Embed Size (px)
Citation preview

LES LIMITATIONS DE LA THEORIE A DEUX PARAM~TRES DANS L’INTERPR~TATION DES RBSULTATS OBTENUS SUR LES SOLUTIONS DE POLYM~RES ET DE COPOLYM~RES
HENRI BENOIT, DANIELLE DECKER, ANASTASIOS DONDOS ET PAUL REMPP
Ceritre de Recherches sur les Mucrornol~cules C.N.R.S. Strusbourg, France
1NTRODUCTlON
Depuis dCji un certain nombre d’annies un travail considirable tant thkorique qu’expkrimental a i t6 consacrC aux solutions diluCes de molicules en chafnes.’ D’importants progrks ont CtC rCalisCs dans ce domaine et il est possible d’affirmer que I’on comprend maintenant les propridtis tliermodynamiques de ces solutions et que l’interpritation de leurs propriitts rhtologiques telles que viscositi et sbdimentation est satisfaisante. Ceci ne veut pas dire qu’il n’y ait pas de progr6s B faire mais I’essentiel des risultats a i t6 obtenu et leur utilisation est maintenant couran te .
Dans cet expos6 je voudrais, non pas rappeler les succks des tlidories actuelles mais a u contraire mettre en Cvidence les domaines dans lesquels il est difficile de les utiliser ou plus exactment les domaines dans lesquels elles ne permettent pas d’interprdter les rCsultats. I1 me semble en effet plus intiressant de chercher les limitations des thiories et les nouvelles voies de recherche plut6t que d’essayer de parfaire et d’amtliorer les thtories existantes.
Mon expost comprendra deux parties. Dam la premiire je m’appuierai sur les travaux de D. Decker et de P. Rempp sur les polymires ramifiis pour montrer que d6s que le degrC de ramification devient important il est impossible d’appliquer les idCes classiques.
Dans la deuxikme je montrerai, en me basant essentiellement sur les travaux de A . Dondos que la gCnCralisation des rksultats concernant les solutions diluies d’uii polymkre dam un solvant est tr6s difficile d6s qu’on a affaire B trois constituants, soit qu’il s’agisse d’un copolymire en solution dans un solvant unique, soit qu’il s’agisse d’un homopolym6re en solution dans un melange de solvan ts.
0 1970 by John Wiley & Sons, Inc. J. Polymer Sci., Part C No. 30, pp. 27-46 (1970)
27

BENOIT. DECKER, DONDOS, ET REMPP
RAPPEL DES THEORIES RELATIVES AUX SOLUTIONS DILUEES
Avant de prisenter ces resultats il est bon de rappeler brievement les principales propriitis des solutions diludes de polymbres et la faqon dont on les interprete.
Si on neglige les interactions entre chainons iloignks, une chaine polymkrique obeit lorsqu‘elle est suffsamment longue i la statistique de Gauss. De ce fait l’tcart quadratique moyen entre ses extrimit& cz ainsi que son rayon de giration G2 sont proportionnels au degrC de polymirisation et l’on peut Ccrire:
la quantiti b2 depend de la nature de la chaine et de nombreaux travaux ont BtC consacris a son calcul en tenant compte des interactions entre atomes voisins de la cha ine .
Quant la chaine est en solution, il faut tenir compte des interactions likes i la presence du solvant. Pratiquement on introduit un parametre caractirisant les interactions entre segments Bloignis. Ce paramitre P dipend Bvidemment du couple solvant-solute consider6 et est defini par la relation
u(r) + ( I --e - - ) d r P = j m KT +
oh u(r) est l’inergie moyenne de deux chainons i la distance T . Si ce parametre est positif il y a repulsion entre les chainons, la chaine se dilate et n’obeit plus A la statistique de Gauss; on Bcrit alors:
(Y T~ o t ~ (Y est un coefficient appel6 coefficient d’expansion dependant ii la fois de la longueur de la chaine et du parambtre 0.
Toutes ces theories actuelles sont basBes sur le fait que la connaissance de ar et de b2 doit permettre de calculer toutes les quantitks thermodynamiques et hydrodynamiques caracteristiques de la chaine en solution.
De nombreux calculs ont Bt6 effectuis pour relier le coefficient d’expansion, le second coefficient du viriel, et la viscositi intrindque i ces deux parametres; ils ont conduit i des formules qui sont souvent assez compliquies, elles permettent de representer de faqon cohCrente la majoriti des risultats expirimentaux.
F = 2 1
Sans entrer dans ces dCtails, rappelons les points les plus importants. Quant 0 = 0, les interactions i longue distance disparaissent; la chahe obiit
donc 5 la statistique de Gauss. De ce fait le coefficient d’expansion est Cgal i l’unite et on a en solution 7 = GSine. Les dimensions que l’on peut mesurer sont les dimensions non perturbies, elles sont proportionnelles au degr6 de polymCrisation et en principe indipendantes du solvant. De plus, le second coefficient du viriel est nul; la solution, se comporte comme une solution idiale, on est au point 0 de Flory’ Enfin la viscositi intrindque obiit i la loi de Flory
28

LA THEORIE A DEUX PARAMETRES
ou Q0 est une constate universelle; sa ditermination permet donc de mesurer des dimensions non perturbies.
Dans un solvant oh 0 est positif c’est-idire dans un bon solvant, (Y est supirieur i un, le second coefficient du viriel est positif et la viscositi intrindque n’obiit plus i la formule (2).
Comme les deux paramQtres les plus importants sont T~~ et QI et que les mesures les plus faciles a rialiser sont celles de viscositi intrindque dans des bons solvants, on a cherchi une reprisentation graphique qui permet de determiner ces grandeurs i partir de mesures de viscositis intrindques en fonction de la masse. Parmi les diffirents procidis priconisis, celui qui semble le plus simple est celui qui a i t6 proposi par Burchard3 ainsi que Stockmayer et Fixman4 qui consiste i porter [77] /M’/’ en fonction de MI/’
On constate expirimentalement que les points pour des masses pas trop ilevies, disons entre 20.000 et 360.000, se placent en giniral sur une droite.’ Comme le coefficient d’expansion croit avec la masse, l’extrapolation i masse nulle permet d’appliquer la formule (2) et l’on obtient ce qu’on appelle K , qui est difini par la relation
K , =Qo [ - 3‘2
ou m est le poids moliculaire de l’unit6 monomdre. La pente permet de calculer la quantiti 0 par la formule
Le procidi que nous venons de rappeler est justifii par le fait qu’il permet d’obtenir une valeur de K,O, indipendante du solvant, c’est-idire des dimensions non perturbies, ce qui est privu par la thiorie.
On voit donc que si les expressions mathimatiques rigoureuses permettant d’icrire des relations pricises entre les grandeurs expirimentales que nous venons de rappeler font encore difaut, on dispose d’un outil suffisamment au point pour interpriter les risultats expirimentaux.
ETUDE DES POLYMBRES RAMIFIBS
L’itude des polymQres ramifiis est beaucoup moins avancie que celle des polymeres liniaires car, pour la mener i bien, il faut connaitre la structure exacte de l’ichantillon ce qui n’est pas toujours facile. De plus les calculs de statistique deviennent beaucoup plus compliquis dds que la chaine n’est plus linkaire ce qui rend l’interpritation des mesures de viscosit6 et de second coefficient du viriel trQs difficile. On a cependant toujours envisage l’interpretation des risultats obtenus sur ce type de polymdre en leur
29

RENO11’. DECKER, DONDOS. ET REMPP
generalisant les notiuiis itablics pour les cliaiiies liiiiaires; nous voudrions montrcr un exemple oil cctte gdndralisation est impossible.
Dcs polyiiiires greffis polystyrhnc sur polystyrine, ont i t e preparks par D. DECKEK-FKEYSS” de la f a p n suivantc. On priparc d’abord, par polymirisation radicalaiie en solution, 1111 copolymhre styrinc-niCtliacrylatc de niitliyle conteiiaiit moins de 15% dc mithacrylate de mCthylc.
Your obtenir des produits aussi bicn difinis que possible, le t a m dc convcrsion a Cti limitis ce qui riduit les fluctuations de composition. 011 sait que si I’on fait agir un polystyrine vivant sur la fonction estcr d’une chainc de poly~iietliacrylate de niithyle il y a fixation par rtaction de I’cxtrimiti carbanioniyue .7
On a donc opirC dc cette f a p i en faisant riagir un polystyrhie vivant monofonctionnel sur ces copolymires statisticlues bien caracteris6s.
I I se fornie toujours un peu d’homopolymere car la riaction dc greffagc n’est jamais totalc, on pcut lc mettre en evidence par chromatographic de partage e t I’eliniiner assez facilenient par fractionnement. Comme on comai t , g r k e ri un prelivement, la masse moliculaire du polys tyrhe greffi, cornme on connait aussi la masse moliculaire du copolymire de dipar t , on peut, en mesurant la masse d u produit final, determiner le noinbre des greffons.
Ces produits ne sont pas i (Tableau I) proprement parler des homopolym6res “en peigne” puisqu’ils contiennent quelques groupes mkthacrylate n’ayant pas riagi ainsi clue des fonctions cCtone aux points d’accrochage des greffons. Cependant la teneur en o x y g h e n’est jamais suptrieure i 1% e t toute I’itude physicochimique qui a Ct6 rialisie sur ces polymires montre que lcs erreurs qu’entraine leur assimilation i un homopolymire sont ntgligeables.
Comme ces polymeres sont assez polydispersis puisqu’on est parti d’un troiic pr ipar i par voie rddicalaire, on peut les fractionner e t on obtient, comme Ic greffage est statistique, une sirie de polymires homologues portant des greffons, tous de mbme longueur, en nombre proportionnel i la masse du squelette.
En principe puisqu’il s’agit d’homopolystyr6ne, lorsque le paraniitre quc nous avons difini plus haut est nul, il est nu1 quclle que soit la structure de la c h i n e ; on doit donc observer la mbme temtrature 0 pour tous les Cchantillons.
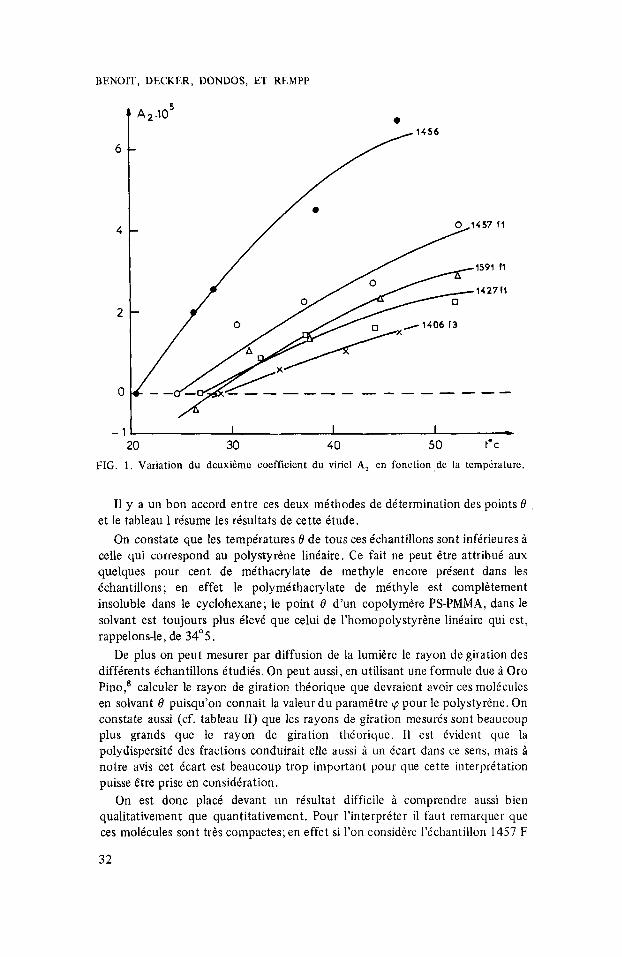
Mme Decker a mesure le point 0 de ses Cchantillons en chercliant le point oii le second coefficient d u viriel s’annule. Sur la figure (1) now avom report6 quelques uns de ses risultats. I1 s’agit de mesures effectuies dans le cyclohexane en fonction de la tempirature par diffusion de la luniiire sur un appareil Sofica. Bien que la prkcision ne soit pas trbs elevie, on voit que les tempiratures, pour lesquelles A2 s’annule, dipendent du type de copolyniire considiri.
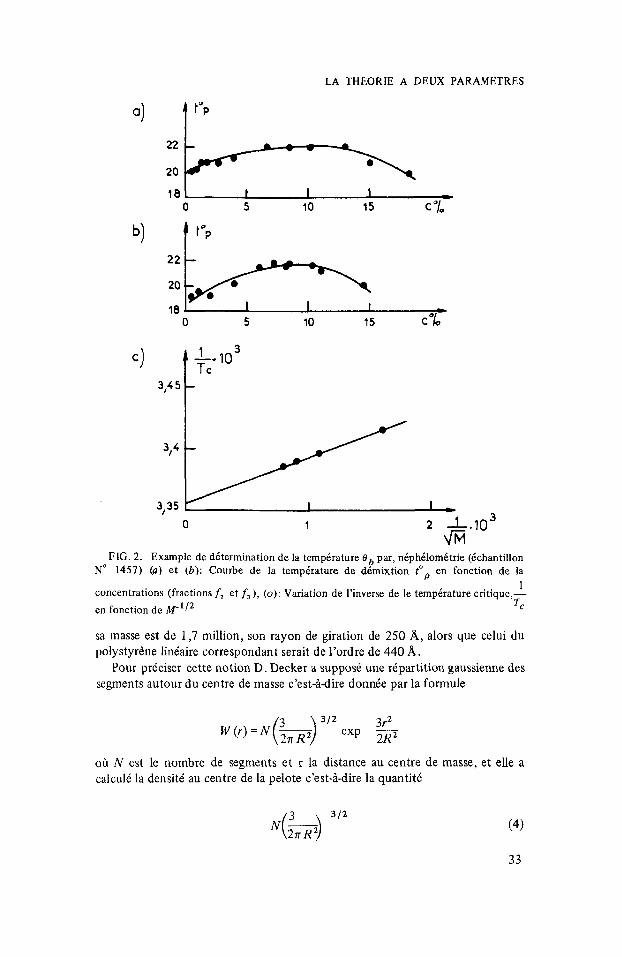
Ce resultat i t an t assez imprivu, nous avons essay6 de le confirmer en cherchant le point critique de dimixtion pour une masse infinie. En utilisant un niphelomitre enregistrement automatique on a determiilk la temptrature critique de deinixtion correspondant i diffirentcs fractions. E n portant selon la mithode priconisie par Flory’ 7/T, en fraction de M-’ les points s’aligncnt sur une droite dont l’ordonnie a l’origine permet de calculer la tempirature 0 (fig. 2 )
30
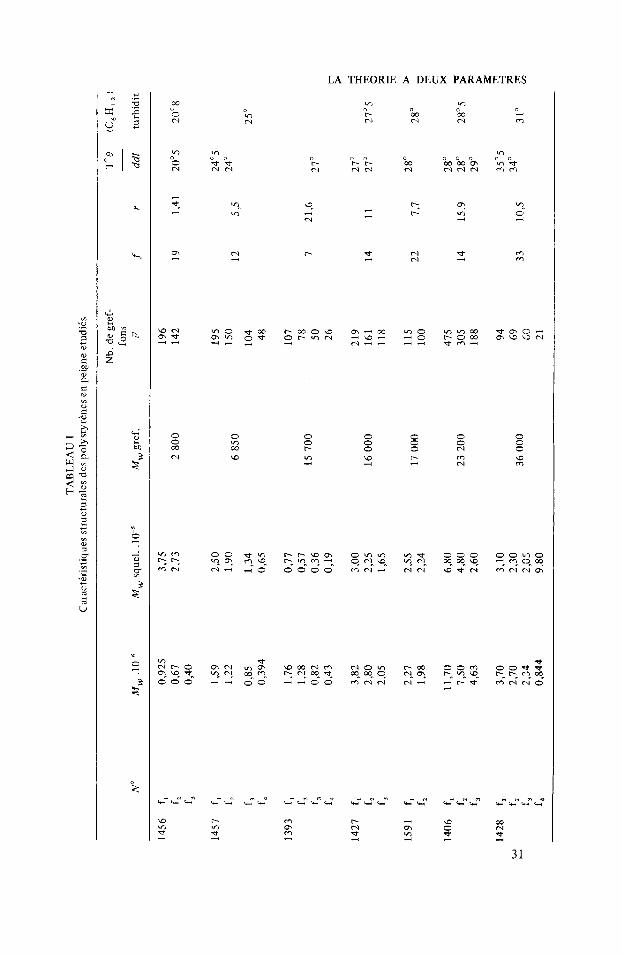
TA
BL
EA
U I
C
arac
tkri
stiq
ues
stm
ctur
ales
des
pol
ysty
rehe
s en
pei
gne
etud
iks
Toe
(C,H
,,)
ddl
turb
idit
Nb.
de
gref
- fo
ns
-
r w
M
, .1
0-6
M,
sque
l. .l
o-'
M,
gref
. i'
f
1456
f,
f,
f,
f,
f, f.4
1457
f,
1393
f,
f2
f,
f,
f2
f,
f2
f2
f,
w
f *
f'l
1427
f,
1591
f,
1406
f,
1428
f,
c
4- '3
0,92
5 0,
67
0,40
1,59
1,
22
0,85
0,
394
1 ,I6
1,
28
0,82
0,
43
3,82
2,
80
2,05
2,21
1,
98
11,7
0 I ,s
o 4,
63
3,70
2,
70
0,84
4 191
A,,-
3,15
2,
13
2,50
1,
90
1,34
0,
651
0,l
l 0,
57
0,36
0,
19
3,OO
2,25
1,
65
2,55
2,
24
6 $0
4,80
2,
60
3,lO
2,
30
9.80
.? n
c
L,"
J
2 80
0
6 85
0
15 7
00
16 0
00
17 0
00
23 2
00
36 0
00
196
142
19
195
150
104 48
12
107 78
50
26
7
2 19
161
14
118
22
115
100
475
305
14
188 94
21
1,41
20
"5
20"8
24"5
24
" 5,
5
27"
21,6
27"
11
27"
2 8"
7,7
28"
15,9
28
" 29
"
25"
F cl
X
2705
2 m
3
r!
C
X
28"
BENOIT, DECKER, DONDOS, ET REMPP
6
4
2
0
- 1
5 A 2.10
20 30 40 50 to c
FIG. 1 . Variation du deuxi6me coefficient du viriel A, en fonction.de la tempdrature.
I1 y a un bon accord entre ces deux mithodes de dttermination des points d et le tableau I resume les rbsultats de cette Ctude.
On constate que les temperatures 0 de tous ces Bchantillons sont infirieures B celle qui correspond au polystyrkne linbaire. Ce fait ne peut Ctre attribui aux quelques pour cent de mithacrylate de metliyle encore present dans les Cchantillons; en effet le polymithacrylate de mbthyle est complktement insoluble dans le cyclohexane; le point 0 d’un copolymere PS-PMMA, dans le solvant est toujours plus BlevC que celui de l’homopolystyrene liniaire qui est, rappelons-le, de 34” 5 .
De plus on peut mesurer par diffusion de la lumikre le rayon de giration des diffkrents Cchantillons Ctudiis. On peut aussi, en utilisant une formule due i Oro Pino,’ calculer le rayon de giration thiorique que devraient avoir ces molCcules en solvant 0 puisqu’on connait la valeur du parametre cp pour le polystyrene. On constate aussi (cf. tableau 11) que les rayons de giration mesurCs sont beaucoup plus grands que le rayon de giration thborique. 11 est Cvident que la polydispersiti des fractions conduirait elle aussi B un Ccart dans ce sens, mais B notre avis cet Ccart est beaucoup trop important pour que cette interprktation puisse Ctre prise en considCration.
On est donc placi devant un risultat difficile i comprendre aussi bien qualitativement que quantitativement. Pour l’interpriter il faut remarquer que ces molicules sont trts compactes; en effet si l’on considkre l’tchantillon 1457 F
32
LA THEORIE A DEUX PARAMETRES
c =/a 18 0 5 10 15
C% 0 5 10 15
FIG. 2. Example de dktermination de la tempirature 0, p,ar, n6phdlom6trie (khantillon No 1457) (a) et (b): Courbe de la temperature de demixtion top en fonction de la
1 concentrations (fractions f, et f, ), (0): Variation de I'inverse de le tempkature critique,- en fonction de M-'l2 =, sa rnasse est de 1,7 million, son rayon de giration de 250 A, alors que celui du polystyrtine liniaire correspondant serait de l'ordre de 440 A .
Pour pricker cette notion D. Decker a supposi une ripartition gaussienne des segments autour du centre de masse c'est-idire donnie par la formule
3r2 w (r) = N (b) 3/2 exp -- 2R
oh N est le nombre de segments et r la distance au centre de rnasse, et elle a calculi la densiti au centre de la pelote c'est-&dire la quantiti
(4)
33
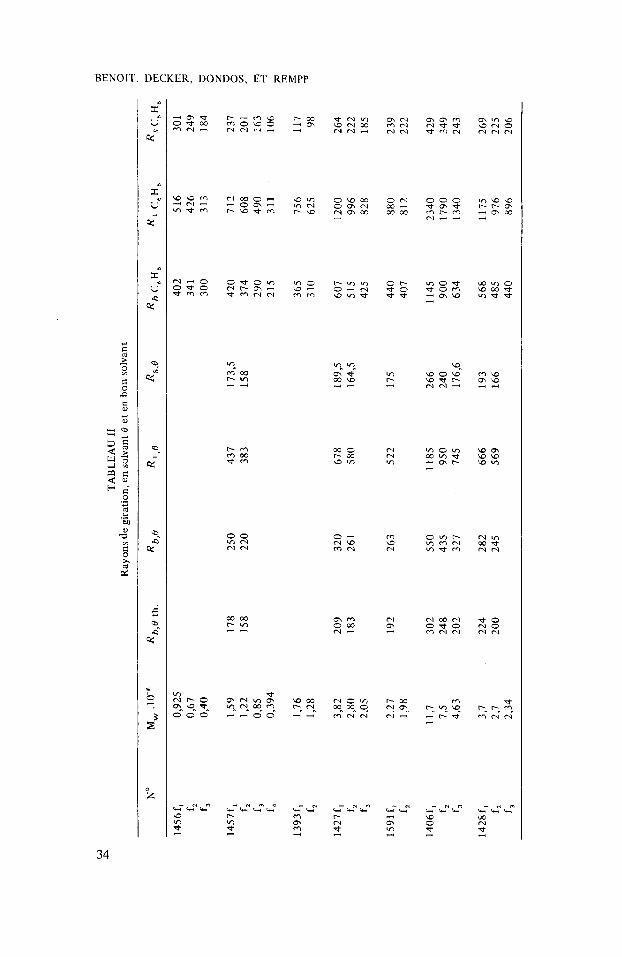
w
P
TA
BL
EA
U I
1 R
ayon
s de
gir
atio
n, e
n so
lvan
t 0 e
t en
bon
solv
ant
0 4
t
No
M,
'b,O
th
. b.
0 R
1,e
4,e
R
b C
,H,
R1
C
,H,
RsC
,H,
m
c
x f*
0,
67
34 1
426
249
-?= z
1456
f,
0,92
5 40
2 5
16
301
E
0,40
30
0 31
3 18
4 C
I €3
f2
1,22
15
8 2 2
0 38
3 15
8 37
4 60
8 20
1
1457
f,
1,59
17
8 25
0 43
7 17
3,5
420
712
237
g E f,
0,85
29
0 49
0 16
3 2
€4
0,39
4 21
5 31
1 10
6
1393
€,
1,76
36
5 75
6 11
7 5
f2
1,28
3 1
0 62
5 98
*
1427
€,
3,82
20
9 32
0 67
8 18
9,s
607
1200
26
4 €2
2,
80
183
26 1
5 8
0 16
4,5
5 15
996
222
€3
2,
05
425
828
185
1591
f,
2,27
19
2 26
3 5 2
2 17
5 44
0 88
0 23
9 €2
1,98
40
7 81
2 22
2
€2
7 $
5 24
8 43
5 95
0 24
0 90
0 17
90
349
1406
€,
11,7
30
2 55
0 11
85
266
1145
23
40
429
€3
4,
63
202
327
7 45
176,
6 6 3
4 13
40
243
1428
f,
3,7
224
282
666
193
568
1175
26
9 f,
2,
7 20
0 24
5 56
9 16
6 48
5 97
6 22
5 f,
2,34
44
0 89
6 20
6
LA Ik160KIE A D t U X PARAMETRES
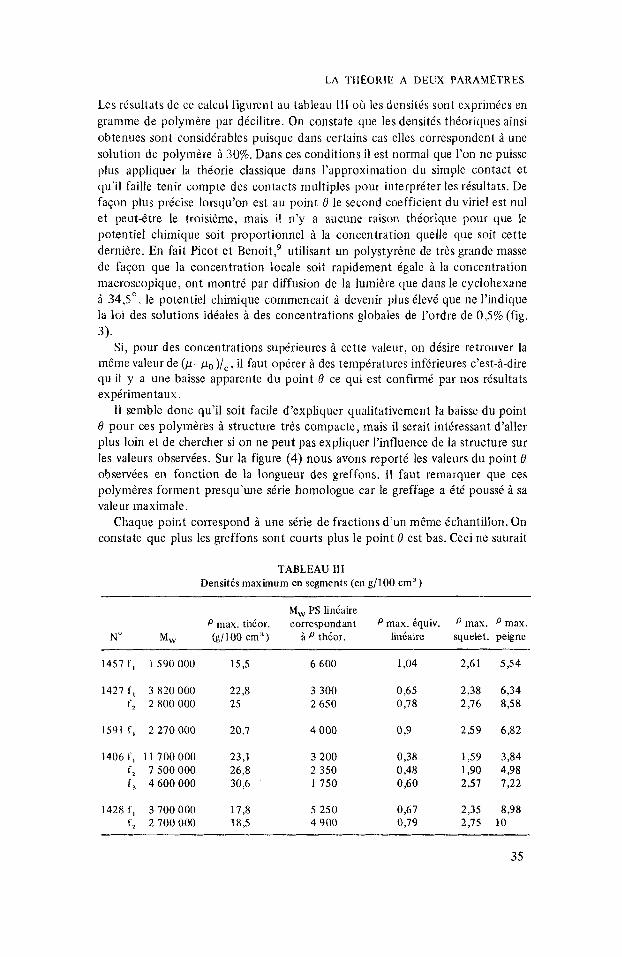
Les rksultnts dc cc calcul figurcnt au tableau 111 oh les densitis sont cxprimies en gramme de polymkre par dicilitre. On constate que les densit& thioriques ainsi obtcnucs sont considdrables puisque dans certains cas elles correspondent B une solution de polymire ti 30%. Dans ces conditions il est normal que l’on ne puisse plus appliquer la theorie classique dans I’approximation du simple contact et qu’il faille tenir compte des contacts multiples pour interpriter Ies rtsultats. De f a y 1 plus pricise lorsqu’on est au point 0 le second coefficient du viriel est nu1 et peut-6tre le troisieme, mais il n’y a aucune raison thioriquc pour que lc potentiel chimique soit proportionnel 2 la concentration quelle que soit cette derni2re. En fait Picot et Benoit,’ utilisant un polystyrkne de tris grande masse de faqon que la concentration locale soit rapidement igale i la concentration macroscopique, ont montri par diffusion de la lumiire clue dans le cyclohexane i 34,5” le potentiel chimique commencait i devenir plus Clevi que ne I’indique la loi des solutions idiales a des concentrations globales de l’ordre de 0,5% (fig. 3).
Si, pour des concentrations supirieurcs A cette valeur, on disire retrouver la m&me valeur de (p po)/ , , il faut operer ti des tenipiratures infirieures c’est-&dire qu il y a une baisse apparente du point 0 ce qui est confirm6 par nos rksultats experimen taux.
11 semble donc qu’il soit facile d’expliquer qu~litativement la baisse du point B pour ces polymires ii structure tris compacte, mais il serait intgressant d’aller plus loin et de chercher si on ne peut pas expliquer I’influence de la structure sur 1es valeurs observies. Sur la figure (4) nous avons report6 les valeurs du point 0 observies en fonction de la longueur des greffons. 11 faut remarquer que ces polymeres forment presqu’une sirie homologue car le greffage a it6 poussi a sa valeur maximale.
Chaque point correspond i une sirie de fractions d’un m&me Cchantillon. On constate que plus les greffons sont courts plus le point 0 est bas. Ceci ne saurait
TABLEAU Il l Densitds maximum en segments (en g/100 cm’)
~~
M, PS hniaire p mdx. thbor. correspondant p max. 6quiv. p max. p max.
N” Mw (g/ 100 cm ’) i P thior . hnCaire squelet. peigne
1457 f , 1 590 000 15,s 6 600 1,04 2,61 5 3 4
1427 f , 3 820 000 22,8 3 300 0,65 2,38 6,34 f, 2800000 25 2 650 0,78 2,76 8 3 8
1591 f , 2 270000 20,7 4 000 0,9 2,59 6,82
I406 f , 11 700 000 23,l 3 200 0,38 1,59 3,84 f, 7500000 26,8 2 350 0,48 1,90 4,98 f , 4 600 000 30,6 1750 0,60 2,57 7,22
1428 f , 3 700 000 17,8 5 250 0,67 2,35 8,98 f , 2700000 18,s 4 900 0,79 2,15 10
35
BENOIT, DECKER, DONDOS, ET REMPP
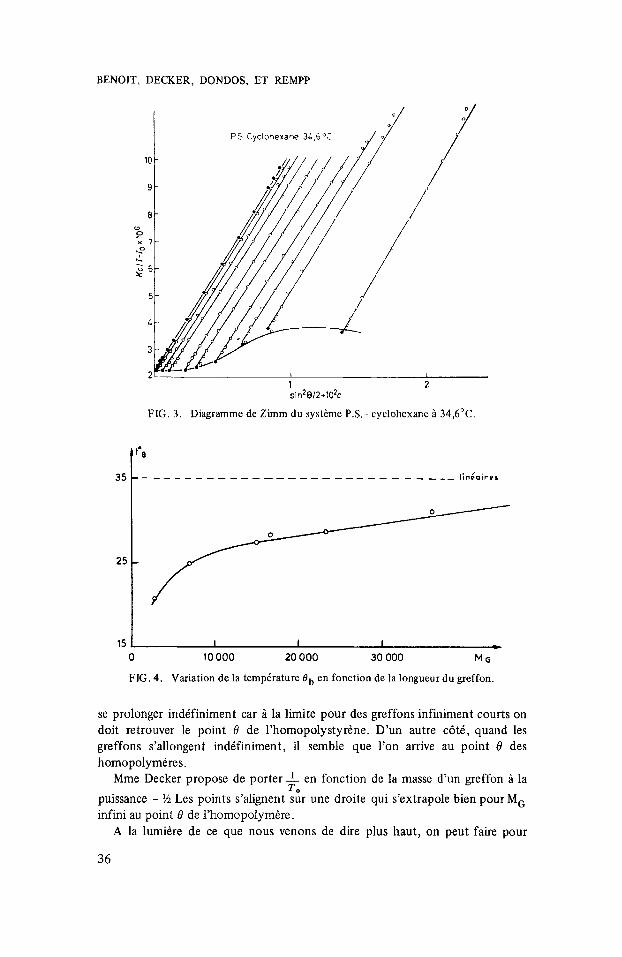
FIG. 3. Diagramme de Zimm du systkme P.S. - cyclohexanc B 34,6”C.
- 0 10 000 20 000 30 000 M G
FIG. 4. Variation de la temp6rature Bb en fonction de la longueur du greffon.
se prolonger indifiniment car a la limite pour des greffons infiniment courts on doit retrouver le point 8 de l’homopolystyr6ne. D’un autre c6tC, quand les greffons s’allongent indifiniment, il semble que l’on arrive au point 0 des homopolymires.
Mme Decker propose de porter 1 en fonction de la masse d’un greffon a la puissance - $5 Les points s’alignent sur une droite qui s’extrapole bien pour M, infini au point 8 de l’homopolymire.
A la lumi&re de ce que nous venons de dire plus haut, on peut faire pour
To
LA THEOKIE A DEUX PARAMETRES
essayer de coniprendrc cette courbe du nioins qualitativement ’la remarque suivante.
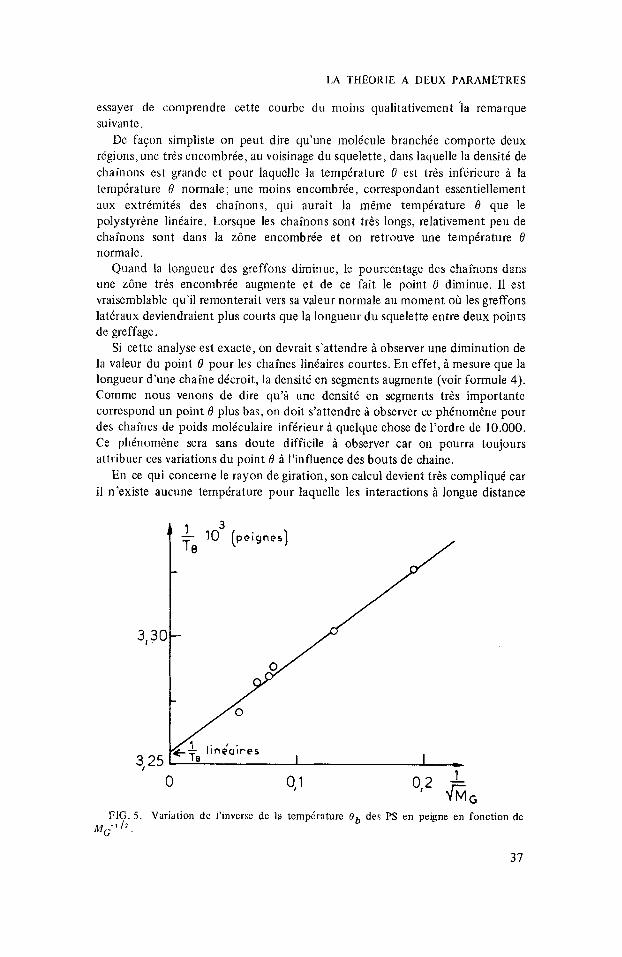
De faqon simpliste on peut dire qu’une mol6cule branchle comporte deux regions, une trts encombree, au voisinage du squelette, dans laquelle la densite de chainons est grande et pour laquelle la tempkrature 0 est trbs inferieure i la temptrature 6’ normale; une moins encombree, correspondant essentiellement aux extrimitks des chainons, qui aurait la m&me temperature 0 que le polystyrine lintaire. Lorsyue les chainons sont tr is longs, relativement peu de chainons sont dans la z6ne encombree et on retrouve une ternpiratwe 6’ normale.
Quand la longueur des greffoiis diminue, le pourcentage des chainons dans une zcine trbs encombree augmente et de ce fait le point 6’ diminue. I1 est vraiscmblable qu il remonterait vers sa valeur normale au moment oh les greffons lat6raux deviendraient plus courts que la longueur du squelette entre deux points de greffage.
Si cette analyse est exacte, on devrait s’attendre A observer une diminution de la valeur du point 6 pour les chaines linkaires courtes. En effet, 5 mesure que la longueur d’une chaine dtcroit, la densitt en segments augmente (voir formule 4). Comme nous venons de dire qu’i une densite en segments trbs importante correspond un point 6 plus bas, on doit s’attendre 6 observer ce phCnom6ne pour des chaines de poids moltculaire inferieur ti quelque chose de I’ordre de 10.000. Ce phhomene sera sans doute difficile a observer car on pourra toujours attribuer ces variations du point 6 i I’influence des bouts de chaine.
En ce qui concerne le rayon de giration, son calcul devient tris compliquC car il n’existe aucune tempkrature pour laquelle les interactions 1 longue distance
FIG. 5. Variation dc I’invcrsc dc la temp6rature O b des PS en peigne en fonction de MG- ’ l2 .
37

BENOIT, DECKER, DONDOS, ET REMPP
disparaissent riellement. On ne pourra donc jamais utiliscr I’approximation gaussienne.
ETUDE DES COPOLYM~RES STATISTIQUES
I1 a i t6 souvent admis qu’un copolymbre statistique avait un comportement voisin de celui d’un homopolymbre, c’est-$dire qu’on pouvait lui appliquer la thiorie ri deux paramitres dont nous avons rappeli les grandes lignes dans la premiere partie de notre exposi. Ceci revient A dire qu’ils sont caractirisis par leurs dimensions non perturbies et un coefficient d’expansion a qui est en giniral plus ilevk que celui que l’on pourrait calculer en utilisant une rbgle d’additivitd, du fait de l’incompatibiliti entre polymire et des rkpulsions qu’elle entraine. Dans un rdcent travail, A . Dondos’ a i tudii systkmatiquement dans diffirents solvants un copolymire statistique polystyrine-polymithacrylate de mithyle; ce sont les risultats de cette etude que je voudrais bribvement rapporter ici. Ces copolymbres ont i t6 prdpari par voie radicalaire dans les conditions aziotropes 46,5% PMM-55,5% PS. 11s ont it6 fractionnis, leurs masses ont kt6 diterminees par diffusion de la lumiire; leur polydispersitk est assez faible car en interpritant les diagrammes de chromatographie sur gel sans tenir compte de la diffusion axiale on trouve des valeurs de M,M, de l’ordre de 1,3.
Les viscositis intrinsQques des fractions et des homopolymbres parents ont kt6 mesuries dans le benzbne, le titrahydrofuranne i 25”, l’acitate d’isoamyle et le paraxylbne B 25” et 75°C.
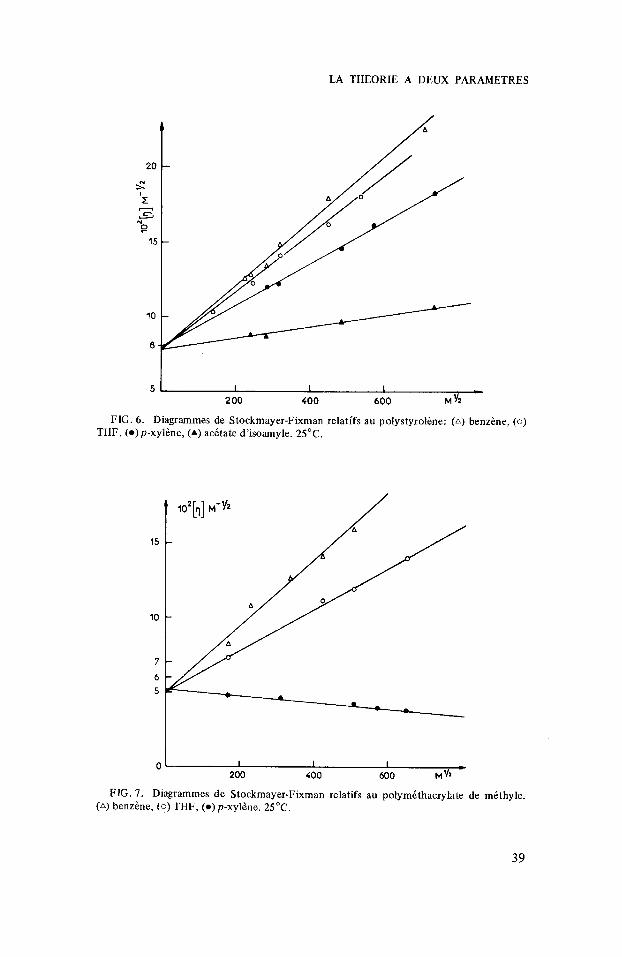
Comme nous nous intiressons spicialement aux dimensions non perturbies, nous les avons Cvalutes en utilisant le diagramme de Stockmayer-Fixman dont j’ai rappel6 le principe.
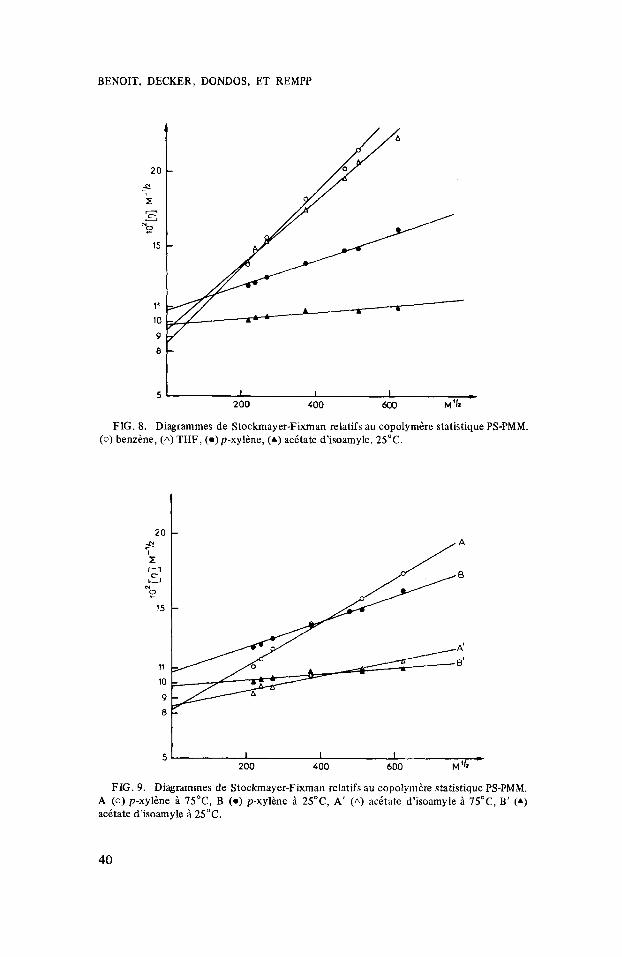
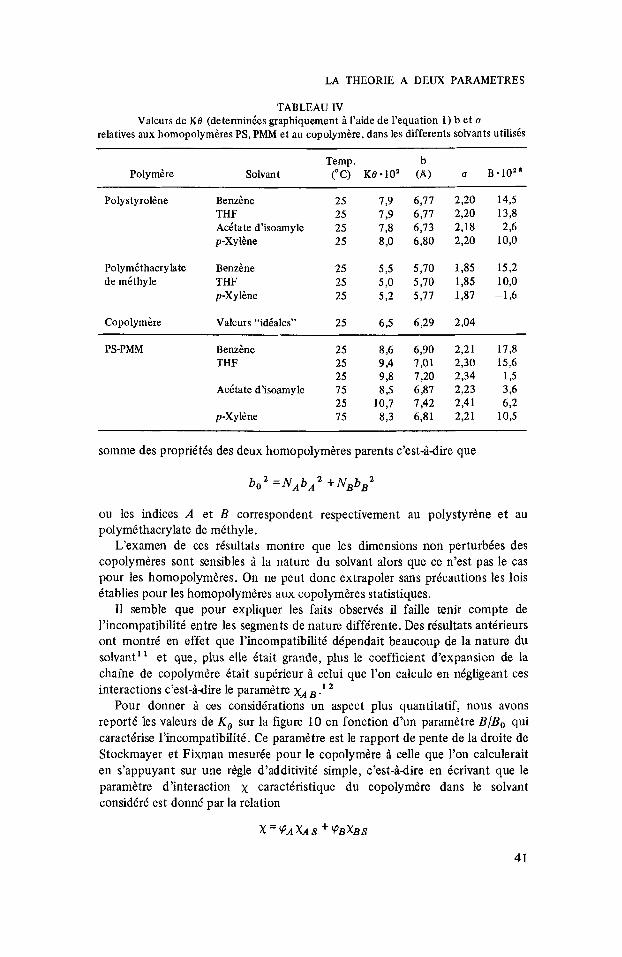
Sur les figures 6 et 7 figurent les risultats obtenus pour les homopolymbres PS et PMM. On constate que les droites obtenus convergent au m6me point, confirmant ainsi que les dimensions non perturbies ne dkpendent pas du solvant. Par contre on obtient dans le cas du copolymbre statistique un comportement tout a fait diffirent. Les points correspondant aux diffirentes fractions (mesuris A 25”) dans les diffkrents solvants se placent bien sur des droites mais l’ordonnie a I’origine de ces droites dipend du solvant considiri (fig. 8). On ne peut pas mettre en cause le mode de reprisentation utilisi car il n’y a pas corrblation entre la pente et I’ordonnie A I’origine. Le m6me risultat se retrouve Q 75”. Sur la figure 9 nous avons reporti les risultats obtenus dans le paraxylene A 75” et 25°C (courbes A et B), et dans I’acitate d’isoamyle aux m6mes tempiratures (courbes A‘ et B‘). On constate aussi des changements importants de pente et d ordonnie A l’origine.
Le tableau IV rassemble les risultats de cette etude, il y figure la grandeur K6
de I’iquation ( 3 ) , le coefficient b et le rapport u =? T~~~ 6taiit l’icart 7 0 f
quadratique moyen que l’on mesurerait si toutes les rotations itaient libres. Sur ce tableau figurent aussi (soulignies) les valeurs idCales c’est-A-dire celles
que I’on obtiendrait en supposant que le copolymbre a des propriitis qui sont la
T o = 112 -
38
LA THEORIE A DEUX PARAMETRES
5 I I I - 2 00 400 600 M '12
FIG. 6. Diagramrnes de Stockrnayer-Fixrnan relatifs au polystyroline: (A) benzine, (0)
THF, ( 0 ) p-xylhe, (A) acetate d'isoamyle. 25°C.
15
10
7 6 5
0 I I I 200 400 600 M% *
FIG. 7 . Diagramrnes de Stockmayer-Fixrnan relatifs au polyrnethacrylate de methyle. (a) benzine, (0 ) THF, ( 0 ) p - x y l h . 25°C.
39
BENOIT, DECKER, DONDOS, ET REMPP
I I I 2 00 400 600 M ' f r --
FIG. 8. Diagrammes de Stockmayer-Fixman relatifs au copolymkre statistique PS-PMM. (0 ) benzkne, (A) THF, (o)p-xyl&ne, (A) acktate d'isoamyle. 25°C.
I I 1 200 400 600 M'h
FIG. 9. Diagrammes de Stockmayer-Fixman relatifs au copolymkre statistique PS-PMM. 25"C, A' (A) acetate d'isoamyle B 75"C, B' (A) A ( 0 ) p-xylhe
acetate d'isoamyle B 25°C. 75"C, B (0) p-xylkne
40
LA THEORIE A DEUX PARAMETRES
TABLEAU IV Valcurs de Ke (determin6es graphiquement i I’aide de l’equation 1) b et u
relatives aux homopolymgres PS, PMM et au copolymere, dans les differents solvants utilises
Temp. b Polymkre Solvan t (“C) KB.10’ (A) u B.10”
Polystyrolkne Benzene THF Acetate d’isoamyle p-Xyline
Polym6thacrylate Benzene de methyle THF
p-xylkne
Copolymere Valcurs “idkalcs”
25 7,9 6,77 2,20 14,s 35 7,9 6,17 2 2 0 1 3 3 25 7,8 6,13 2,18 2,6 25 8,O 6,80 2,20 10,o
25 5,5 5,70 1,85 15,2 25 5,O 5,70 1,85 10,O 25 5,2 5,77 1,87 -1,6
25 6,5 6,29 2,04
PS-PMM Benzene 25 8,6 6,90 2,21 17,8
25 9,8 7,20 2,34 1,s Acetate d’isoamyle 75 8,5 6,87 2,23 3,6
25 10,7 7,42 2,41 6,2 p-Xylgne 15 8,3 6,81 2,21 10,s
THF 25 9,4 7,ai 2,30 i5,6
somme des propriktks des deux homopolym6res parents c’est-iidire que
ou les indices A et B correspondent respectivement au polystyrkne et au polymkthacrylate de mkthyle.
L’examen de ces rksultats montre que les dimensions non perturbkes des copolymeres sont sensibles i la nature du solvant alors que ce n’est pas le cas pour les homopolymkres. On ne peut donc extrapoler sans prkcautions les lois Ctablies pour les homopolymkres aux copolymkres statistiques.
I1 semble que pour expliquer les faits observks il faille tenir compte de l’incompatibilitk entre les segments de nature diffkrente. Des rksultats antkrieurs ont montrk en effet que l’incompatibilitk dkpendait beaucoup de la nature du solvant” et que, plus elle ktait grande, plus le coefficient d’expansion de la chaine de copolymkre ktait supkrieur ii celui que l’on calcule en nkgligeant ces interactions c’est-idire le paramktre xAB .’
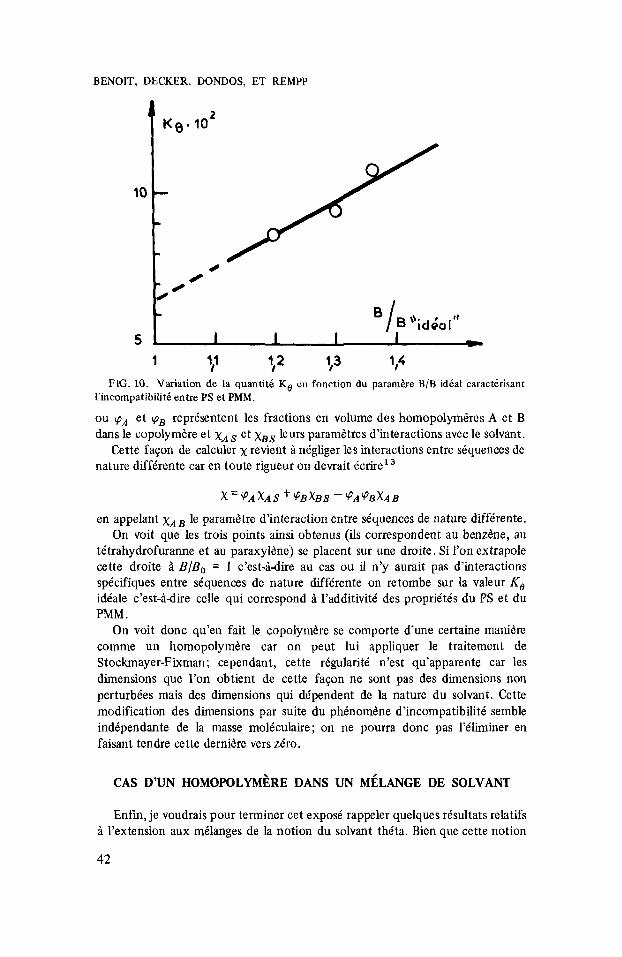
Pour donner 5 ces considkrations un aspect plus quantitatif, nous avons report6 les valeurs de K , sur la figure 10 en fonction d’un paramktre B/Bo qui caractkrise l’incompatibilitk. Ce parambtre est le rapport de pente de la droite de Stockmayer et Fixman mesurke pour le copolymkre ii celle que l’on calculerait en s’appuyant sur une rkgle d’additivitk simple, c’est-iidire en Ccrivant que le parametre d’interaction x caractkristique du copolym6re dans le solvant considkrk est donnC par la relation
41
BENOIT, DECKER, DONDOS, ET REMPP
10
5
0 /
1 l? lr ’13 ’14 FIG. 10. Variation de la quantiti K, en fonction du paramere B/B id6aI caracterisant
l’incompatibiliti entre PS et PMM.
ou pA et qB representent les fractions en volume des homopolymires A et B dans le copolymkre et xAs et xBS leurs parametres d’interactions avec le solvant.
Cette fagon de calculer x revient i nkgliger les interactions entre sequences de nature diffirente. car en toute rigueur on devrait Ccrire‘
en appelant x A B le paramitre d’interaction entre sequences de nature diffirente. On voit que les trois points ainsi obtenus (ils correspondent au benzine, au
tktrahydrofuranne et au paraxylbne) se placent sur une droite. Si I’on extrapole cette droite h BIB, = 1 c’est-hdire au cas ou il n’y aurait pas d’interactions specifiques entre skquences de nature diffe’rente on retombe sur la valeur KO idkale c’est-idire celle qui correspond i l’additivite’ des propriete’s du PS et du PMM .
On voit donc qu’en fait le copolymkre se comporte d’une certaine manidre comme un homopolymkre car on peut lui appliquer le traitement de Stockmayer-Fixman ; cependant, cette regularitk n’est qu’apparente car les dimensions que l’on obtient de cette fagon ne sont pas des dimensions non perturbees mais des dimensions qui de‘pendent de la nature du solvant. Cette modification des dimensions par suite du phknomtne d’incompatibiliti semble independante de la masse molkculaire; on ne pourra donc pas l’e’liminer en faisant tendre cette derniire vers zero.
CAS D’UN HOMOPOLYM~RE DANS UN MBLANGE DE SOLVANT
Enfin, je voudrais pour terminer cet expose rappeler quelques rtsultats relatifs i I’extension aux melanges de la notion du solvant thkta. Bien que cette notion
42
LA THEORIE A DEUX PARAMETRES
ne soit pas fondee thioriquement de fagon rigoureuse, on a souvent utilis6, quand on ne disposait pas de solvant 8 convenable, des melanges solvant-pricipitant en ajustant leur composition de fagon soit que la viscositd varie comme Mi/*, soit que le second coefficient de viriel A 2 soit nu1 et on a appeli par extrapolation ces milanges des melanges 8.
Nous voudrions montrer sur quelques exemples que cette extrapolation est trhs dangereuse.
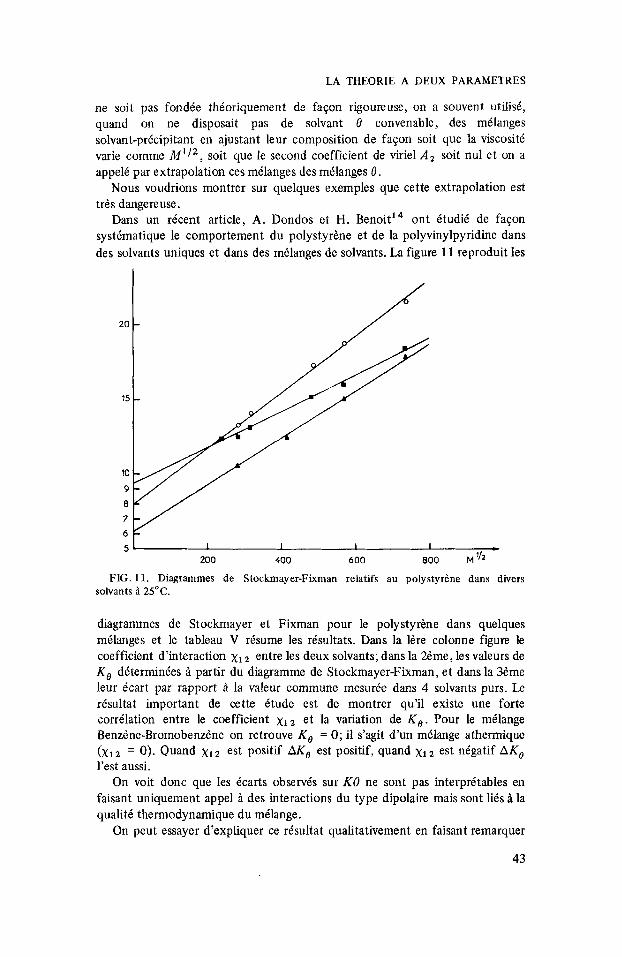
Dans un recent article, A. Dondos et H. Benoit14 ont BtudiC de fagon systimatique le comportement du polystyrhne et de la polyvinylpyridine dans des solvants uniques et dans des melanges de solvants. La figure 1 1 reproduit les
I I I I
200 400 600 800 M ‘12
FIG. 11. Diagrammes de Stockmayer-Fixman relatifs au polystyrene dans divers solvants B 25°C.
diagrammes de Stockmayer et Fixman pour le polystyrene dans quelques milanges et le tableau V risume les rdsultats. Dans la ldre colonne figure le coefficient d’interaction x1 entre les deux solvants; dans la 2hme, les valeurs de K , determinies ;i partir du diagramme de Stockmayer-Fixman, et dans la 3dme leur k a r t par rapport A la valeur commune mesurde dans 4 solvants purs. Le resultat important de cette Btude est de montrer qu’il existe une forte correlation entre le coefficient x l z et la variation de K , . Pour le milange Benzhe-BromobenzCne on retrouve K , = 0; il s’agit d’un melange athermique (x, = 0). Quand xI2 est positif AK, est positif, quand x1 est nBgatif A K , I’est aussi.
On voit donc que les Bcarts observe’s sur KO ne sont pas interprktables en faisant uniquement appel ;i des interactions du type dipolaire mais sont liCs A la qualiti thermodynamique du melange.
On peut essayer d’expliquer ce resultat qualitativement en faisant remarquer
43

BENOIT, DECKER, DONDOS, ET REMPP
TABLEAU V
Solvant X I 2 Ke AK5
Benzine Cyclohexane Chloroforme Dioxanne
Benz. 50 - Cyclohex. 50 HCCl, 80 - EtOH 20 HCC1, 66 - EtOH 34 Benz. 61 - EtOH 33 HCC1, 22 - Acetone 18 HCCI, 50 - Diox. 50 Benz. 78 - MeOH 22 Benz. 50 - Br. Benz. 50
+ 046 + 1,20 + 1,20 + 1,7 - 0,9 - l ,o + 2,o
0 ,o
Polys tyrine
+ 19 + 12 + 21 + 20
- 8 - 21 + 5
0
que quand x12 est positif il y a repulsion entre les deux types de molecules du solvant; cette repulsion s’exerce aussi entre molecules de solvant adsorbees sur la macromolecule et tend Q augmenter ses dimensions d’oh la valeur de K , trop Blevie. lnversement quand x I 2 est negatif les deux solvants ont tendance i s’associer et cette tendance a pour resultat de faire diminuer les dimensions de la pelote macromoleculaire .
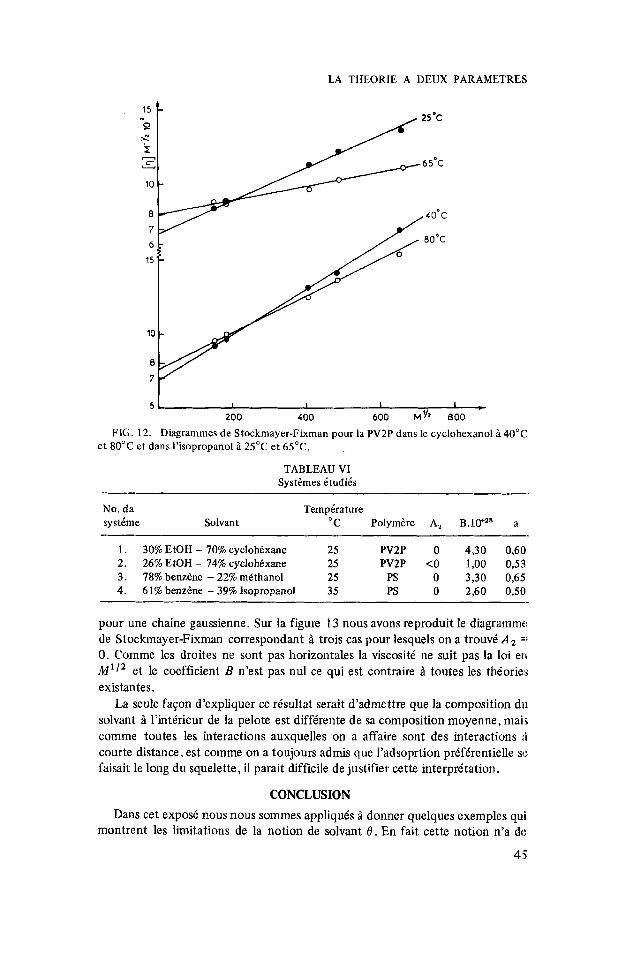
Ce phinomine que nous venons de decrire dans le cas des melanges de solvant se retrouve dans le cas des solvants associis. Sur la figure 12 nous avons reproduit des risultats de viscositk intrinsbque d’une skrie de fractions de polyvinyl-2 pyridine dans l’isopropanol. Les deux droites correspondent I’une Q 25”C, l’autre Q 65°C. On constate une variation importante de K , , variation qui ne peut btre expliquee par le coefficient de temperature des dimensions non perturbke s.
A 25°C on trouve un K , infkrieur au K , obtenu dans les solvants classiques alors, qu’a 65” on retrouve une valeur qui tend vers la valeur commune Q tous les solvants, (87l o-2). On sait que l’isopropanol est un solvant associe Q temperature ordinaire et que son degre d’association diminue beaucoup avec la temperature; il semble donc que l’explication que nous avions suggkrie pour les melanges de solvants permette aussi de rendre compte des resultats aberrants que l’on peut obtenir dans les solvants associks.
Enfin en terminant cet expose je voudrais encore attirer l’attention sur les rksultats suivants. Le solvant 19 a toujours etd defini comme celui pour lequel soit la viscosite intrindque est proportionnelle Q soit le second coefficient du viriel est nul.
Dans tout ce qui prtcide nous nous sommes bases pour dkfinir K, sur les risultats de viscosit6. Dans le tableau VI figurent quelques rtsultats obtenus sur du polystyrhe et de la polyvinyl 2-pyridine dans diffkrents melanges de solvants. Dans ce tableau figurent la valeur de A 2 , celle du coefficient B de Stockmayer-Fixman et l’exposant a de la loi de viscositi. On constate que dans un certain nombre de cas A2 = 0 alors que les dimensions croissent plus vite que
44
LA THEORIE A DEUX PARAMETRES
n
0
I F n r Y
10 -
7
15
0
I
n
F n r Y
10 -
6 ;
5 1 I 1 I I , 200 400 600 MYz 800
FIG. 12. Diagrammes de Stockmayer-Fixman pour la PV2P dans le cyclohexanol i 40°C , et 80°C et dam I’isopropanol i 25°C et 65°C.
TABLEAU VI Systkmes dtudiis
25%
.6 5OC
, 40°C
80°C
No. da Tempirature systime Solvant “C Polymbre A, B.lOtz* a
1 . 30% EtOH - 70% cyclohdxane 25 PV2P 0 4,30 0,60 2. 26% EtOH - 74% cyclohkxane 25 PV2P <O 1,00 0,53 3 . 78% benzene - 22% mkthanol 25 PS 0 3,30 0,65 4. 61% benzdne - 39% Isopropanol 35 PS 0 2,60 0,50
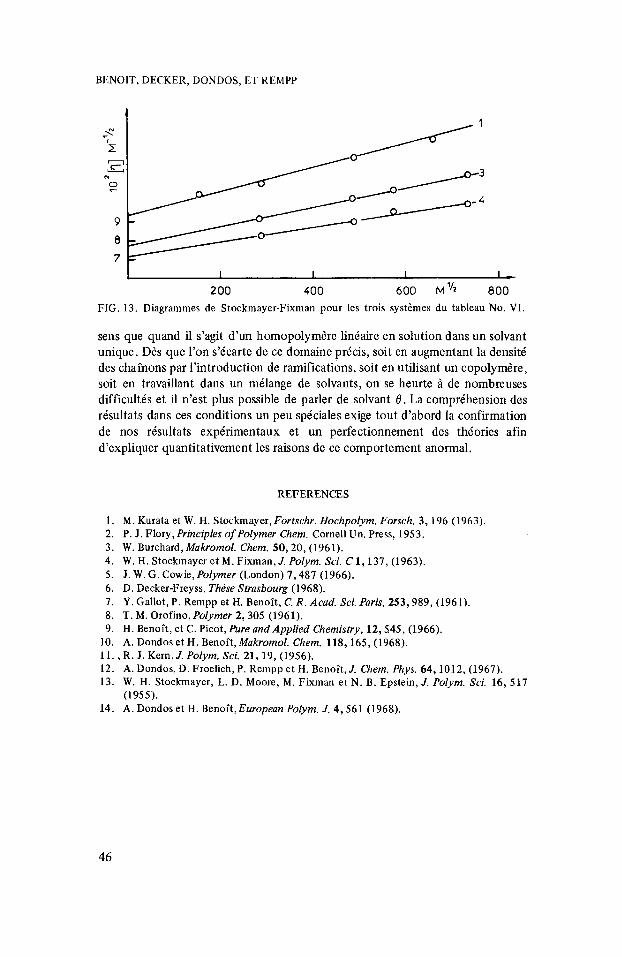
pour une chaine gaussienne. Sur la figure 13 nous avons reproduit le diagramme de Stockmayer-Fixman correspondant i trois cas pour lesquels on a trouvi A =: 0. Comme les droites ne sont pas horizontales la viscositt ne suit pas la loi en M’I2 et le coefficient B n’est pas nu1 ce qui est contraire i toutes les th6orie.s existantes.
La seule faGon d’expliquer ce resultat serait d’admettre que la composition dii solvant i l’intkrieur de la pelote est diffkrente de sa composition moyenne, mai3 comme toutes les interactions auxquelles on a affaire sont des interactions ;i courte distance, est comme on a toujours admis que l’adsoprtion prifirentielle SI:
faisait le long du squelette, il parait difficile de justifier cette interpktation.
CONCLUSION Dans cet expos6 nous nous sommes appliquts 1 donner quelques exemples qui
montrent les limitations de la notion de solvant 0 . En fait cette notion n’a de
4.5
BENOIT, DECKER, DONDOS, ET KEMYP
I 1 1 1 1
200 400 600 M”2 800 FIG. 13. Diagrammes de Stockmayer-Fixman pour les trois systkmes du tableau No. VI.
sens que quand il s’agit d’un homopolym6re liniaire en solution dans un solvant unique. Dks que l’on s’icarte de ce domaine pricis, soit en augmentant la densitt des chainons par l’introduction de ramifications, soit en utilisant un copolymGre, soit en travaillant dans un melange de solvants, on se heurte de nombreuses difficultis et il n’est plus possible de parler de solvant 8 . La compr6hension des risultats dans ces conditions un peu spdciales exige tout d’abord la confirmation de nos rdsultats expirimentaux et un perfectionnement des theories afin d’expliquer quantitativement les raisons de ce comportement anormal.
REFERENCES
1 . M. Kurata et W. H. Stockmayer, Fortschr. Hochpolym. Forsch. 3, 196 (1963). 2. P. J . Flory,Principles of Polymer Chem. Cornell Un. Press, 1953. 3. W. Burchard, Makromol. Chem. 50,20 , (1961). 4 . W. H. Stockmayer et M. Fixman,J. Polym. Sci. C 1,137, (1963). 5 . J . W. G. Cowie,Polymer (London) 7,487 (1966). 6. D. Decker-Freyss, These Strusbourg (1968). 7. Y. Gallot, P. Rempp et H. Benoit, C. R. Acad. Sci. Paris, 253,989, (1961). 8. T. M. Orofino, Polymer 2,305 (1961). 9. H. Benoit, et C. Picot, Pure and Applied Chemistry, 12,545, (1966).
10. A. Dondos et H. Benoit, Mukromol. Chem. 118,165, (1968). 11. , R. J. Kern,J. Polym. Sci. 21,19, (1956). 12. A. Dondos, D. Froelich, P. Rempp et H. Benoit,J. Chem. Phys. 64,1012, (1967). 13. W. H . Stockmayer, L. D. Moore, M. Fixman et N . B. Epstein, J. Polym. Sci. 16, 517
(1 955). 14. A . Dondos et H. Benoit,European Polym. J. 4,561 (1968).
46





























