Embed Size (px)
Citation preview

PRISE EN CHARGE DES NMP
LA MYELOFIBROSE PRIMITIVE
Brigitte Dupriez. Hématologie Clinique. Centre Hospitalier de Lens
GFHC mai 2017

GFHC mai 2017
EPIDEMIOLOGIE de la MFP
parmi les NMP phi neg
• la + rare incidence : 4 à 7/106/an
• la + grave médiane de survie : 5 à 6 ans
• sujets âgés âge médian au diagnostic : 65 ans
(mais 20 % < 55 ans dont 1/2 < 45 ans)
(MF post TE/PV ? ≈ 10% à 15 ans)

GFHC mai 2017
PARTICULARITES parmi NMP:
la + complexe
• Myélofibrose cytopénies
BOM
• Hématopoïèse extramédullaire
splénomégalie ++ et autres sites
• Hyperproduction cytokines
signes généraux

LE DIAGNOSTIC
Est-ce UNE MFP ?
GFHC mai 2017

LE DIAGNOSTIC est le + souvent facile dans la forme « classique » devant une
splénomégalie avec anomalies caractéristiques de l’hémogramme
GFHC mai 2017
• Rouge anémie (3/4)
dacryocytes (déformation des GR)
érythroblastes
• Blanche hyperleucocytose modérée (10 à 25.109/l)
myélémie blastes (peu)
monocytes normaux
• Plaquettes taux ↑↓ ( N 1/2, 1/4 , 1/4 )
myelo pauvre; BOM confirme; mutations vectrices
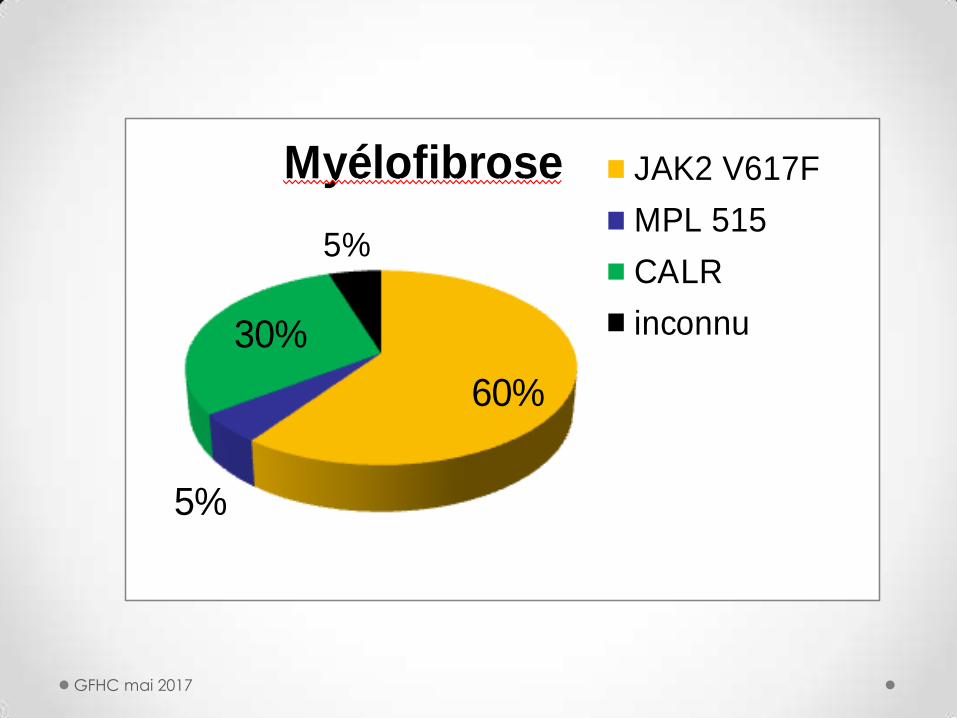
60%
5%
30%
5%
Myélofibrose JAK2 V617F
MPL 515
CALR
inconnu
GFHC mai 2017

Myélofibrose Primitive « classique »
GFHC mai 2017
OMS 2008 ( +préfibrose) OMS 2016 Critères majeurs
1
Prolifération mégacaryocytaire avec atypie + fibrose réticulinique et/ou collagène En absence de fibrose réticulinique, modifications morphologiques MK + une augmentation de la cellularité (prolifération granulocytaire et diminution de l’érythropoïèse)
Prolifération mégacaryocytaire avec atypie + fibrose réticulinique et/ou collagène (grade 2 ou 3)
2
Absence des critères OMS de LMC, PV, TE, SMD ou d’une autre hémopathie maligne de la lignée myéloïde
Absence des critères OMS de LMC, PV, TE, SMD ou d’une autre hémopathie maligne de la lignée myéloïde
3 Mutation JAK2 ou autre marqueur clonalité ou absence de cause de fibrose réactionnelle
Mutation JAK2, CALR ou MPL ou autre marqueur ou absence de cause de fibrose réactionnelle
Critères mineurs
1 Erythromyélémie Erythromyélémie
2 Augmentation des LDH Augmentation des LDH
3 Anémie Anémie sans autre cause
4 Splénomégalie palpable Splénomégalie palpable
5 Leucocytose > 11 G/L …
D 3 critères Maj + 1 mineur 3 critères Majeurs et au moins 1 critère mineur

hématologiques non hématologiques
Hémopathies myéloïdes Hémopathies lymphoïdes
NMP
• Myélofibrose primitive
• MF post PV
• MF post TE
• LMC
• SHE
• Mastocytose systémique
SMD et SMD/NMP++
Hémopathies aiguës
• LAM7, myélofibrose aiguë
• Histiocytose maligne
Leucémie à Tricholeucocytes
Maladie de Hodgkin
Myélome
Maladie de Waldenström
Extension médullaire de LNH
Autres : tt par analogue TPO
malignes
• Carcinome métastatique
immunologiques
• Myélofibrose autoimmune
(lupus systémique,…)
métaboliques
• hyperparathyroïdie
• rachitisme
• ostéodystrophie rénale
infectieuses
• VIH
• leishmaniose viscérale
• tuberculose
autres
• maladie de Paget
• maladie des plaquettes grises
Fibrose médullaire n’est pas synonyme de MFP…
GFHC mai 2017

GFHC mai 2017
DIAGNOSTIC DIFFERENTIEL
parfois difficile surtout quand myélo pauvre
• essentiellement les SMD avec MF
++ attention qd triple nég splénomégalie modérée ou absente,pancytopénie,CD34+ normales
• et les formes frontières SMD/NMP
d’autant que mutation JAK2 possible
nota LMMC : monocytose ; richesse médullaire , dysplasie au myélo
CD 34+
cytogénétique et NGS peuvent aider mais non discriminants
(anomalies communes bien que de fréquences différentes)

LA PREFIBROSE (Thiele J et al, classification OMS 2001)
séries allemandes de thrombocytoses avec BOM
• présentation initiale évoquant TE
• y compris absence de fibrose médullaire (ou grade 1)
• mais anomalies médullaires des MGK TE
• évolution fréquente vers le tableau classique de la MFP… mais inconstante …
à distinguer
de la TE : intérêt pratique de la distinction ? du « MDS/ MPN- RS-T » anciennement ARS-T ( svt JAK2+)
GFHC mai 2017

LA PREFIBROSE était incluse dans MFP OMS 2008
OMS 2008 OMS 2016 Critères majeurs
1
En absence de fibrose réticulinique, modifications morphologiques MK + augmentation de la cellularité (prolifération granulocytaire et diminution de l’érythropoïèse)
Prolifération mégacaryocytaire avec atypie augmentation de la cellularité pour l’âge sans fibrose réticulinique et/ou collagène grade 0 ou 1
2
Absence des critères OMS de LMC, PV, TE, SMD ou d’une autre hémopathie maligne de la lignée myéloïde
Absence des critères OMS de LMC, PV, TE, SMD ou d’une autre hémopathie maligne de la lignée myéloïde
3
Mutation JAK2 ou autre marqueur clonalité ou absence de cause de fibrose réactionnelle
Mutation JAK2, CALR ou MPL ou autre marqueur ou absence de cause de fibrose réactionnelle
Critères mineurs
1 Anémie Anémie sans autre cause
2 Splénomégalie palpable Splénomégalie palpable
3 Augmentation des LDH Augmentation des LDH
4 Erythromyélémie Leucocytose > 11 G/L
D 3 critères Maj + 1 mineur 3 critères Majeurs et au moins 1 critère mineur
GFHC mai 2017

TRAITEMENT ? oui ou non ?
Lequel ?
2 critères de décision :
le pronostic ( pour la décision d’allogreffe)
et les symptômes sinon
GFHC mai 2017
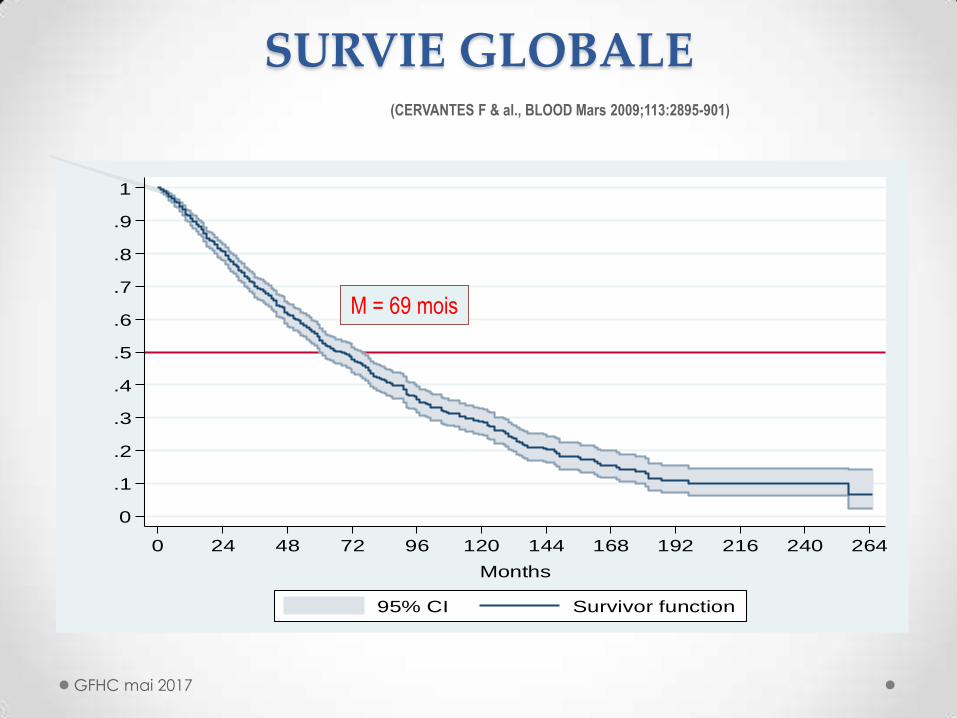
SURVIE GLOBALE
GFHC mai 2017
0
.1
.2
.3
.4
.5
.6
.7
.8
.9
1
Pro
babi
lity
0 24 48 72 96 120 144 168 192 216 240 264
Months
95% CI Survivor function
M = 69 mois
(CERVANTES F & al., BLOOD Mars 2009;113:2895-901)

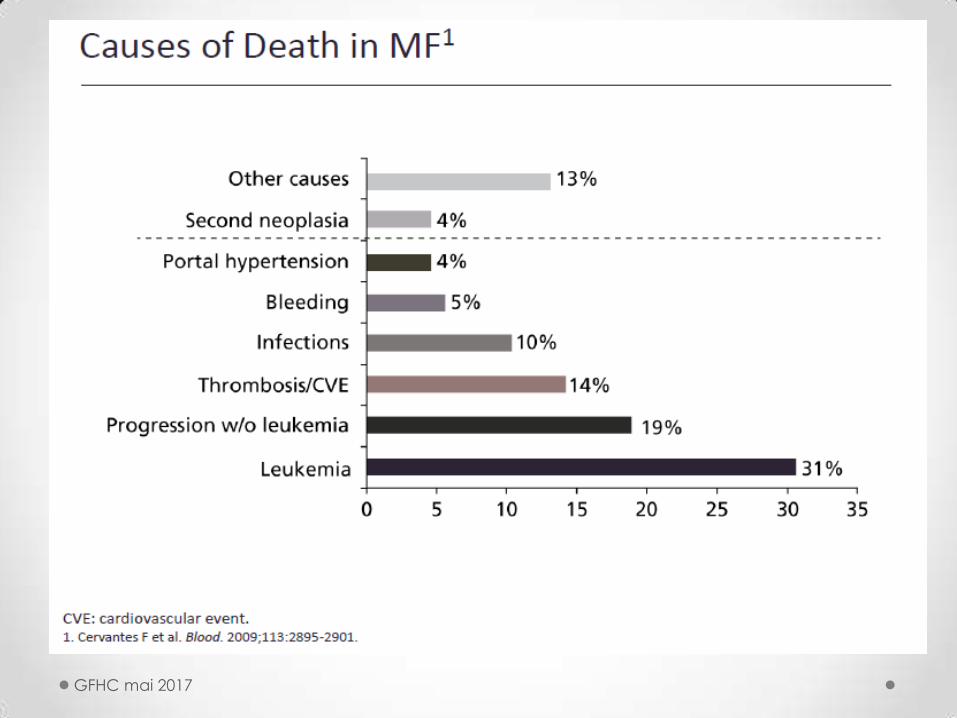
L’EVOLUTION
Longévité très variable : 1 à 25 ans (médiane 5 à 6)
• Thromboses et complications cardiovasculaires
• Hémorragies et infections
• Hypertension porte
• Foyers d’hématopoïèse ectopiques
• Signes généraux : amaig, Ht°,sueurs (« B ») , autres
(asthénie, prurit) … cachexie
• Transformation aiguë : 5 à 30% , survie réduite ( attitude palliative +/ vidaza sauf si greffable : induction type LA avant greffe)
GFHC mai 2017
GFHC mai 2017
GFHC mai 2017
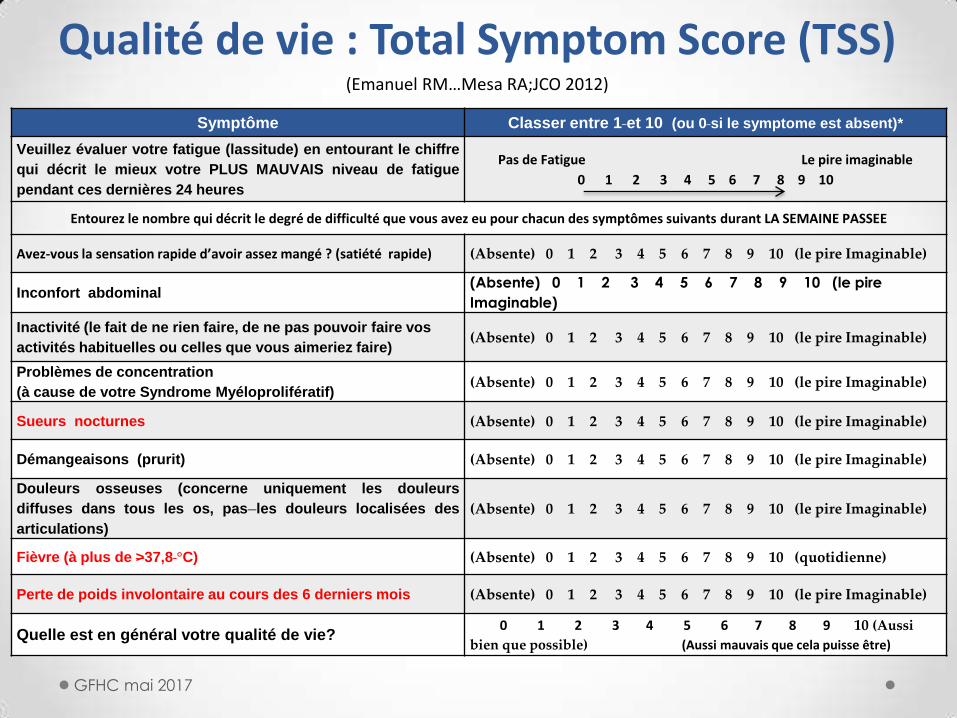
Symptôme Classer entre 1 et 10 (ou 0 si le symptome est absent)*
Veuillez évaluer votre fatigue (lassitude) en entourant le chiffre
qui décrit le mieux votre PLUS MAUVAIS niveau de fatigue
pendant ces dernières 24 heures
Pas de Fatigue Le pire imaginable
0 1 2 3 4 5 6 7 8 9 10
Entourez le nombre qui décrit le degré de difficulté que vous avez eu pour chacun des symptômes suivants durant LA SEMAINE PASSEE
Avez-vous la sensation rapide d’avoir assez mangé ? (satiété rapide) (Absente) 0 1 2 3 4 5 6 7 8 9 10 (le pire Imaginable)
Inconfort abdominal (Absente) 0 1 2 3 4 5 6 7 8 9 10 (le pire
Imaginable)
Inactivité (le fait de ne rien faire, de ne pas pouvoir faire vos
activités habituelles ou celles que vous aimeriez faire) (Absente) 0 1 2 3 4 5 6 7 8 9 10 (le pire Imaginable)
Problèmes de concentration
(à cause de votre Syndrome Myéloprolifératif) (Absente) 0 1 2 3 4 5 6 7 8 9 10 (le pire Imaginable)
Sueurs nocturnes (Absente) 0 1 2 3 4 5 6 7 8 9 10 (le pire Imaginable)
Démangeaisons (prurit) (Absente) 0 1 2 3 4 5 6 7 8 9 10 (le pire Imaginable)
Douleurs osseuses (concerne uniquement les douleurs
diffuses dans tous les os, pas les douleurs localisées des
articulations)
(Absente) 0 1 2 3 4 5 6 7 8 9 10 (le pire Imaginable)
Fièvre (à plus de >37,8 °C) (Absente) 0 1 2 3 4 5 6 7 8 9 10 (quotidienne)
Perte de poids involontaire au cours des 6 derniers mois (Absente) 0 1 2 3 4 5 6 7 8 9 10 (le pire Imaginable)
Quelle est en général votre qualité de vie? 0 1 2 3 4 5 6 7 8 9 10 (Aussi
bien que possible) (Aussi mauvais que cela puisse être)
Qualité de vie : Total Symptom Score (TSS) (Emanuel RM…Mesa RA;JCO 2012)

LE PRONOSTIC
du score de Lille au score
clinico-moléculaire…
GFHC mai 2017
GFHC mai 2017
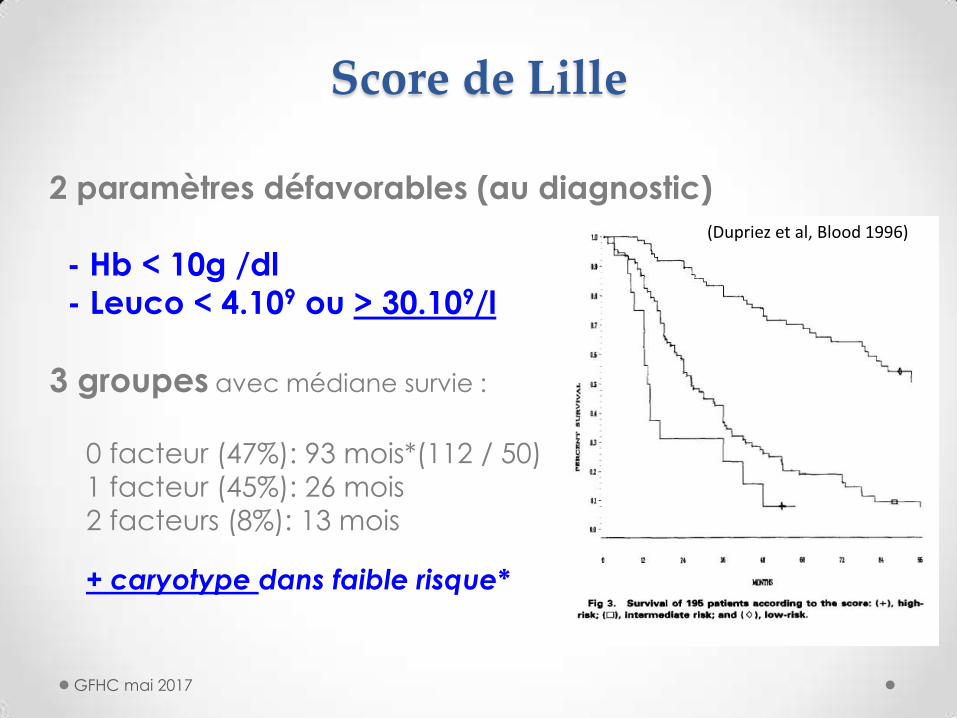
Score de Lille
2 paramètres défavorables (au diagnostic)
- Hb < 10g /dl
- Leuco < 4.109 ou > 30.109/l
3 groupes avec médiane survie :
0 facteur (47%): 93 mois*(112 / 50)
1 facteur (45%): 26 mois
2 facteurs (8%): 13 mois
+ caryotype dans faible risque*
(Dupriez et al, Blood 1996)
GFHC mai 2017
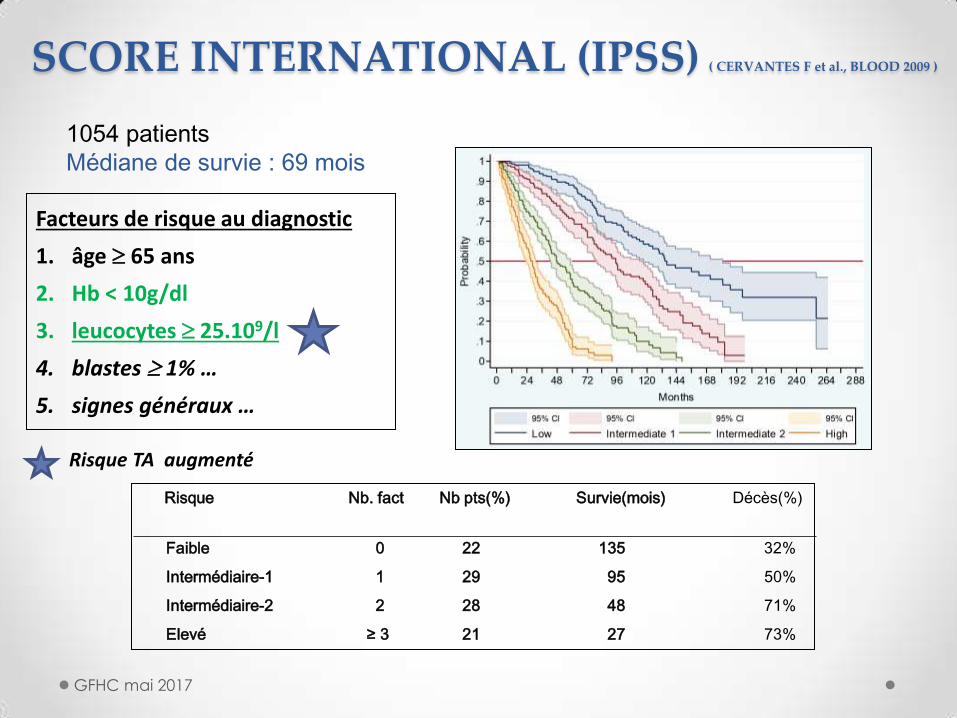
Facteurs de risque au diagnostic
1. âge 65 ans
2. Hb < 10g/dl
3. leucocytes 25.109/l
4. blastes 1% …
5. signes généraux …
Risque Nb. fact Nb pts(%) Survie(mois) Décès(%)
Faible 0 22 135 32%
Intermédiaire-1 1 29 95 50%
Intermédiaire-2 2 28 48 71%
Elevé ≥ 3 21 27 73%
1054 patients
Médiane de survie : 69 mois
Risque TA augmenté
SCORE INTERNATIONAL (IPSS) ( CERVANTES F et al., BLOOD 2009 )
GFHC mai 2017
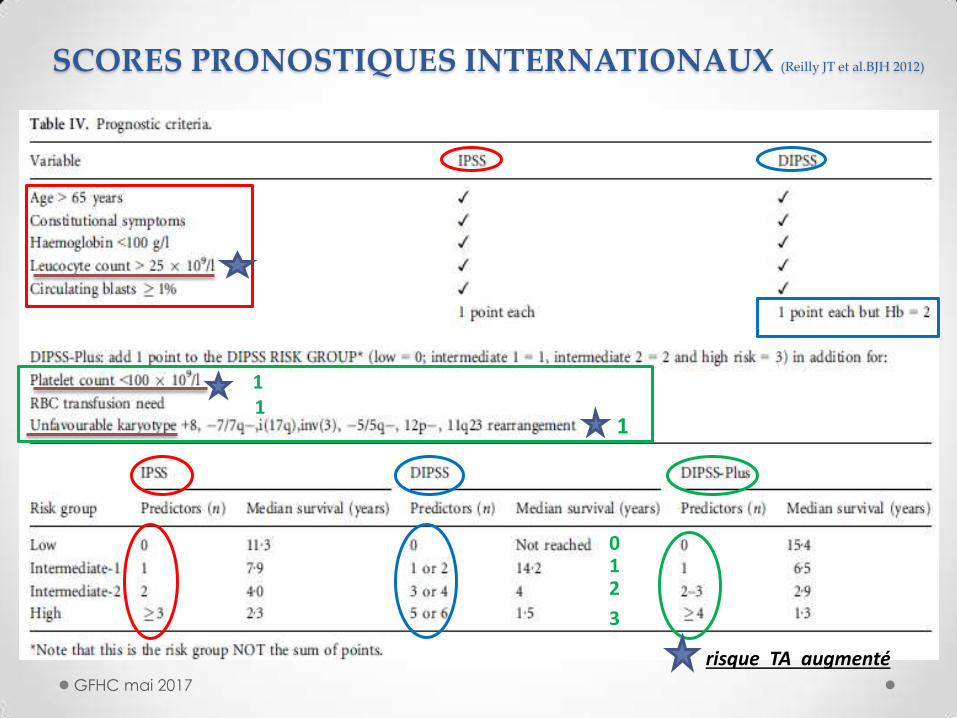
SCORES PRONOSTIQUES INTERNATIONAUX (Reilly JT et al.BJH 2012)
1 1
1
0 1 2
3
risque TA augmenté
GFHC mai 2017
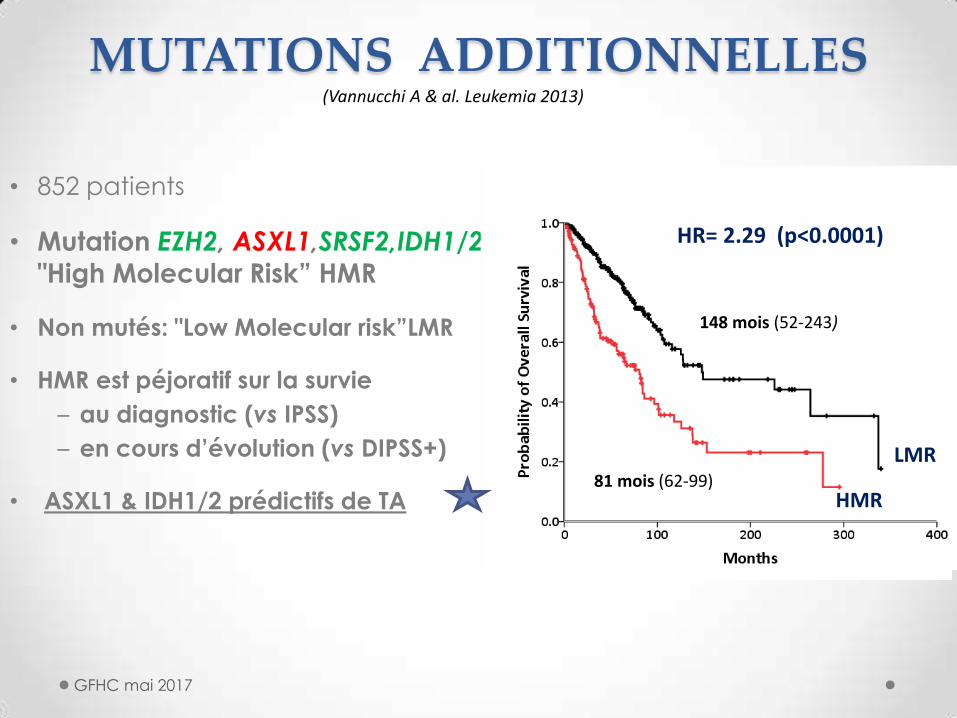
MUTATIONS ADDITIONNELLES
• 852 patients
• Mutation EZH2, ASXL1,SRSF2,IDH1/2
"High Molecular Risk” HMR
• Non mutés: "Low Molecular risk”LMR
• HMR est péjoratif sur la survie
– au diagnostic (vs IPSS)
– en cours d’évolution (vs DIPSS+)
• ASXL1 & IDH1/2 prédictifs de TA
LMR
HMR
(Vannucchi A & al. Leukemia 2013)
148 mois (52-243)
81 mois (62-99)
HR= 2.29 (p<0.0001)
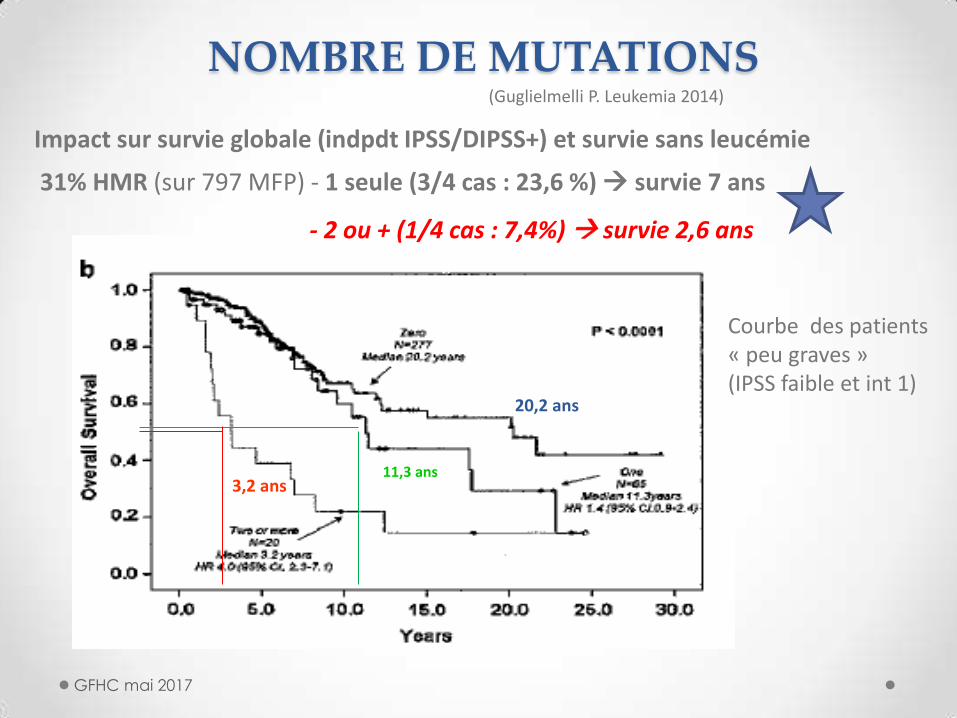
NOMBRE DE MUTATIONS
GFHC mai 2017
(Guglielmelli P. Leukemia 2014)
Impact sur survie globale (indpdt IPSS/DIPSS+) et survie sans leucémie
31% HMR (sur 797 MFP) - 1 seule (3/4 cas : 23,6 %) survie 7 ans
- 2 ou + (1/4 cas : 7,4%) survie 2,6 ans
3,2 ans 11,3 ans
20,2 ans
Courbe des patients « peu graves » (IPSS faible et int 1)
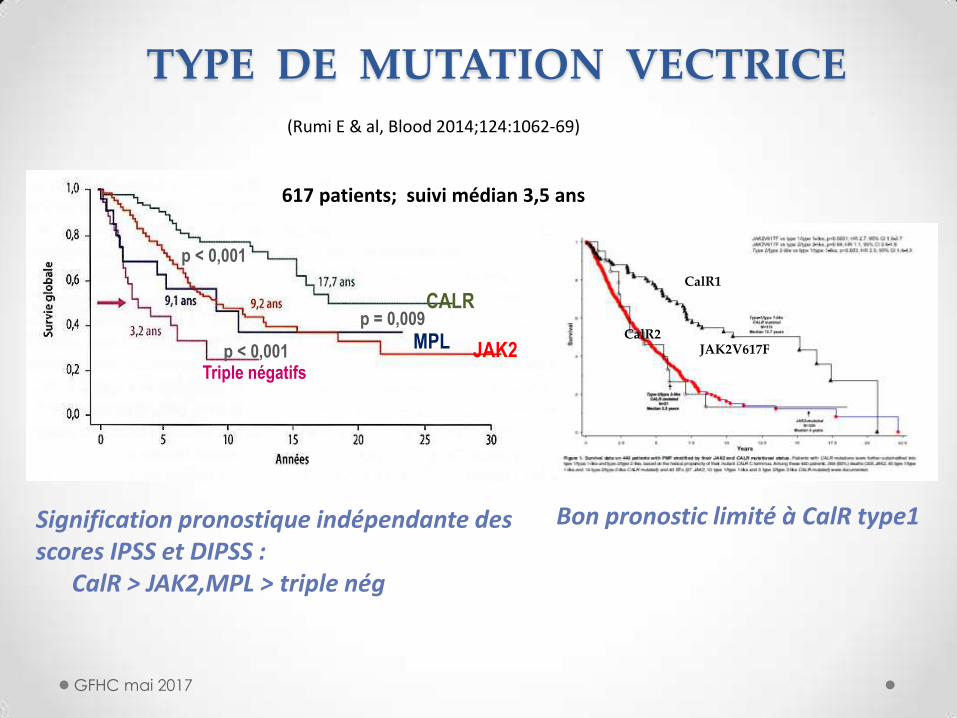
TYPE DE MUTATION VECTRICE
GFHC mai 2017
Signification pronostique indépendante des scores IPSS et DIPSS : CalR > JAK2,MPL > triple nég
p < 0,001
p = 0,009
p < 0,001 JAK2 MPL
CALR
Triple négatifs
(Rumi E & al, Blood 2014;124:1062-69)
617 patients; suivi médian 3,5 ans
CalR1
CalR2 JAK2V617F
Bon pronostic limité à CalR type1
GFHC mai 2017
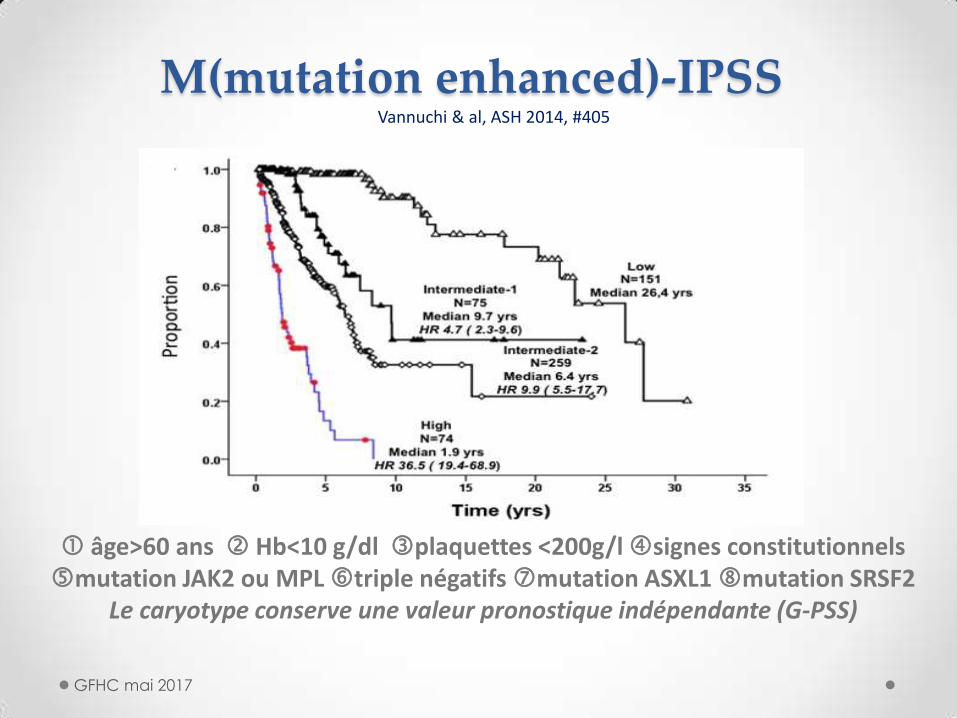
M(mutation enhanced)-IPSS
âge>60 ans Hb<10 g/dl plaquettes <200g/l signes constitutionnels mutation JAK2 ou MPL triple négatifs mutation ASXL1 mutation SRSF2
Le caryotype conserve une valeur pronostique indépendante (G-PSS)
Vannuchi & al, ASH 2014, #405

INDICATIONS THERAPEUTIQUES
de Bousser à la greffe…
« dans cette affection,il y a peu de traitements utiles mais
il y en a de nuisibles » (Bousser,Le Sang,1952)
GFHC mai 2017

GFHC mai 2017
COMPLEXITE THERAPEUTIQUE
• Intrication de cytopénies (anémie++ / thrombopénie / neutropénie)
et de myéloprolifération (splénomégalie et autres foyers d’hmp
ectopique / hyperleucocytose / thrombocytose)
• Intrication de la toxicité du ttt et de l’évolution de la maladie
• Intrication de l’âge et des comorbidités
rapport bénéfice / risque délicat
critères de réponse difficiles à établir

OBJECTIFS THERAPEUTIQUES
GFHC mai 2017
améliorer symptômes et qualité de vie diminuer ou ne pas augmenter le risque de TA améliorer la survie ?
CIBLES PRONOSTIQUES
• Signes constitutionnels « B » (amg,sueurs,Ht°)
• Anémie
• Hyperleucocytose
• Thrombopénie
CIBLES NON PRONOSTIQUES
• Rate : taille(>10cm)et/ou sympt
• Autres signes généraux
(prurit, asthénie…)
• Thrombocytose

GFHC mai 2017
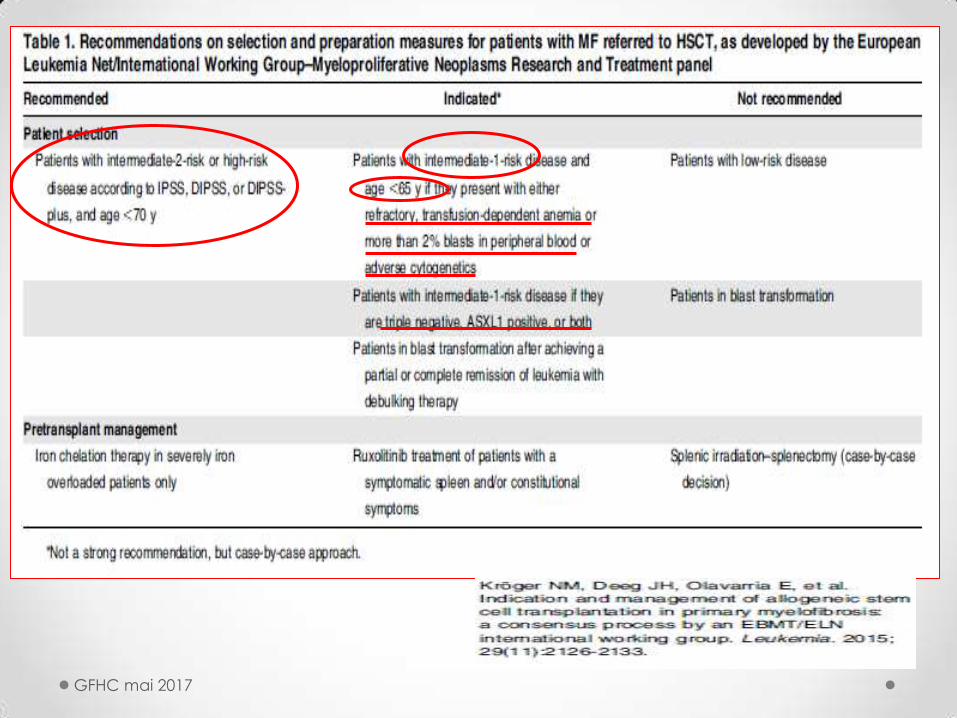
ALLOGREFFE MEDULLAIRE
Conditionnement myéloablatif
thérapeutique risquée réservée aux patients jeunes avec pronostic péjoratif
dilemme :
75% des < 55 ans sont faible risque au diagnostic (survie méd 15 ans sans greffe)
et mêmes facteurs péjoratifs pour l’allogreffe et pour la maladie
Allo-greffe avec conditionnement atténué:
recul de la limite d’âge à 65-70 ans voire +
(cf comorbidités : score de Sorror)

SPECIFICITES ALLOGREFFE dans MF
• Patient : souvent limite d’âge
• Maladie
prise + difficile et + tardive à cause de
la splénomégalie et de la fibrose
complications thrombotiques et
gestion du ttt anticoag
MVO et défaillance hépatique
GFHC mai 2017

RUXOLITINIB (1é inhibiteur de JAK)
GFHC mai 2017
Avec splénomégalie et/ou symptômes en Europe
De score pronostique intermédiaire ou élevé aux USA
En pratique: PO, 2 prises par jour ; 15 mg ou 20 mg x 2 en fonction plaquettes ; Bonne tolérance ; Suivi NFS en raison anémie et thrombopénie
Efficacité ++ sur rate et symptômes (pas sur cytopénies et peu
sur hyperleuco) ; allongement de la survie?
Approbation pour les MF primitives ou secondaires
Which patients with myelofibrosis should receive ruxolitinib therapy? ELN-SIE evidence-based recommendations. Marchetti M et al. Leukemia. 2017 Apr;31(4):882-888.
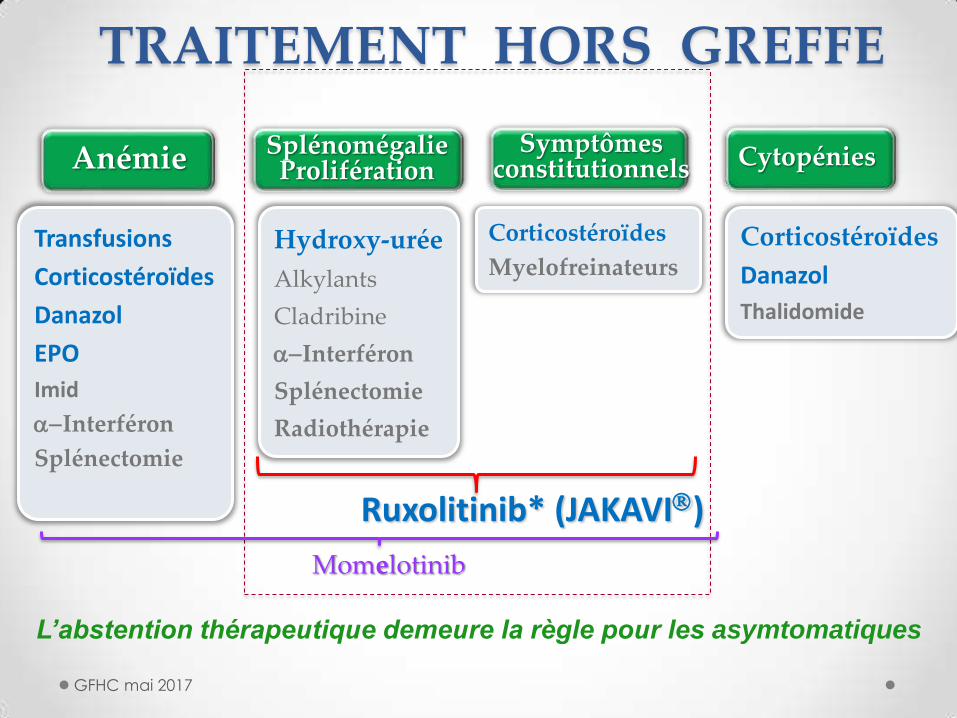
Transfusions
Corticostéroïdes
Danazol
EPO
Imid
a-Interféron
Splénectomie
Hydroxy-urée
Alkylants
Cladribine
a-Interféron
Splénectomie
Radiothérapie
Symptômes constitutionnels
Splénomégalie Prolifération Anémie Cytopénies
Corticostéroïdes
Myelofreinateurs Corticostéroïdes
Danazol
Thalidomide
Ruxolitinib* (JAKAVI)
Momelotinib
GFHC mai 2017
TRAITEMENT HORS GREFFE
L’abstention thérapeutique demeure la règle pour les asymtomatiques
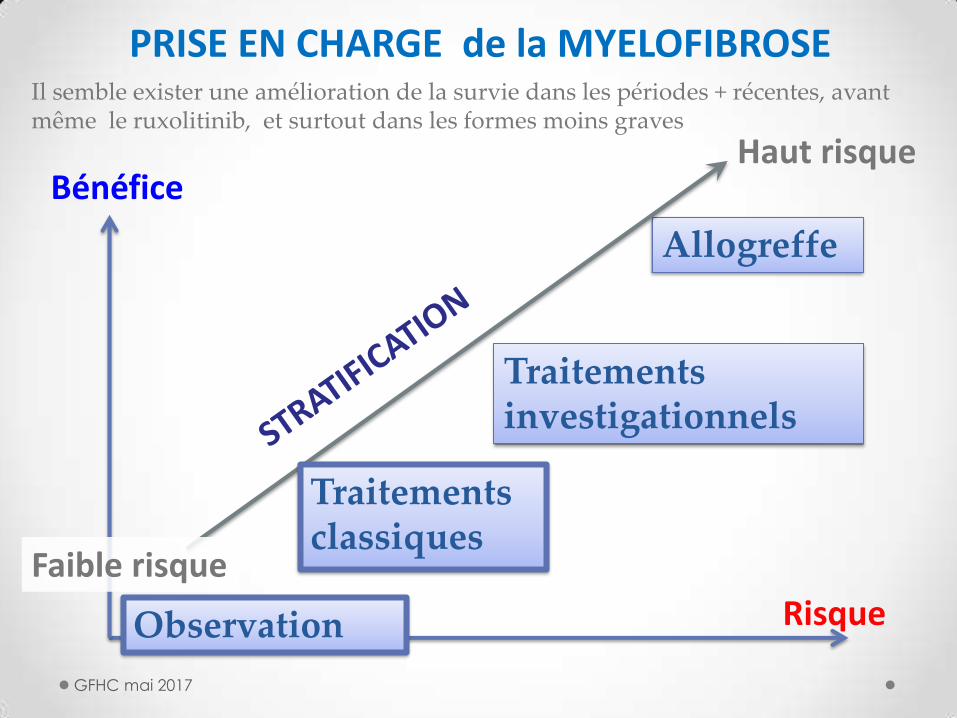
GFHC mai 2017
Bénéfice
Observation
Traitements investigationnels
Allogreffe
Haut risque
Faible risque
Traitements classiques
PRISE EN CHARGE de la MYELOFIBROSE
Risque
Il semble exister une amélioration de la survie dans les périodes + récentes, avant même le ruxolitinib, et surtout dans les formes moins graves
GFHC mai 2017
Avenir : associations thérapeutiques
GFHC mai 2017

GFHC mai 2017

GFHC mai 2017
CONCLUSION
• Maladie rare et complexe mais de mieux en mieux comprise • Plus de traitements disponibles ,+ ciblés mais coûteux
• Amélioration de la survie chez les malades + récents
impact d’une meilleure prise en charge?
• Encore nombreuses interrogations
Suivi étroit des patients (évaluation pronostique) nota jeunes
Attention au retard de l’indication de greffe
( aucun ttt ne réduit le risque de TA )
essais cliniques

Indispensable de connaitre « la vraie vie »…
BASE DE DONNEES MF du FIM avec + de 600 inclus depuis fin 2013
+ Collection biologique FIMBANK (AAP BCB INCa 2013 ; V Ugo)
+ Séquençage NGS (AAP Force Hémato 2016 ; V Ugo)
+ Relecture centralisée des BOM (GEBOM ; B Burroni)
GFHC mai 2017





























