperature, after mixing1 mg of the glass with 200 mg of dried KBr.
2.5. Morphological characterization
The small angle X-ray diffraction (SAXRD) is devoted to detectmesopores organization at nanometer scale. Data were collectedusing the same diffractometer than for wide angle X-ray diffraction(step 0.02� and counting time 1.8 s per step) over a range of1� < 2h < 8�.
Scanning Electron Microscopy (SEM) was performed on a JEOL-JSM-6301F instrument. Transmission Electron Microscopy (TEM)investigations were performed on a FEI Tecnai G2 30 (LaB6 cath-ode) microscope operating at 300 kV. A small quantity of BI andBNI powders were crushed in an agate mortar to obtain small frag-ments that were put in a suspension in alcohol. A drop of the sus-pension was then deposited and dried on a copper grid with a thinfilm of amorphous carbon. BET surface area measurements wereperformed with nitrogen N2 adsorption–desorption at 77 K usinga Micromeritics ASAP-2010. Both the adsorption and desorptionbranches of the isotherms were analyzed. From these data the spe-cific surface area and the pore size distribution of the samples werecalculated. The pore size distribution was determined using theBJH method (desorption) [18].
2.6. In vitro bioactivity test
It has been accepted that the bioactive feature of biomaterials isrelated to their ability to develop chemical reactions betweenimplant and its surrounding medium, e.g. body fluids or tissues,resulting in layers formation at their surfaces. This mechanismallow the further chemical bonding between implant and hosttissues.
The in vitro bioactivity of glasses was assessed by soaking thepowdered glasses into simulated body fluid (SBF), at 37 �C, undercontinuous stirring (50 rpm), for 2, 7, 15, 21 and 30 days. We useda solid/liquid ratio of 1 mg/2 ml and the SBF solution was notrenewed. After soaking, samples were removed from the SBF andwashed with distilled water. The powder samples were analyzedby XRD, BET, FTIR, and SEM. The evolutions, versus soaking time,of concentrations of phosphorous [P], silicon [Si] and calcium[Ca] in SBF solutions were evaluated using (ICP-OES) (Spectro CirosVision).
Table 2Textural parameters of glasses before soaking in SBF: Specific surface area (SBET),mesopore volume (Vp) and pore size (Dp).
Samples SBET (m2/g) Vp (cm3/g) Dp (nm)
BNI 376.7 0.66 5.7BI 332 0.34 4.4
3. Results and discussion
3.1. Morphological, textural and structural analyses of BI and BNIglasses before soaking in SBF
3.1.1. Nitrogen sorption measurements (porosity of the material)The isotherm shape was dependent on the glass porous texture.
According to the IUPAC classification, the majority of physiosorp-tion isotherms may be grouped into six basic types [35]. This clas-sification is formal in principle.
Fig. 1 shows the results of the nitrogen adsorption isotherm ofsample, which have been synthesized with two different surfac-tants. The nitrogen adsorption and desorption isotherm of sampleswas identified as type IV isotherm with an H4 hysteresis loop for BIglass (Fig. 1a) and an H1 hysteresis loop for the BNI glass (Fig. 1b)[36]. Type IV isotherm is typical of mesoporous materials. The H1hysteresis loop is consistent with agglomerates or compacts ofapproximately uniform spheres in fairly regular array, and henceto have narrow distribution of pore size [36] with cylindrical pores
open at both ends [16]. The H4 hysteresis loop is often associatedwith narrow slit-like pores [36]. For BI glass, mesopores fillingoccurs in a slightly larger range of P/P0 values, in that the step ofisotherms appears less pronounced. This suggests a lower homoge-neity of pore size, in agreement with a lower mesoscopic order, asevidenced by SAXRD results [37].
The BET surface area (SBET), total pore volume (Vp) and meanpore diameter (Dp) for our samples are listed in Table 2.
BJH analysis gives a pore size distribution of BI and BNI glasscentered at 4.4 and 5.7 nm, respectively. Accordingly, mesoporousvolume results indicate that sample synthesized with non ionicsurfactant P123 present a much larger volume of mesopores(0.66 cm3 g�1) than that of sample synthesized with cationicsurfactant CTAB (0.34 cm3 g�1). This result may be related to thecylindrical shape of pores for BNI glass: pore elongation increasesthe pore volume, as suggested by the hysteresis shape (Fig. 1b).
In the same way, SBET value of BNI glass (376.7 m2/g) is higherthan that obtained for the BI glass (332 m2/g). These resultsdemonstrate that the textural features of glasses are related tothe surfactant ability to form lyotropic liquid crystals during
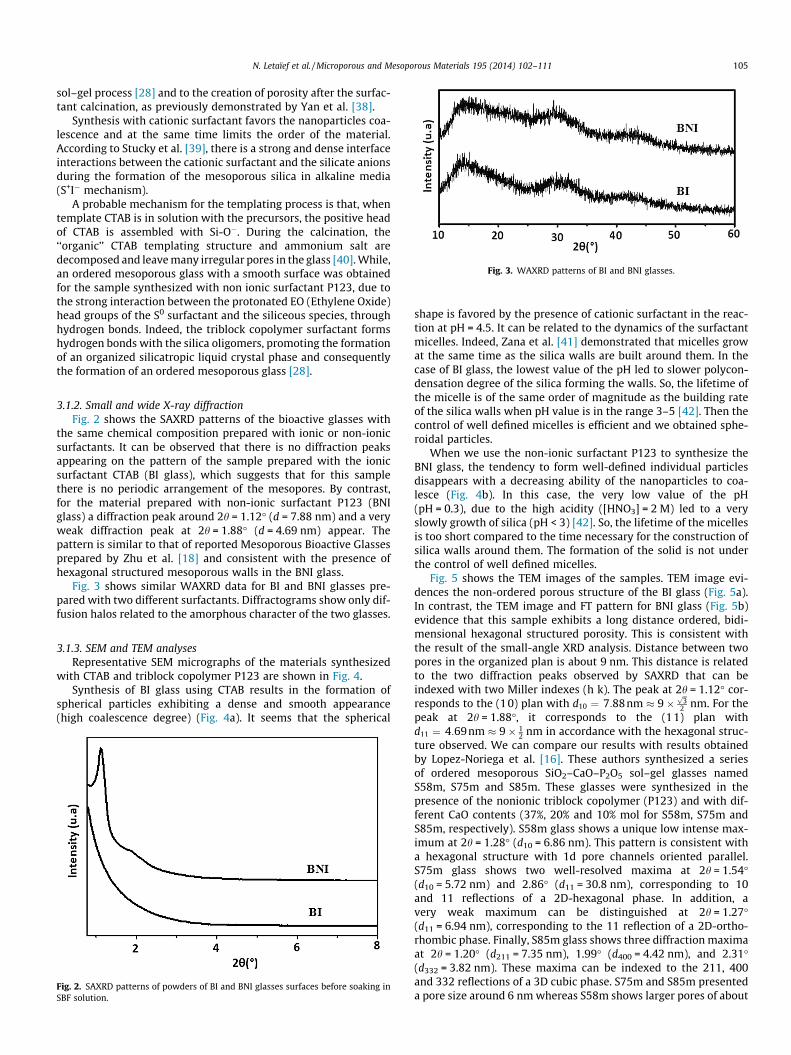
Fig. 3. WAXRD patterns of BI and BNI glasses.
N. Letaïef et al. / Microporous and Mesoporous Materials 195 (2014) 102–111 105
sol–gel process [28] and to the creation of porosity after the surfac-tant calcination, as previously demonstrated by Yan et al. [38].
Synthesis with cationic surfactant favors the nanoparticles coa-lescence and at the same time limits the order of the material.According to Stucky et al. [39], there is a strong and dense interfaceinteractions between the cationic surfactant and the silicate anionsduring the formation of the mesoporous silica in alkaline media(S+I� mechanism).
A probable mechanism for the templating process is that, whentemplate CTAB is in solution with the precursors, the positive headof CTAB is assembled with Si-O�. During the calcination, the‘‘organic’’ CTAB templating structure and ammonium salt aredecomposed and leave many irregular pores in the glass [40]. While,an ordered mesoporous glass with a smooth surface was obtainedfor the sample synthesized with non ionic surfactant P123, due tothe strong interaction between the protonated EO (Ethylene Oxide)head groups of the S0 surfactant and the siliceous species, throughhydrogen bonds. Indeed, the triblock copolymer surfactant formshydrogen bonds with the silica oligomers, promoting the formationof an organized silicatropic liquid crystal phase and consequentlythe formation of an ordered mesoporous glass [28].
3.1.2. Small and wide X-ray diffractionFig. 2 shows the SAXRD patterns of the bioactive glasses with
the same chemical composition prepared with ionic or non-ionicsurfactants. It can be observed that there is no diffraction peaksappearing on the pattern of the sample prepared with the ionicsurfactant CTAB (BI glass), which suggests that for this samplethere is no periodic arrangement of the mesopores. By contrast,for the material prepared with non-ionic surfactant P123 (BNIglass) a diffraction peak around 2h = 1.12� (d = 7.88 nm) and a veryweak diffraction peak at 2h = 1.88� (d = 4.69 nm) appear. Thepattern is similar to that of reported Mesoporous Bioactive Glassesprepared by Zhu et al. [18] and consistent with the presence ofhexagonal structured mesoporous walls in the BNI glass.
Fig. 3 shows similar WAXRD data for BI and BNI glasses pre-pared with two different surfactants. Diffractograms show only dif-fusion halos related to the amorphous character of the two glasses.
3.1.3. SEM and TEM analysesRepresentative SEM micrographs of the materials synthesized
with CTAB and triblock copolymer P123 are shown in Fig. 4.Synthesis of BI glass using CTAB results in the formation of
spherical particles exhibiting a dense and smooth appearance(high coalescence degree) (Fig. 4a). It seems that the spherical
Fig. 2. SAXRD patterns of powders of BI and BNI glasses surfaces before soaking inSBF solution.
shape is favored by the presence of cationic surfactant in the reac-tion at pH = 4.5. It can be related to the dynamics of the surfactantmicelles. Indeed, Zana et al. [41] demonstrated that micelles growat the same time as the silica walls are built around them. In thecase of BI glass, the lowest value of the pH led to slower polycon-densation degree of the silica forming the walls. So, the lifetime ofthe micelle is of the same order of magnitude as the building rateof the silica walls when pH value is in the range 3–5 [42]. Then thecontrol of well defined micelles is efficient and we obtained sphe-roidal particles.
When we use the non-ionic surfactant P123 to synthesize theBNI glass, the tendency to form well-defined individual particlesdisappears with a decreasing ability of the nanoparticles to coa-lesce (Fig. 4b). In this case, the very low value of the pH(pH = 0.3), due to the high acidity ([HNO3] = 2 M) led to a veryslowly growth of silica (pH < 3) [42]. So, the lifetime of the micellesis too short compared to the time necessary for the construction ofsilica walls around them. The formation of the solid is not underthe control of well defined micelles.
Fig. 5 shows the TEM images of the samples. TEM image evi-dences the non-ordered porous structure of the BI glass (Fig. 5a).In contrast, the TEM image and FT pattern for BNI glass (Fig. 5b)evidence that this sample exhibits a long distance ordered, bidi-mensional hexagonal structured porosity. This is consistent withthe result of the small-angle XRD analysis. Distance between twopores in the organized plan is about 9 nm. This distance is relatedto the two diffraction peaks observed by SAXRD that can beindexed with two Miller indexes (h k). The peak at 2h = 1.12� cor-responds to the (10) plan with d10 ¼ 7:88nm � 9�
ffiffi
3p
2 nm. For thepeak at 2h = 1.88�, it corresponds to the (11) plan withd11 ¼ 4:69nm � 9� 1
2 nm in accordance with the hexagonal struc-ture observed. We can compare our results with results obtainedby Lopez-Noriega et al. [16]. These authors synthesized a seriesof ordered mesoporous SiO2–CaO–P2O5 sol–gel glasses namedS58m, S75m and S85m. These glasses were synthesized in thepresence of the nonionic triblock copolymer (P123) and with dif-ferent CaO contents (37%, 20% and 10% mol for S58m, S75m andS85m, respectively). S58m glass shows a unique low intense max-imum at 2h = 1.28� (d10 = 6.86 nm). This pattern is consistent witha hexagonal structure with 1d pore channels oriented parallel.S75m glass shows two well-resolved maxima at 2h = 1.54�(d10 = 5.72 nm) and 2.86� (d11 = 30.8 nm), corresponding to 10and 11 reflections of a 2D-hexagonal phase. In addition, avery weak maximum can be distinguished at 2h = 1.27�(d11 = 6.94 nm), corresponding to the 11 reflection of a 2D-ortho-rhombic phase. Finally, S85m glass shows three diffraction maximaat 2h = 1.20� (d211 = 7.35 nm), 1.99� (d400 = 4.42 nm), and 2.31�(d332 = 3.82 nm). These maxima can be indexed to the 211, 400and 332 reflections of a 3D cubic phase. S75m and S85m presenteda pore size around 6 nm whereas S58m shows larger pores of about
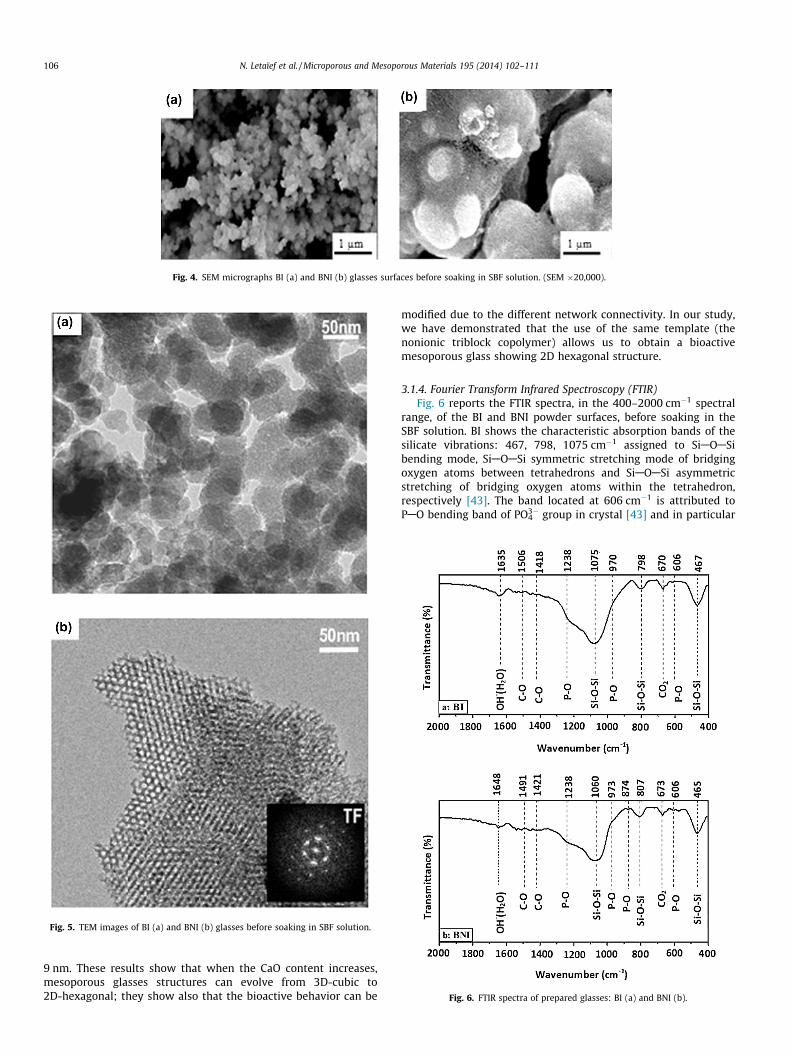
Fig. 4. SEM micrographs BI (a) and BNI (b) glasses surfaces before soaking in SBF solution. (SEM �20,000).
Fig. 5. TEM images of BI (a) and BNI (b) glasses before soaking in SBF solution.
Fig. 6. FTIR spectra of prepared glasses: BI (a) and BNI (b).
106 N. Letaïef et al. / Microporous and Mesoporous Materials 195 (2014) 102–111
9 nm. These results show that when the CaO content increases,mesoporous glasses structures can evolve from 3D-cubic to2D-hexagonal; they show also that the bioactive behavior can be
modified due to the different network connectivity. In our study,we have demonstrated that the use of the same template (thenonionic triblock copolymer) allows us to obtain a bioactivemesoporous glass showing 2D hexagonal structure.
3.1.4. Fourier Transform Infrared Spectroscopy (FTIR)Fig. 6 reports the FTIR spectra, in the 400–2000 cm�1 spectral
range, of the BI and BNI powder surfaces, before soaking in theSBF solution. BI shows the characteristic absorption bands of thesilicate vibrations: 467, 798, 1075 cm�1 assigned to SiAOASibending mode, SiAOASi symmetric stretching mode of bridgingoxygen atoms between tetrahedrons and SiAOASi asymmetricstretching of bridging oxygen atoms within the tetrahedron,respectively [43]. The band located at 606 cm�1 is attributed toPAO bending band of PO4
3� group in crystal [43] and in particular
N. Letaïef et al. / Microporous and Mesoporous Materials 195 (2014) 102–111 107
in carbonated hydroxyapatite (CHA) [3]. A weak band appearing asshoulder at 970 cm�1 may be related to PAO stretching vibrationsin tetracalcium phosphate [44]. It was probably due to a glass sur-face reaction with air water and oxygen to form an amorphousapatite-like phase. The band observed at 1238 cm�1 is not associ-ated to the presence of apatite-like phase (see comparison onFig. 9) but related to the presence of PO4
3� groups in the glassstructure. For glasses with low content of P2O5, phosphorous isnot considered as a glass former like silicon, but phosphorous ispresent in the glass structure as PO4
3� ions like a glass modifier[45]. The peaks at 1418 and 1506 cm�1 are assigned to CAO vibra-tions in carbonate CO3
2� group. The band at 670 cm�1 is due to thepresence of some adsorbed CO2 at the glass surface [46]. The bandaround 1638 cm�1 is the deformation mode of OAHAO groupattributed to the adsorbed water molecules [47]. Comparativelyto BI glass, BNI glass includes all above mentioned bands. Supple-mentary band is observed at 874 cm�1. This band may be linked toCO3
2� groups. This phenomenon was observed by Mezahi et al., andthey attributed this vibration to the carbonate group in carbonatedapatite [48].
So it is clear that the surfactant nature did not influence themain structural features of the synthesized glass. In both case theglasses were synthesized using sol–gel method without changingcomposition. If the composition or the synthesis mode (meltingversus sol–gel) change, some differences in the position of vibra-tions for SiO4 tetrahedrons are observed [45,49]. This phenomenonis not observed here and the glass structure is not surfactant-dependent.
3.2. In vitro analysis
Variations versus soaking time of ionic concentrations and solu-tion pH, as well as changes in the glass surface are related to therate of biomineralization of biomaterials after immersion in simu-lated body fluid. The evolution of the glass surface was analyzed byXRD, FTIR and SEM techniques. The variations of phosphorus,
Fig. 7. Evolution of Si (a), Ca (b) and P (c) concentrations in SBF, and e
silicon and calcium concentrations in SBF, as a function of soakingtime, were measured by ICP-OES.
3.2.1. [Si] evolutionThe silicon concentration in SBF increases with a high rate after
the first day for the two glasses BI and BNI (Fig. 7a). Afterwards, forBNI glass, it continues to rising from 41.3 to reach 59.6 ppm after30 days. This phenomenon is related to glass dissolution. For BIglass, Si concentration remains constant between 2 and 30 days.It reaches 41.01 ppm after 30 days. Compared to BNI glass, the con-centration of silicon in SBF for BI glass is lower. It can be explainedby the increased textural characteristics (surface area, porosity) ofBNI glass supplied by the P123 template.
3.2.2. [Ca] and [P] evolutionsThe concentrations of calcium and phosphorus in SBF solution
followed the same tendency in the case of both BI and BNI glasses(Fig. 7b and c). When the BI glass powder was introduced in SBFsolution, Ca and P were released in SBF from the glass surface for2 days. This release could be attributed to the transfer of a largequantity of the glass modifiers (calcium and phosphate ions) fromthe surface of 92S6. Between 2 and 15 days, there is a fluctuation inCa and P concentrations. After that, Ca and P concentrationsdecrease in SBF. However, for BNI glass powder, Ca and P concen-trations decreased in SBF up to 15 days. After 15 days of immersionin SBF, Ca and P concentrations increased from 103.3 and 6.8 ppmto 107.3 and 10.2 ppm, respectively. Then, the calcium and phos-phorus ions concentrations decreased very strongly from 21 days.After 30 days, the concentrations of calcium and phosphorus inSBF were 100 and 4.9 ppm, respectively.
The decrease of the phosphorous concentration in SBF indicatedthat there was a phosphorous uptake by the glass surface. Thisdecrease combined with the decrease of Ca concentrations in theSBF solution confirmed the growth of an apatite-like phase withthe soaking time. The growth from the apatite starts from the ionicexchange between the material surface and the surrounding fluid.
volution of SBF pH (d), versus soaking time for BI and BNI glasses.
108 N. Letaïef et al. / Microporous and Mesoporous Materials 195 (2014) 102–111
The obtained results are consistent with those obtained for the60S glass obtained by Mami et al. [50] and for 52S4 glass obtainedby Mezahi et al. [51]: a release of calcium from surface glass, a lim-ited dissolution of the glass surface and an uptake of phosphorousearly after immersion.
3.2.3. pH evolutionThe pH of sample solutions was measured (Brinkmann 632 dig-
ital pH meter) before and after immersion in SBF during 2, 7, 15, 21and 30 days. The pH of the sample solutions of BNI and BI glassesvaried between 7.4 and 7.8 and between 7.4 and 7.5, respectivelyfor the entire experiment duration (Fig. 7d). Thus, pH slightlyincreased during the two first days for the two glasses. It continuesto rising from 7.7 to 7.8 after 15 days for BNI glass. After that, itdecreased between the 15 and 21 days to 7.6. However, the pHvalue of SBF solution of BI glass decreased between two and21 days soaking time. After 21 days, the pH values of SBF for BNIand BI glasses increase slowly to reach a value of 7.8, 7.5 pH units,respectively, after 30 days assay.
The intense ionic exchange during the first stages of the bioac-tive process leads to local pH increase. Therefore, the initialincrease in pH may be related to the release in Ca ions from glassto SBF and the uptake of H+ present in the SBF by the glass surface.Depending on the sink conditions of the area (blood perfusion,mainly) the pH increase can be toxic for surrounding tissues [16].BI and BNI glasses produce a slight initial increase in pH as the
Fig. 8. WAXRD patterns of glass surfaces before and after different soaking times inSBF solution: BI (a) and BNI (b).
Ca2+ release is very low. So these samples are excellent candidatesas grafts for bone tissue regeneration.
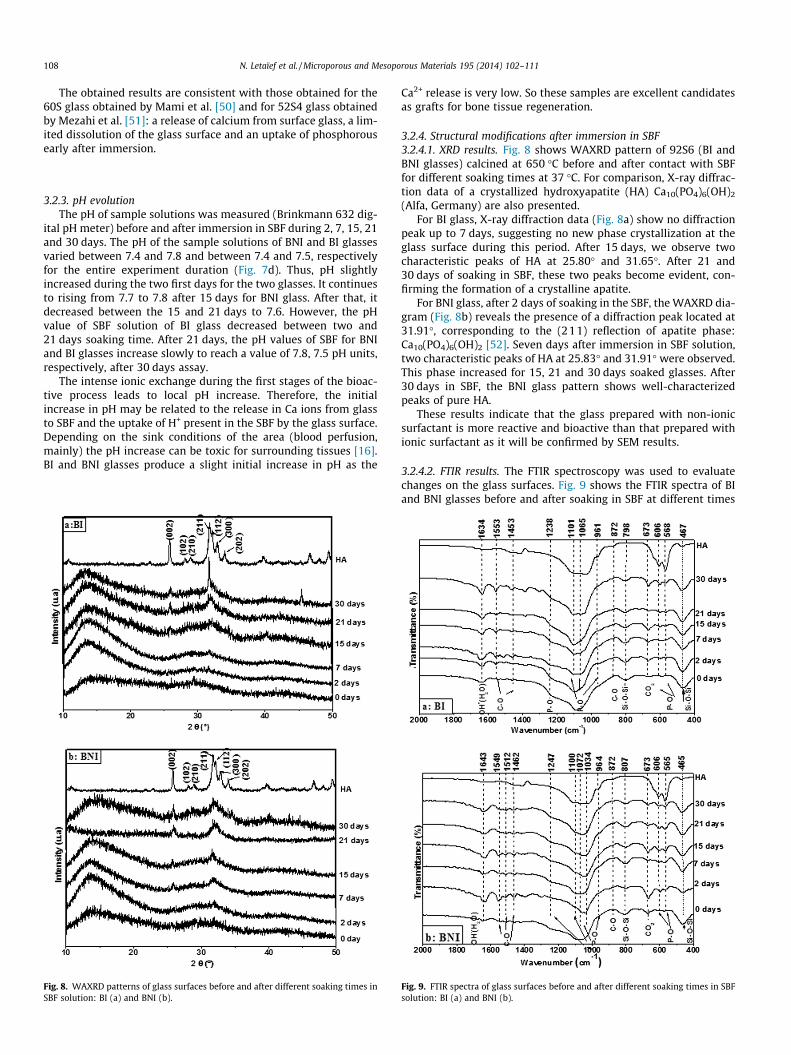
3.2.4. Structural modifications after immersion in SBF3.2.4.1. XRD results. Fig. 8 shows WAXRD pattern of 92S6 (BI andBNI glasses) calcined at 650 �C before and after contact with SBFfor different soaking times at 37 �C. For comparison, X-ray diffrac-tion data of a crystallized hydroxyapatite (HA) Ca10(PO4)6(OH)2
(Alfa, Germany) are also presented.For BI glass, X-ray diffraction data (Fig. 8a) show no diffraction
peak up to 7 days, suggesting no new phase crystallization at theglass surface during this period. After 15 days, we observe twocharacteristic peaks of HA at 25.80� and 31.65�. After 21 and30 days of soaking in SBF, these two peaks become evident, con-firming the formation of a crystalline apatite.
For BNI glass, after 2 days of soaking in the SBF, the WAXRD dia-gram (Fig. 8b) reveals the presence of a diffraction peak located at31.91�, corresponding to the (211) reflection of apatite phase:Ca10(PO4)6(OH)2 [52]. Seven days after immersion in SBF solution,two characteristic peaks of HA at 25.83� and 31.91� were observed.This phase increased for 15, 21 and 30 days soaked glasses. After30 days in SBF, the BNI glass pattern shows well-characterizedpeaks of pure HA.
These results indicate that the glass prepared with non-ionicsurfactant is more reactive and bioactive than that prepared withionic surfactant as it will be confirmed by SEM results.
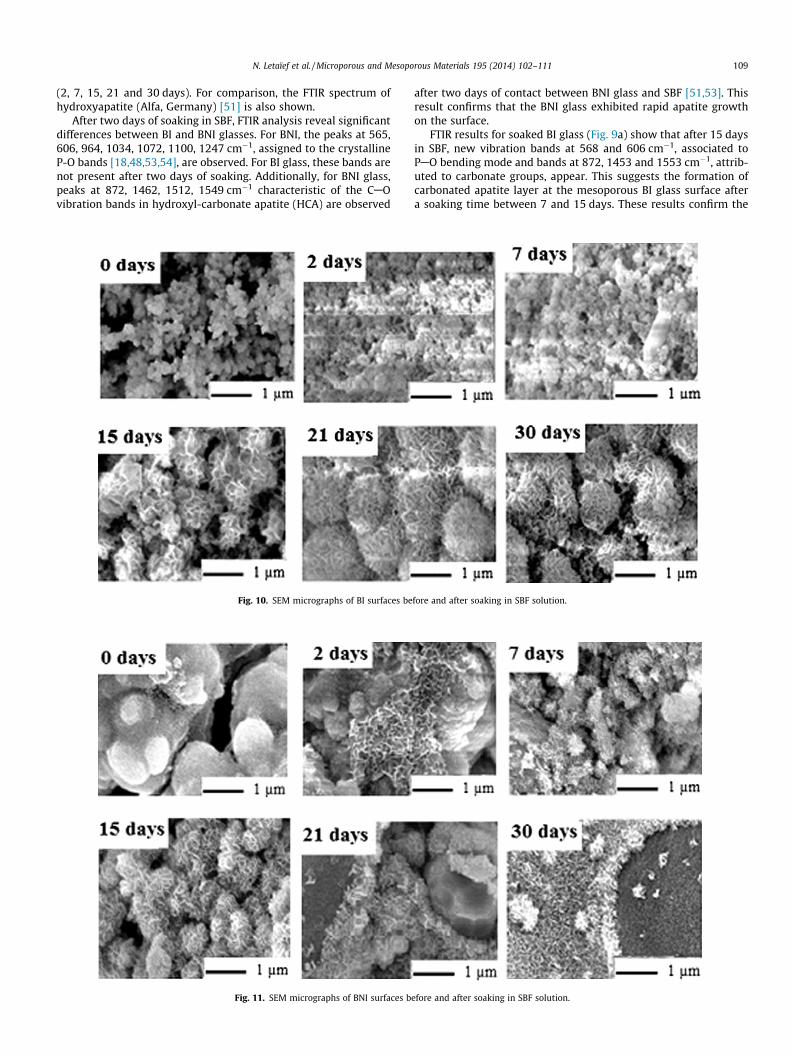
3.2.4.2. FTIR results. The FTIR spectroscopy was used to evaluatechanges on the glass surfaces. Fig. 9 shows the FTIR spectra of BIand BNI glasses before and after soaking in SBF at different times
Fig. 9. FTIR spectra of glass surfaces before and after different soaking times in SBFsolution: BI (a) and BNI (b).
N. Letaïef et al. / Microporous and Mesoporous Materials 195 (2014) 102–111 109
(2, 7, 15, 21 and 30 days). For comparison, the FTIR spectrum ofhydroxyapatite (Alfa, Germany) [51] is also shown.
After two days of soaking in SBF, FTIR analysis reveal significantdifferences between BI and BNI glasses. For BNI, the peaks at 565,606, 964, 1034, 1072, 1100, 1247 cm�1, assigned to the crystallineP-O bands [18,48,53,54], are observed. For BI glass, these bands arenot present after two days of soaking. Additionally, for BNI glass,peaks at 872, 1462, 1512, 1549 cm�1 characteristic of the CAOvibration bands in hydroxyl-carbonate apatite (HCA) are observed
Fig. 10. SEM micrographs of BI surfaces be
Fig. 11. SEM micrographs of BNI surfaces b
after two days of contact between BNI glass and SBF [51,53]. Thisresult confirms that the BNI glass exhibited rapid apatite growthon the surface.
FTIR results for soaked BI glass (Fig. 9a) show that after 15 daysin SBF, new vibration bands at 568 and 606 cm�1, associated toPAO bending mode and bands at 872, 1453 and 1553 cm�1, attrib-uted to carbonate groups, appear. This suggests the formation ofcarbonated apatite layer at the mesoporous BI glass surface aftera soaking time between 7 and 15 days. These results confirm the
fore and after soaking in SBF solution.
efore and after soaking in SBF solution.

110 N. Letaïef et al. / Microporous and Mesoporous Materials 195 (2014) 102–111
formation of HCA layer on the surface of BNI glass faster than on BIglass.
3.2.4.3. SEM results. After soaking in SBF, changes of glass surfacemorphology and particularly formation of apatite layer wereevaluated by SEM microscopy. Untreated glass surface of BI glass(Fig. 10) has structured morphology constituted by spheres withan average diameter of 0.4 lm. The surface morphology changeswith incubation periods. 15-days surface shows the formation ofindividual apatite grains with 0.5 lm size. With increasingincubations periods, apatite grains grow like spherical balls withan average diameter of 1.5 lm at 21 days. After 30 days surfaceshows 3 lm size clusters with cauliflower like morphologycovering the entire surface.
For the sample BNI (Fig. 11), the surface of the glass is smoothand we do not observe spheres like for BI glass. The surface ofBNI glass exhibits important changes after soaking in SBF after2 days. A layer composed of needle-shaped crystallites covers thesurface. After 7 days, the layer is constituted by spherical particlescoating the whole surface of the BNI glass.
So, the clusters and the grains observed at these surfaces areattributed to the formation of phosphate phases (HCA as confirmedby XRD and FTIR) for both BI and BNI glasses. Furthermore, thethickness of the HCA layer and the spherical particles withneedlelike crystallites increase with the soaking time in SBF. Theabove results indicate that the BNI glass can induce the formationof a HCA layer on its surface even for short (2 days) soaking periodsin SBF. Our results indicate that the difference between BI and BNIglasses is noticeable. BI glass, a SiO2–CaO–P2O5 sol–gel glass, with acomplete non-ordered mesoporous structure, exhibits a slowerkinetics of bioactivity than BNI glass. In contrast, BNI glass, thesame SiO2–CaO–P2O5 sol–gel glass, with a highly ordered mesopor-ous structure, allows a fast nucleation and rapid apatite growth onits surface. In fact, the textural studies demonstrate that BNI glasspossess better textural properties than those of BI glass for a rapidkinetics of bioactivity. So, we can conclude that, for a same compo-sition, the main factor that contributes to the crystallization rate ofthe apatite phase on the surface is the textural properties, as it waspreviously demonstrated by Lopez-Noriega et al. [16]. Theseauthors demonstrated that S85m, composition close to our glasscomposition (% mol 85 SiO2-10 CaO-5 P2O5) shows the best crystal-line apatite rate formation due to its excellent textural propertiesand structural characteristics: crystalline CaP is detected after 4 hin SBF for S85m, whereas S58m and S75m show the first tracesof crystalline CaP after 24 and 8 h, respectively.
4. Conclusion
A mesoporous bioactive glass 92SiO2–6CaO–2P2O5 has beensynthesized by sol–gel method, using surfactant CTAB or triblockcopolymer P123 as template. A study of the textural and structuralproperties of theses glasses has been carried out.
The sample synthesized with the cationic surfactant (BI glass)presents a mesoporous structure without further organization. Asalready mentioned in Section 3.1, the use of the cationic surfactantis probably responsible for the lack of regular and periodic arrange-ment. Well-defined and dense spherical mesoporous glass particlesare obtained. SBF immersion tests reveal that BI glass required15 days in SBF to form a new apatite phase, indicating the low bio-active behavior of this sol–gel glass. After 21 days in SBF, the sur-face appears fully covered by crystallites of calcium phosphatecharacteristic of the growth of apatite phase on a bioactive mate-rial surface. However, the use of the triblock copolymer P123(BNI glass) induces a smooth morphology and the formation ofan ordered mesoporous glass. Additionally, higher specific area,
pore volume and pore size lead to fast ionic exchanges with sur-rounding medium, and contribute to a high bioactive feature BNIglass. Indeed, BNI glass develops a thin apatite layer at the surfaceafter only 2 days of soaking in SBF. This apatite phase shows a fastnucleation and growth and it becomes crystallized after 15 days inSBF, indicating that the BNI glass shows the best crystalline apatiterate formation. Furthermore, the ordered mesopores will favorhomogeneous distribution of encapsulated therapeutic substancein a potential composite.
So, our mesoporous glasses are bioactive. An apatite-like phaseis formed at the glasses surface when soaked in SBF, as confirmedby FTIR spectroscopy results and SEM observations. These charac-teristic mesoporous glasses will display good future applications inbone tissue repairing and engineering.
Acknowledgments
The authors would like to acknowledge Vincet Dorcet fromInstitut des Sciences Chimiques de Rennes, for discussion on poros-ity organization and small angle diffraction, and Joseph Le Lannic,from the CMEBA, Université de Rennes 1, for SEM. OdileMerdrignac-Conanec, from Laboratoire Verres et Céramiques,Université de Rennes 1, for BET studies.
References
[1] M. Alcaide, P. Portoles, A. Lopez-Noriega, D. Arcos, M. Vallet-Regí, M.T. Portoles,Acta Biomater. 6 (2010) 892–899.
[2] L.L. Hench, J.M. Polak, Biomed. Mater. Sci. 295 (2002) 1014–1017.[3] L.L. Hench, J. Am. Ceram. Soc. 74 (1991) 1487–1510.[4] W. Xia, J. Chang, J. Control. Release 110 (2006) 522–530.[5] V. Cannillo, F. Pierli, I. Ronchetti, C. Siligardi, D. Zaffe, Ceram. Int. 35 (2009)
2853–2869.[6] J.M. Oliveira, R.N. Correia, M.H. Fernandes, Biomaterials 23 (2002) 371–379.[7] Q.H. Shi, J.F. Wang, J.P. Zhang, J. Fan, G.D. Stucky, Adv. Mater. 18 (2006) 1038–
1345.[8] M. Vallet-Regí, I. Izquierdo-Barba, A.J. Salinas, J. Biomed. Mater. Res. 46 (1999)
560–565.[9] M. Vallet-Regí, A.J. Salinas, J. Roman, M. Gil, J. Mater. Chem. 9 (1999) 515–519.
[10] A.J. Salinas, A.I. Martin, M. Vallet-Regí, J. Biomed. Mater. Res. 61 (2002) 524–530.
[11] M. Vallet-Regí, C.V. Ragel, A.J. Salinas, J. Inorg. Chem. 6 (2003) 1029–1037.[12] C.T. Kresge, M.E. Leonowicz, W.J. Roth, J.C. Vartuli, J.S. Beck, Nature 359 (1992)
710–714.[13] Y. Guo, Y. Zhou, D. Jia, H. Tang, Micropor. Mesopor. Mater. 118 (2009) 480–488.[14] A. Balamurugan, G. Balossier, S. Kannan, J. Michel, A.H.S. Rebelo, J.M.F. Ferreira,
J. Acta Biomater. 3 (2007) 255–262.[15] X.X. Yan, H.X. Deng, X.H. Huang, G.Q. Lu, S.Z. Qiao, D.Y. Zhao, C.Z. Yu, J. Non-
Cryst. Solids 351 (2005) 3209–3217.[16] A. Lopez-Noriega, D. Acros, I. Izquierdo-Barba, Y. Sakamoto, O. Terasaki, M.
Vallet-Regí, Chem. Mater. 18 (2006) 3137–3144.[17] C.J. Shih, H.T. Chen, L.F. Huang, P.S. Lu, H.F. Chang, I.L. Chang, Mater. Sci. Eng. C
30 (2010) 657–663.[18] Y. Zhu, C. Wu, Y. Ramaswamy, E. Kockrick, P. Simon, S. Kaskel, H. Zreiqat,
Micropor. Mesopor. Mater. 112 (2008) 494–503.[19] M. Vallet-Regí, F. Balas, Open Biomed. Eng. J. 2 (2008) 1–9.[20] K.K. Unger, D. Kumar, M. Grun, G. Buchel, S. Ludtke, T. Adam, K. Schumacher, S.
Renker, J. Chromatogr. A 892 (2000) 47–55.[21] M. Gruen, I. Lauer, K.K. Unger, Adv. Mater. 9 (1997) 254–257.[22] P. Sepulveda, J.R. Jones, L.L. Hench, J. Biomed. Mater. Res. 59 (2002) 340–344.[23] Q. Yang, S.H. Wang, P.W. Fan, L.F. Wang, Y. Di, K.F. Lin, F.S. Xiao, Chem. Mater.
17 (2005) 5999–6005.[24] S. Wang, Micropor. Mesopor. Mater. 117 (2009) 1–9.[25] Y. Yamashita, A. Uchida, T. Yamakawa, Y. Shinto, N. Araki, K. Kato, Int. Orthop.
22 (1998) 247–254.[26] I.I. Slowing, J.L. Vivero-Escoto, C.W. Wu, V.S.Y. Lin, Adv. Drug. Deliv. Rev. 60
(2008) 1278–1288.[27] M. Vallet-Regí, C. R. Chem. 13 (2010) 174–185.[28] L. Sierra, S. Valange, J. Barrault, J.L. Guth, Micropor. Mesopor. Mater. 113 (2008)
352–361.[29] T.W. Kim, R. Ryoo, M. Kruk, K.P. Gierszal, M. Jaroniec, S. Kamiya, O. Terasaki, J.
Phys. Chem. B 108 (2004) 11480–11487.[30] L. Wang, J. Fan, B. Tian, H. Yang, Ch. Yu, B. Tu, D. Zhao, Micropor. Mesopor.
Mater. 67 (2004) 135–141.[31] Á. Szegedi, Z. Kónya, D. Méhn, E. Solymár, G. Pál-Borbély, Z.E. Horváth, L.P. Biró,
I. Kiricsi, Appl. Catal. A 272 (2004) 257–266.[32] D. Zhao, J. Feng, Q. Huo, N. Melosh, G.H. Fredrickson, B.F. Chmelka, G.D. Stucky,
Science 279 (1998) 548–552.

N. Letaïef et al. / Microporous and Mesoporous Materials 195 (2014) 102–111 111
[33] T. Kokubo, H. Hushitani, S. Sakka, S.T. Yamamuro, J. Biomed. Mater. Res. 24(1990) 721–727.
[34] A. Saboori, M. Rabiee, F. Moztarzadeh, M. Sheikhi, M. Tahriri, M. Karimi, Mater.Sci. Eng. C 29 (2009) 335–340.
[35] J. Ma, C.Z. Chen, D.G. Wang, J.Z. Shi, Mater. Sci. Eng. C30 (2010) 886–890.[36] K.S.W. Sing, D.H. Everett, R.A.W. Haul, L. Moscou, R.A. Pierotti, J. Rouquérol, T.
Siemienniewska, Pure Appl. Chem. 57 (1985) 603–619.[37] R. Mortera, B. Onida, S. Fiorilli, V. Cauda, C. Vitale Brovarone, F. Baino, E. Vernè,
E. Garrone, J. Chem. Eng. 137 (2008) 54–61.[38] X.X. Yan, C.Z. Yu, X.F. Zhou, J.W. Tang, D.Y. Zhao, Angew. Chem. Int. 43 (2004)
5980–5984.[39] G.D. Stucky, D. Zhao, P. Yang, W. Lukens, N. Melosh, B.F. Chmelka, in: L.
Bonneviot, F. Bélarid, C. Danumah, S. Giasson, S. Kaliagine (Eds.), MesoporousMolecular Sieves 1998, Studies in Surface Science and Catalysis, vol. 117,Elsevier, Amsterdam, 1998, pp. 1–12.
[40] H. Wang, L. Zhai, Y. Li, T. Shi, J. Mater. Res. Bull. 43 (2008) 1607–1614.[41] R. Zana, J. Frasch, M. Soulard, B. Lebeau, J. Patarin, Langmuir 15 (1999) 2603–
2610.[42] L. Sierra, B. Lopez, J.-L. Guth, Micropor. Mesopor. Mater. 39 (2000) 519–527.[43] H.A. Elbatal, M.A. Azooz, E.M.A. Khalil, A. Soltan Monem, Y.M. Hamdy, Mater.
Chem. Phys. 80 (2003) 599–603.
[44] A. Jillavenkatesa, R.A. Condrate, Spec. Lett. 31 (1998) 1619–1627.[45] A. Lucas-Girot, F.Z. Mezahi, M. Mami, H. Oudadesse, A. Harabi, M. Le Floch, J.
Non-Cryst. Solids. 357 (2011) 3322–3327.[46] S.G. Kazarian, M.F. Vincent, F.V. Bright, C.L. Liotta, C.A. Eckert, J. Am. Chem. Soc.
118 (1996) 1729–1738.[47] C.Y. Kim, A.E. Clark, L.L. Hench, J. Biomed. Mater. Res. 26 (1992) 1147–
1153.[48] F.Z. Mezahi, A. Lucas-Girot, H. Oudadesse, A. Harabi, J. Non-Cryst, Solids 361
(2013) 111–118.[49] M. Mami, A. Lucas-Girot, H. Oudadesse, R. Dorbez-Sridi, F. Mezahi, E. Dietrich,
Appl. Surf. Sci. 254 (2008) 7386–7393.[50] V.C. Farmer, Mineralogical Society, SW7 5HR London, 1974.[51] F.Z. Mezahi, H. Oudadesse, A. Harabi, A. Lucas-Girot, Y. Le Gal, H. Chaair, G.
Cathelineau, J. Therm. Anal. Calorim. 95 (1) (2009) 21–29.[52] Powder diffraction File, No. 09–0432, JCPDS, International Centre for
Diffraction Data, 2002.[53] M. Vallet-Regí, A.M. Romero, C.V. Ragel, R.Z. LeGeros, J. Biomed. Mater. Res. A
44 (1999) 416–421.[54] C. Garcia, S. Cere, A. Duran, J. Non-Cryst, Solids 348 (2004) 218–224.












