Embed Size (px)
Citation preview

R E S E A R C H A R T I C L E
MicrodiversityofBurkholderiales associatedwithmycorrhizal andnonmycorrhizal rootsofMedicago truncatulaPierre Offre1, Barbara Pivato1,2, Sylvie Mazurier1, Severine Siblot1, Graziella Berta2, PhilippeLemanceau1 & Christophe Mougel1
1INRA, Universite de Bourgogne, UMR1229 ‘Microbiologie du Sol et de l’Environnement’, CMSE, BP, Dijon, France; and2Universita del Piemonte Orientale ‘Amedeo Avogadro’, Dipartimento di Scienze dell’Ambiente e della Vita, Alessandria, Italy
Correspondence: Philippe Lemanceau,
INRA, Universite de Bourgogne, UMR1229
‘Microbiologie du Sol et de l’Environnement’,
CMSE, 17 rue Sully, BP 86510, F-21065 Dijon,
France. Tel.: 133 3 80 69 30 56; fax: 133 3
80 69 32 24; e-mail: [email protected]
Received 15 September 2007; revised 25 March
2008; accepted 2 April 2008.
First published online 28 May 2008.
DOI:10.1111/j.1574-6941.2008.00504.x
Editor: Jim Prosser
Keywords
arbuscular mycorrhizas; rhizosphere; bacterial
diversity; Comamonadaceae ;
Oxalobacteraceae .
Abstract
The genetic diversity of bacterial communities associated with mycorrhizal and
nonmycorrhizal roots of Medicago truncatula was characterized by two ap-
proaches. Firstly, phylogenetic analysis was performed on 164 partial 16S rRNA
gene–intergenic spacer (IGS) sequences from operational taxonomic units pre-
viously shown to be preferentially associated with mycorrhizal roots. These
sequences were distributed into three branches corresponding to Comamonada-
ceae, Oxalobacteraceae and Rubrivivax subgroups. Most sequences were obtained
from mycorrhizal roots, indicating the preferential association of the correspond-
ing families with mycorrhizal roots. A second phylogenetic analysis was performed
on the partial 16S rRNA gene–IGS sequences of 173 isolates among a large
collection of isolates, from mycorrhizal and nonmycorrhizal roots, belonging to
Comamonadaceae and Oxalobacteraceae on the basis of their positive hybridization
with a partial 16S rRNA gene–IGS probe obtained in this study. Sequence analysis
confirmed the affiliation of 166 isolates to Comamonadaceae and seven to
Oxalobacteraceae. Oxalobacteraceae isolates were more abundant in mycorrhizal
(five) than in nonmycorrhizal (two) roots, whereas Comamonadaceae isolates were
more abundant in nonmycorrhizal (109) than mycorrhizal roots (57). Further
analysis of Comamonadaceae isolates by BOX-PCR showed that the genetic
structure of culturable populations belonging to this family differed significantly
in mycorrhizal and nonmycorrhizal roots, as indicated by distributions in different
BOX types, differences being significantly explained by BOX types only including
isolates from mycorrhizal roots. These data are discussed in an ecological context.
Introduction
Arbuscular mycorrhizas (AM) are symbiotic associations
between most land plants (Wang & Qiu, 2006) and obligate
biotrophic AM fungal species belonging to the Glomeromy-
cota phylum (Schußler et al., 2001). The AM association
represents an ancient symbiosis with fossil evidence dating
back 400 millions years (Remy et al., 1994). The roots of
earliest land plants contained arbuscular mycorrhizal struc-
tures and the symbiosis of ancestral plants with the Glomer-
omycota may have enabled plants to colonize land
(Brundrett, 2002). AM have a central position in terrestrial
nutrient-cycling processes and are of particular interest
because of their positive effects on plant growth and health
(Smith & Read, 1997).
Because of the long coevolution of plants and AM fungi,
reciprocal interactions based on feedbacks between them
have been proposed (Bever et al., 2002). This proposal is
based on the observation of differential responses of plant
species to individual isolates and species of AM fungi
(Streitwolf-Engel et al., 1997) leading to variations in plant
composition and productivity according to AM fungal
diversity and identity (van der Heijden et al., 1998; Klirono-
mos et al., 2000). In return plant species, even those that are
closely related, differentially affect the AM fungal commu-
nity associated with mycorrhizal roots (Vandenkoornhuyse
et al., 2003; Pivato et al., 2007).
Coevolution of plants and AM fungi is not expected
to have occurred independently from the associated
bacterial communities. However, information on bacteria
FEMS Microbiol Ecol 65 (2008) 180–192c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved

preferentially associated with mycorrhizal roots appears to
be scarce. Few studies have addressed the relationship
between free-living bacteria and AM fungi (Meyer & Linder-
man, 1986; Andrade et al., 1997; Marschner et al., 2001;
Artursson et al., 2005). Until recently (Offre et al., 2007),
none of these studies has identified taxa explaining varia-
tions of bacterial community structure associated with AM.
Furthermore, these studies did not take into account the
natural diversity of AM fungi, because they were based
on inoculation of selected strains of Glomeromycota.
Progress in our knowledge on bacteria associated with
mycorrhizal roots is required. Indeed, from an evolutionary
point of view, one may expect that these bacteria would at
least be nondeleterious or even beneficial to mycorrhization,
as has been shown for a fluorescent pseudomonad repre-
sentative of populations preferentially associated with
ectomycorrhizas (Frey et al., 1997), and with Paenibacillus
sp. strain B2, isolated from the mycorrhizosphere of
Sorghum bicolor–Glomus mosseae and promoting AM (Budi
et al., 1999).
Based on these statements, a research was undertaken to
characterize bacteria preferentially associated with mycor-
rhizal roots. The first step of this research was to compare
the bacterial communities associated with mycorrhizal and
nonmycorrhizal plant genotypes when cultivated in a fallow
soil from the Mediterranean area (Mas d’Imbert, France),
corresponding to the diversification zone of annual Medics.
The genetic structure of these communities, assessed by
Automated-Ribosomal Intergenic Spacer Analysis (A-RISA)
from DNA directly extracted from root tissues, appeared to
differ significantly. The molecular markers [partial 16S
rRNA gene and 16S–23S intergenic spacer (IGS) sequences]
explaining these differences were cloned and sequenced. The
corresponding sequences, grouped into operational taxo-
nomic units (OTUs), showed a high level of similarity with
partial 16S rRNA gene sequences of Methylibium petrolei-
philum and Collimonas fungivorans belonging to Comamo-
nadaceae and Oxalobacteraceae (Burkholderiales),
respectively (Offre et al., 2007).
The second step and the aim of this study were to
characterize further the diversity of Burkholderiales preferen-
tially associated with mycorrhizal roots using molecular and
cultivation-based approaches. The molecular approach was
based on phylogenetic analysis of partial 16S rRNA gene–IGS
sequences belonging to OTUs identified previously (Offre
et al., 2007). The cultivation-based approach involved analy-
sis of a large collection of bacterial isolates from root tissues of
Medicago truncatula J5 and its AM defective mutant TRV25,
cultivated in the Mas d’Imbert soil. Among them, isolates
belonging to Comamonadaceae and Oxalobacteraceae were
expected to be preferentially associated with mycorrhizal
roots (Offre et al., 2007) and were detected by dot-blot
hybridization with a polynucleotide probe. The genetic
diversity of these isolates was characterized by analysing their
partial 16S rRNA gene–IGS sequences and bacterial repetitive
BOX element (BOX)-PCR fingerprinting.
Materials and methods
Plant growth conditions and bacterial isolation
Experiments were conducted with M. truncatula Gaertn. cv.
Jemalong line J5 (Myc1/Nod1) and its symbiosis-defective
mutant TRV25 (Myc�/Nod�) (Sagan et al., 1995). Seeds of
the two plant genotypes were scarified and surface-sterilized
by gently shaking in 98% sulphuric acid for 2 min, 95%
ethanol for 5 min and 3.5% sodium hypochlorite solution
for 10 min and rinsed successively six times for 5 min in
sterile demineralized water. Sterilized seeds were germinated
on a 0.7% (w/v) water agar plate at 25 1C for 48 h. Twelve
plants were cultivated per M. truncatula genotype by sowing
one germinated seed per cylindrical polyvinyl chloride
container (diameter: 9 cm, height: 20 cm) filled with 1.5 L
of a silt-clay loam soil (Mas d’Imbert, France). They were
cultivated in a growth chamber at a 16 : 8 h light/dark
photoperiod, a 23 : 18 1C light/dark thermoperiod, photo-
synthetic active radiation (PAR) = 500 mE m�2 s�1 at pot
height and 55% relative humidity. Water was added daily
to maintain humidity at 55% of the water-holding capacity.
Containers were randomly distributed in the growth cham-
ber and moved each day. The plants were sampled after 34
days, corresponding to the appearance of shoot ramifica-
tions of order 2 (Mougel et al., 2006).
Bacteria were isolated from four root samples per plant
genotype, each sample resulting from pooling three root
systems. Adhering soil was removed from the roots by
shaking them twice in a flask containing 100 mL of sterile
distilled water at 200 r.p.m. for 10 min (Agitests, Bioblock
Scientific, Illkirch, France). The washed roots were ground
with an Ultra-Turax (Janke and Kunkel GmbH and Co.KG,
Staufen, Germany) in 50 mL of sterile distilled water. Root
suspensions were dilution-plated on 0.1� tryptic soy agar
(TSA) (Forbes et al., 1998) supplemented with 200 mg mL�1
of cycloheximide (Sigma, St Louis, MO) and incubated for 5
days at 25 1C. A total of 2412 bacterial colonies were isolated:
1200 from M. truncatula J5 and 1212 from M. truncatula
TRV25 (Table 1). Isolates were subjected to single-colony
isolation and stored at � 80 1C in tryptic soy broth (TSB)
containing 20% glycerol.
Sequences of 16S rRNA gene--IGS
One hundred and sixty-four partial sequences of 16S rRNA
genes and 16S–23S IGS obtained previously from DNA
extracted from root tissues of M. truncatula were analysed.
These sequences were chosen as belonging to OTUs pre-
viously shown to be preferentially associated with
FEMS Microbiol Ecol 65 (2008) 180–192 c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
181Bacterial diversity associated with mycorrhizal roots
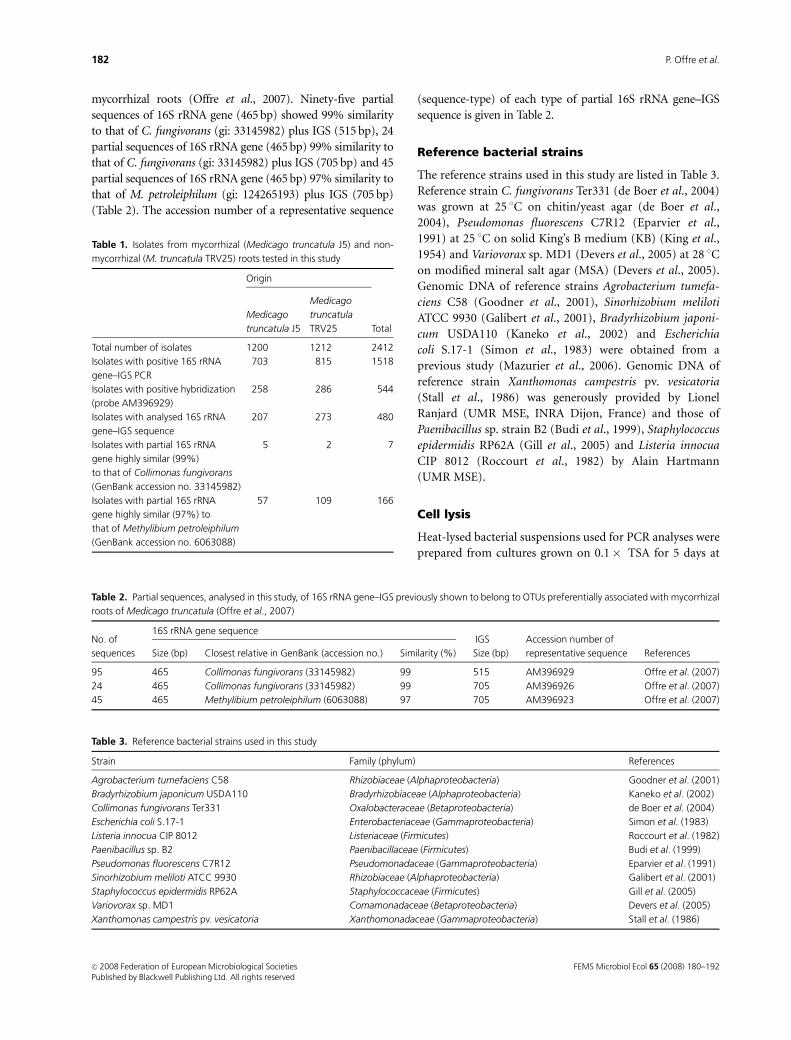
mycorrhizal roots (Offre et al., 2007). Ninety-five partial
sequences of 16S rRNA gene (465 bp) showed 99% similarity
to that of C. fungivorans (gi: 33145982) plus IGS (515 bp), 24
partial sequences of 16S rRNA gene (465 bp) 99% similarity to
that of C. fungivorans (gi: 33145982) plus IGS (705 bp) and 45
partial sequences of 16S rRNA gene (465 bp) 97% similarity to
that of M. petroleiphilum (gi: 124265193) plus IGS (705 bp)
(Table 2). The accession number of a representative sequence
(sequence-type) of each type of partial 16S rRNA gene–IGS
sequence is given in Table 2.
Reference bacterial strains
The reference strains used in this study are listed in Table 3.
Reference strain C. fungivorans Ter331 (de Boer et al., 2004)
was grown at 25 1C on chitin/yeast agar (de Boer et al.,
2004), Pseudomonas fluorescens C7R12 (Eparvier et al.,
1991) at 25 1C on solid King’s B medium (KB) (King et al.,
1954) and Variovorax sp. MD1 (Devers et al., 2005) at 28 1C
on modified mineral salt agar (MSA) (Devers et al., 2005).
Genomic DNA of reference strains Agrobacterium tumefa-
ciens C58 (Goodner et al., 2001), Sinorhizobium meliloti
ATCC 9930 (Galibert et al., 2001), Bradyrhizobium japoni-
cum USDA110 (Kaneko et al., 2002) and Escherichia
coli S.17-1 (Simon et al., 1983) were obtained from a
previous study (Mazurier et al., 2006). Genomic DNA of
reference strain Xanthomonas campestris pv. vesicatoria
(Stall et al., 1986) was generously provided by Lionel
Ranjard (UMR MSE, INRA Dijon, France) and those of
Paenibacillus sp. strain B2 (Budi et al., 1999), Staphylococcus
epidermidis RP62A (Gill et al., 2005) and Listeria innocua
CIP 8012 (Roccourt et al., 1982) by Alain Hartmann
(UMR MSE).
Cell lysis
Heat-lysed bacterial suspensions used for PCR analyses were
prepared from cultures grown on 0.1� TSA for 5 days at
Table 1. Isolates from mycorrhizal (Medicago truncatula J5) and non-
mycorrhizal (M. truncatula TRV25) roots tested in this study
Origin
Total
Medicago
truncatula J5
Medicago
truncatula
TRV25
Total number of isolates 1200 1212 2412
Isolates with positive 16S rRNA
gene–IGS PCR
703 815 1518
Isolates with positive hybridization
(probe AM396929)
258 286 544
Isolates with analysed 16S rRNA
gene–IGS sequence
207 273 480
Isolates with partial 16S rRNA
gene highly similar (99%)
to that of Collimonas fungivorans
(GenBank accession no. 33145982)
5 2 7
Isolates with partial 16S rRNA
gene highly similar (97%) to
that of Methylibium petroleiphilum
(GenBank accession no. 6063088)
57 109 166
Table 2. Partial sequences, analysed in this study, of 16S rRNA gene–IGS previously shown to belong to OTUs preferentially associated with mycorrhizal
roots of Medicago truncatula (Offre et al., 2007)
No. of
sequences
16S rRNA gene sequenceIGS Accession number of
representative sequence ReferencesSize (bp) Closest relative in GenBank (accession no.) Similarity (%) Size (bp)
95 465 Collimonas fungivorans (33145982) 99 515 AM396929 Offre et al. (2007)
24 465 Collimonas fungivorans (33145982) 99 705 AM396926 Offre et al. (2007)
45 465 Methylibium petroleiphilum (6063088) 97 705 AM396923 Offre et al. (2007)
Table 3. Reference bacterial strains used in this study
Strain Family (phylum) References
Agrobacterium tumefaciens C58 Rhizobiaceae (Alphaproteobacteria) Goodner et al. (2001)
Bradyrhizobium japonicum USDA110 Bradyrhizobiaceae (Alphaproteobacteria) Kaneko et al. (2002)
Collimonas fungivorans Ter331 Oxalobacteraceae (Betaproteobacteria) de Boer et al. (2004)
Escherichia coli S.17-1 Enterobacteriaceae (Gammaproteobacteria) Simon et al. (1983)
Listeria innocua CIP 8012 Listeriaceae (Firmicutes) Roccourt et al. (1982)
Paenibacillus sp. B2 Paenibacillaceae (Firmicutes) Budi et al. (1999)
Pseudomonas fluorescens C7R12 Pseudomonadaceae (Gammaproteobacteria) Eparvier et al. (1991)
Sinorhizobium meliloti ATCC 9930 Rhizobiaceae (Alphaproteobacteria) Galibert et al. (2001)
Staphylococcus epidermidis RP62A Staphylococcaceae (Firmicutes) Gill et al. (2005)
Variovorax sp. MD1 Comamonadaceae (Betaproteobacteria) Devers et al. (2005)
Xanthomonas campestris pv. vesicatoria Xanthomonadaceae (Gammaproteobacteria) Stall et al. (1986)
FEMS Microbiol Ecol 65 (2008) 180–192c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
182 P. Offre et al.

25 1C for bacterial isolates, and as described above for the
reference strains. One colony per bacterial isolate/strain was
suspended in 100mL of lysis solution [0.05 M NaOH, 0.25%
sodium dodecyl sulphate (SDS)] and incubated for 15 min
in boiling water. The resulting suspension was centrifuged
for 1 min at 15 294 g and the supernatant was diluted 50-fold
in sterile water.
PCR amplification and sequencing of 16S rRNAgene--IGS
Sequences of partial 16S rRNA gene–IGS from DNA extracted
previously from root tissues (Table 2) and from bacterial
isolates were amplified by PCR with the primers 1055F
(50-ATGGCTGTCGTCAGCTT-30) and L-D-Bact-132-a-
A-18 (50-CCGGGTTTCCCCATTCGG-30) (Normand et al.,
1996). Reactions were performed in a total volume of 25mL,
by mixing 1 ngmL�1 of DNA, 1� of buffer containing MgCl2[10 mM Tris-HCl (pH 9), 50 mM KCl, 1.5 mM MgCl2, 0.1%
Triton X-100, 0.2 mg mL�1 bovine serum albumin solution
(BSA)] (Q-BIOgene, Illkirch, France), 200mM of dNTPs
(Q-BIOgene), 1mM of each primer and 1.5 U of Taq DNA
polymerase (Q-BIOgene). Each reaction was performed in a
thermal cycler (MJ Research PTC-0225 DNA Engine Tetrad,
Bio-rad, Marne la Coquette, France). PCR conditions were as
follows: a first step at 72 1C for 6 min, a second step at 95 1C
for 5 min, followed by 30 cycles of 94 1C for 1 min, 55 1C for
1 min and 72 1C for 1 min, before a final elongation step for
5 min at 72 1C. PCR products were checked on 2% (w/v)
agarose gels in TAE buffer [40 mM Tris (pH 7.8), 20 mM
acetic acid, 2 mM EDTA] for size determination and were
sequenced using the 1055F primer.
Sequencing of partial 16S rRNA gene–IGS was performed by
the GENOME express company (Genome Express, Meylan,
France) with a standard sequencing protocol. Sequences pro-
vided as text files were used for sequence analysis and phyloge-
netic inference. Newly obtained partial sequences of 16S rRNA
gene–IGS were deposited in GenBank under the following
accession numbers: EU074784, EU074785, EU074786,
EU074787, EU074788, and EU074789.
Development of probe and hybridizationconditions
Hybridizations were performed on Pall Biodyne Plus mem-
branes (VWR France, Fontenay-sous-Bois, France). Ten
nanograms of PCR product was spotted onto the membrane
after denaturation for 10 min in a boiling water bath using a
dot-blot vacuum manifold (Schleicher & Schuell, Dassel,
Germany). DNA was fixed on a membrane for 30 min at
80 1C. Attempts were made to prepare probes from the
sequences AM396929 and AM396923 (Table 2), but were
only successful with the sequence AM396929. The probe was
obtained by labelling this sequence with digoxigenin by PCR
performed in the same reaction mixture as described above,
except for the final volume (50 mL) and for the dNTPs,
which were used at the following concentrations: 5 mM of
dATP, 5 mM of dGTP, 5mM of dCTP, 0.5mM of dTTP and
0.2 mM of Dig-11-dUTP (Roche, Molecular Biochemicals,
Meylan, France). DNA hybridization was performed over-
night at 68 1C in 10 mL of hybridization buffer [sodium
saline citrate (SSC) 5� , 0.1% (w/v) sodium lauroyl sarco-
sinate, 0.02% (w/v) SDS and 0.5% (v/v) blocking reagent
(Roche)] containing 10 ng mL�1 of the AM396929 probe.
The washing steps were performed applying high-stringency
conditions: twice for 5 min at room temperature in 0.1%
SDS and SSC 2� , twice for 15 min at 68 1C in 0.1% SDS
and SSC 0.2� and once for 15 min at 68 1C in 0.1% SDS
and SSC 0.02� . Probe detection was performed using the
DIG Luminescent Detection kit (Roche), following the
manufacturer’s instructions.
Phylogenetic analyses
Sequences of partial 16S rRNA gene–IGS from DNA extracted
previously from root tissues (Table 2) and those from bacterial
isolates hybridizing with the AM396929 probe were used as
queries in Basic Alignment Search Tool (BLAST) searches for
standard nucleotide–nucleotide BLAST (Altschul et al., 1990).
Nucleotide sequences corresponding to queries and the most
closely relative sequences identified by BLAST analysis were
applied to construct multiple sequence alignments as deter-
mined by CLUSTAL W (Thompson et al., 1994). Optimization of
the multiple sequence alignments was performed using the
graphical editor SeaView (Galtier et al., 1996).
Phylogenetic inferences were performed using the PHYLO_WIN
graphical interface (Galtier et al., 1996). Phylogenies were
determined by the neighbour-joining method (Saitou & Nei,
1987) using the Kimura ‘two-parameters’ correction (Kimura,
1980) with the pairwise gap removal option. In order to
estimate tree node validity, the result of 1000 bootstrapped data
sets was determined. Reconstructed trees were drawn and
evaluated using the NJPLOT program (Perriere & Gouy, 1996).
BOX-PCR
Isolates having both a positive hybridization with the
AM396929 probe and a high similarity (at least equal to 97%)
of their 16S rRNA gene with the corresponding partial
sequence (465 bp) of C. fungivorans (gi: 33145982) and of
M. petroleiphilum (gi: 6063088) were selected for repetitive
sequences PCR (rep-PCR) characterization. Rep-PCR finger-
printing was achieved using the primer BOXA1R (50-CTACG
GCAAGGCGACGCTGACG-30) (Versalovic et al., 1994).
BOX-PCR reactions were performed in a total volume of
FEMS Microbiol Ecol 65 (2008) 180–192 c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
183Bacterial diversity associated with mycorrhizal roots

25mL, by mixing 3mL of the diluted heat-lysed bacterial
suspension, 5mL of 5� Gitschier buffer [83 mM (NH4)2SO4,
335 mM Tris-HCl (pH 8.8), 33.5 mM MgCl2, 33.5mM EDTA,
150 mM b-mercaptoethanol], 0.4mL of 10 mg mL�1 BSA
(Q-BIOgene), 2.5mL of dimethyl sulphoxide, 11.7mL of sterile
distilled water, 25mM each of dATP, dCTP, dGTP, and dTTP,
1mM of BOXA1R primer (Eurogentec, Seraing, Belgium) and
2.25 U of Taq DNA polymerase (Q-BIOgene). Amplification
reactions were processed in a Perkin Elmer GeneAmp PCR
System 9600 thermal cycler (Applied Biosystems, Foster City,
CA). Cycling conditions for BOX-PCR were as follows: 95 1C
for 2 min and then 35 cycles of 94 1C for 3 s, 92 1C for 30 s,
50 1C for 1 min and 65 1C for 8 min, before a final elongation
for 8 min at 65 1C. Five microlitres of amplified products were
separated on 1.5% (w/v) agarose gels in TAE buffer for 15 h at
40 V. A 1-kb DNA ladder (Invitrogen SARL, Cergy Pontoise,
France) was included in the gels as a size marker. The gels were
stained with ethidium bromide (1 mg mL�1) and DNA band-
ing patterns were visualized under UV (BIOCAPT software
version 99.0385, Vilbert Lourmat, Marne La Vallee, France).
BOX-PCR reactions were performed in triplicate to ensure that
only reliable bands were scored. Visual analysis was performed
to group together similar BOX-PCR fingerprints with subse-
quent further visual analysis allowing, for each BOX-PCR
fingerprint, establishment of a binary data matrix scoring 45
bands as being either present (1) or absent (0). A similarity
matrix was calculated using the pairwise Nei & Li (1979)
similarity coefficient, and an arbitrary similarity coefficient of
0.54 was chosen to delineate BOX types. Cluster analysis was
performed by the unweighted pair group method using
arithmetic averages (UPGMA).
The distributions of isolates from M. truncatula J5 and
TRV25 in these BOX types were compared using Pearson w2
test and the probability that the two distributions differed
significantly was calculated using the general Monte Carlo
algorithm. Computations were performed with the STATXACT
software version 3 (CYTEL Software Corporation, Cam-
bridge). BOX types explaining differences between the two
distributions were identified by analysis of contingency-
table cell contribution to the w2 value.
Results
Diversity of partial 16S rRNA gene--IGSsequences belonging to OTUs preferentiallyassociated with mycorrhizal roots ofM. truncatula
The diversity of the 164 partial sequences of 16S rRNA
gene–IGS described in Table 2 was characterized by phylo-
genetic analyses performed separately on the partial 16S
rRNA gene (Fig. 1a) and IGS (Fig. 1b) sequences. The partial
16S rRNA gene sequences analysed were distributed into
three monophyletic branches, supported by a significant
bootstrap value (100), corresponding to the Rubrivivax
subgroup, the Comamonadaceae and the Oxalobacteraceae
families, all belonging to Burkholderiales (Betaproteobacter-
ia) (Fig. 1a). The Rubrivivax subgroup branch encompassed
36 AM396923-type sequences, showing 97% similarity to
that of M. petroleiphilum (gi: 6063088) (Table 2); 32
originated from M. truncatula J5 and four from TRV25.
The Comamonadaceae branch encompassed nine
AM396923-type sequences, eight from J5 and one from
TRV25. Finally, the Oxalobacteraceae branch encompassed
all sequences (119) showing 99% similarity to C. fungivorans
(gi: 33145982) either associated with IGS (515 bp) corre-
sponding to AM396929-type sequences (95), 65 from J5 and
30 from TRV25, or with IGS (705 bp) corresponding to
AM396926-type sequences (24), 20 from J5 and four from
TRV25.
IGS sequences were distributed into four branches (W, X,
Y and Z) supported by a high bootstrap value (100)
(Fig. 1b). Thirty-one AM396923-type sequences were in-
cluded in branch W, 29 from M. truncatula J5 and two from
TRV25. Three AM396923-type sequences were included in
branch X: two from J5 and one from TRV25. Nine
AM396923-type sequences were included in branch Y: eight
from J5 and one from TRV25. Finally, 95 AM396929-type
sequences, 65 from J5 and 30 from TRV25, and 23
AM396926-type sequences, 20 from J5 and 3 from TRV25,
were included in branch Z (Fig. 1b). IGS branches W and
Fig. 1. Phylogenetic analyses of (i) 95 partial 16S rRNA gene–IGS sequences (980 bp) including partial 16S rRNA gene sequences having 99% similarity
to Collimonas fungivorans (gi: 33145982) (AM396929-type sequences, Table 3), (ii) 24 (1170 bp) including 16S rRNA gene having 99% similarity to C.
fungivorans (gi: 33145982) (AM396926-type sequences) and (iii) 45 (1170 bp) including 16S rRNA gene having 97% similarity to Methylibium
petroleiphilum (gi: 6063088) (AM396923-type sequences). Phylogenetic distances were determined by the neighbour-joining analysis using the Kimura
‘two-parameters’ correction with the pairwise gap removal option. The sequence types (AM396929, AM396926, and AM396923) are indicated in bold
and their numbers in brackets. Branching robustness is expressed as the percentage of reliability after 1000 bootstraps’ resampling (values 4 75 are
indicated). The scale bar represents the expected nucleotide replacement per site. (a) Relationships between partial 16S rRNA gene sequences (465 bp)
from (i) AM396929-, AM396926- and AM396923-type sequences and (ii) sequences extracted from GenBank of Burkholderiales (Betaproteobacteria)
belonging to Comamonadaceae and Oxalobacteraceae families and the Rubrivivax subgroup. (b) Relationships between IGS sequences from AM396929
(515 bp)-, AM396926 (705 bp)- and AM396923 (705 bp)-type sequences. Partial 16S rRNA gene sequences and corresponding IGS related to the Rubrivivax
subgroup, Comamonadaceae and Oxalobacteraceae are highlighted with and respectively. Sequences belonging to DNA extracted
previously from M. truncatula J5 and TRV25 roots (Offre et al., 2007) are indicated by and respectively.
FEMS Microbiol Ecol 65 (2008) 180–192c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
184 P. Offre et al.

X included sequence types corresponding to partial 16S
rRNA gene sequences belonging to the Rubrivivax subgroup,
and Y and Z IGS branches to partial 16S rRNA gene
sequences belonging to Comamonadaceae and Oxalobacter-
aceae, respectively (Fig. 1a). Three sequences were not
clustered in any IGS branch (Fig. 1b): two AM396923-type
sequences corresponding to the 16S rRNA gene Rubrivivax
subgroup branch (Fig. 1a) and one AM396926-type
Aquaspirillum arcticum (gi: 21068882)Duganella zoogloeoides (gi: 1944498)
Sinobacter plicatus (gi: 62240118)
Aquaspirillum anulus (gi: 21068885)Giesbergeria kuznetsovii (gi: 60117041)Rhodoferax ferrireducens (gi: 74023592)
Variovorax paradoxus (gi: 82504534)Variovorax ginsengisoli (gi: 84453033)Variovorax dokdonensis (gi: 74423019)Curvibacter delicatus (gi: 3462592)Simplicispira metamorpha (gi: 3462593)Comamonas aquatica (gi: 20513173)
Rubrivivax benzolyticum (gi: 61657864)Methylibium petroleiphilum (gi: 6063088)Rubrivivax gelatinosus (gi: 303828)
Leptothrix mobilis (gi: 1263136)Ideonella dechloratans (gi: 577726)
Leptothrix discophora (gi: 501166)Sphaerotilus natans (gi: 47280)
100
100
100
0.01
Herbaspirillum putei (gi: 39652274)Herbaspirillum huttiense (gi: 4165372)
Collimonas fungivorans (gi: 33145982)Herminiimonas aquatilis (gi: 75754588)
Duganella violaceinigra (gi: 57996778)Massilia timonae (gi: 4204854)
Oxalobacteraceae
Comamonadaceae 100
100
100
100
0.1(a) (b)
AM396923-type (32)
AM396923-type (4)
AM396923-type (8)AM396923-type (1)
AM396929-type (65) &AM396926-type (20)
AM396929-type (30) &AM396926-type (4)
Rubrivivaxsub-group
W
X
Y
Z
AM396923-type (29)
AM396923-type (2)
AM396923-type (1)AM396923-type (2)
AM396923-type (1)AM396923-type (8)
AM396923-type (1)AM396923-type (1)
AM396929-type (65) &AM396926-type (20)
AM396929-type (30) &AM396926-type (3)
AM396926-type (1)
FEMS Microbiol Ecol 65 (2008) 180–192 c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
185Bacterial diversity associated with mycorrhizal roots

sequence corresponding to the 16S rRNA gene Oxalobacter-
aceae branch (Fig. 1a).
Root isolates belonging to Comamonadaceaeand Oxalobacteraceae
Bacterial isolates expected to be preferentially associated
with mycorrhizal roots were selected on the basis of their
affiliation to Comamonadaceae and Oxalobacteraceae by
dot-blot hybridization and by analysis of their partial 16S
rRNA gene–IGS sequences.
Attempts were made to prepare probes from an
AM396929-type sequence (Table 2) to target sequences
belonging to the Oxalobacteraceae branch (Fig. 1a) and from
two AM396923-type sequences to target those belonging to
the Rubrivivax subgroup and Comamonadaceae branches
(Fig. 1a). However, these attempts were only successful with
the AM396929-type sequence (Table 2). The specificity of the
AM396929-probe was assessed by recording (Fig. 2): (1) a
positive signal when hybridized onto: AM396929-type
sequences (1–3) and partial 16S rRNA gene–IGS from
C. fungivorans Ter331 (4), all belonging to Oxalobacteraceae;
AM396923-type sequences (1–3) and partial 16S rRNA
gene–IGS from Variovorax sp. MD1 (4), all belonging
to Comamonadaceae; one AM396923-type sequence (3)
belonging to the Rubrivivax subgroup; partial 16S rRNA
gene–IGS from A. tumefaciens C58 (1), B. japonicum
USDA110 (2) and S. meliloti ATCC 9930 (3), all belonging
to Alphaproteobacteria; partial 16S rRNA gene–IGS from X.
campestris pv. vesicatoria (3) belonging to Gammaproteobac-
teria; and partial 16S rRNA gene–IGS from S. epidermidis
RP62A (2), belonging to Firmicutes and (2) a negative signal
when hybridized onto two AM396923-type sequences (1–2)
belonging to the Rubrivivax subgroup, onto partial 16S rRNA
gene–IGS from P. fluorescens C7R12 (1) and E. coli S.17–1 (2),
all belonging to Gammaproteobacteria, and onto partial 16S
rRNA gene–IGS from Paenibacillus sp. strain B2 (1) and L.
innocua CIP 8012 (3), all belonging to Firmicutes. Taken
together, these observations indicate that the AM396929
probe was only partially specific because, besides sequences
from Oxalobacteraceae, a positive hybridization was recorded
with those from Comamonadaceae, with one from the
Rubrivivax subgroup and with those from Alphaproteobacter-
ia; a positive signal, although weaker, was also recorded
with one sequence from Gammaproteobacteria and from
Firmicutes.
Partial 16S rRNA gene–IGS sequences were successfully
amplified for 1518 of the 2412 isolates tested (Table 1). A
positive hybridization of partial 16S rRNA gene–IGS se-
quences with the AM396929 probe was obtained for 544
isolates (Table 1). Partial 16S rRNA gene–IGS was sequenced
for 480 of these isolates and 373 of them were analysed.
Taking into account the lack of specificity of the AM396929
probe, partial 16S rRNA gene–IGS of these 373 isolates was
compared with those available in GenBank. Partial 16S
rRNA gene–IGS of seven of these isolates showed a high
level of similarity (99%) to C. fungivorans (gi: 33145982)
belonging to Oxalobacteraceae, that of 166 isolates a high
Fig. 2. Dot-blot analyses of the specificity of the AM396929 probe. The
AM396929 probe was deposited onto (i) 10 ng of partial 16S rRNA
gene–IGS sequences analysed in this study (1–3) and Collimonas fungi-
vorans Ter 331 (4), all belonging to the Oxalobacteraceae, (ii) 10 ng of
partial 16S rRNA gene–IGS sequences analysed in this study (1–3) and of
Variovorax sp. DM1 (4), all belonging to Comamonadaceae, (iii) 10 ng of
partial 16S rRNA gene–IGS sequences analysed in this study (1–3)
belonging to the Rubrivivax subgroup, (iv) 10 ng of partial 16S rRNA
gene–IGS sequences of Agrobacterium tumefaciens C58 (1), Bradyrhi-
zobium japonicum USDA110 (2) and Sinorhizobium meliloti ATCC 9930
(3) all belonging to Alphaproteobacteria, (v) 10 ng of partial 16S rRNA
gene–IGS of Pseudomonas fluorescens C7R12 (1), Escherichia coli S.17-1
(2) and Xanthomonas campestris pv. vesicatoria (3) all belonging to
Gammaproteobacteria, and (vi) 10 ng of partial 16S rRNA gene–IGS
sequences of Paenibacillus sp. strain B2 (1), Staphylococcus epidermis
RP62A (2) and Listeria innocua CIP 8012 (3) all belonging to Firmicutes.
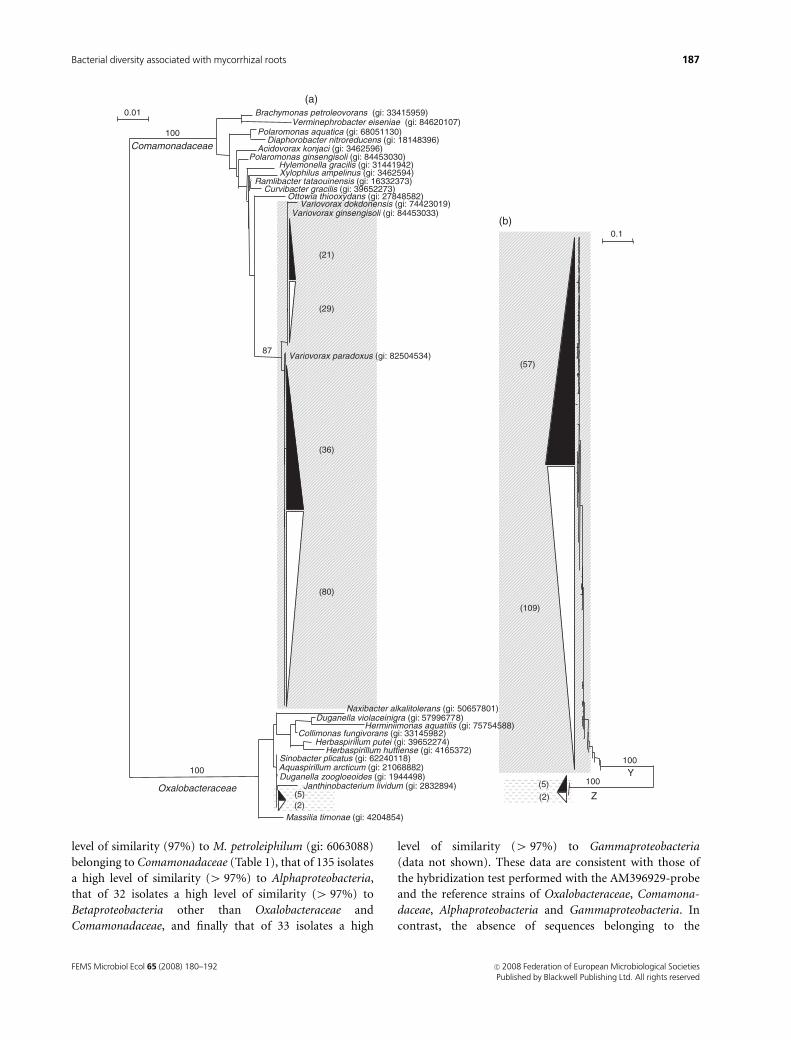
Fig. 3. Phylogenetic analyses of partial 16S rRNA gene–IGS sequences from 62 and 111 isolates from Medicago truncatula J5 ( ) and TRV25 ( ),
respectively. Phylogenetic distances were determined by neighbour-joining analysis using the Kimura ‘two-parameters’ correction with the pairwise gap
removal option. The sequence number for each M. truncatula genotype is indicated in brackets. Branching robustness is expressed as the percentage of
reliability after 1000 bootstraps’ resampling (values 4 75 were indicated). The scale bar represents the expected nucleotide replacement per site. (a)
Relationships between partial 16S rRNA gene sequences from isolates of the present study and extracted from GenBank of Burkholderiales
(Betaproteobacteria) belonging to Comamonadaceae and Oxalobacteraceae families. (b) Relationships between corresponding IGS sequences. Partial 16S
rRNA gene and corresponding IGS related to the Comamonadaceae and Oxalobacteraceae are highlighted with and respectively.
FEMS Microbiol Ecol 65 (2008) 180–192c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
186 P. Offre et al.
level of similarity (97%) to M. petroleiphilum (gi: 6063088)
belonging to Comamonadaceae (Table 1), that of 135 isolates
a high level of similarity (4 97%) to Alphaproteobacteria,
that of 32 isolates a high level of similarity (4 97%) to
Betaproteobacteria other than Oxalobacteraceae and
Comamonadaceae, and finally that of 33 isolates a high
level of similarity (4 97%) to Gammaproteobacteria
(data not shown). These data are consistent with those of
the hybridization test performed with the AM396929-probe
and the reference strains of Oxalobacteraceae, Comamona-
daceae, Alphaproteobacteria and Gammaproteobacteria. In
contrast, the absence of sequences belonging to the
Massilia timonae (gi: 4204854)
Janthinobacterium lividum (gi: 2832894)
Aquaspirillum arcticum (gi: 21068882)Duganella zoogloeoides (gi: 1944498)
Sinobacter plicatus (gi: 62240118)
Herbaspirillum putei (gi: 39652274)Herbaspirillum huttiense (gi: 4165372)
Collimonas fungivorans (gi: 33145982)Herminiimonas aquatilis (gi: 75754588)
Duganella violaceinigra (gi: 57996778)Naxibacter alkalitolerans (gi: 50657801)
Variovorax paradoxus (gi: 82504534)
Variovorax ginsengisoli (gi: 84453033)Variovorax dokdonensis (gi: 74423019)
Ottowia thiooxydans (gi: 27848582)Curvibacter gracilis (gi: 39652273)
Ramlibacter tataouinensis (gi: 16332373)Xylophilus ampelinus (gi: 3462594)Hylemonella gracilis (gi: 31441942)
Polaromonas ginsengisoli (gi: 84453030)Acidovorax konjaci (gi: 3462596)
Diaphorobacter nitroreducens (gi: 18148396)Polaromonas aquatica (gi: 68051130)
Verminephrobacter eiseniae (gi: 84620107)Brachymonas petroleovorans (gi: 33415959)0.01
(29)
(21)
(36)
(2)(5)
Oxalobacteraceae
Comamonadaceae
0.1
(109)
(57)
(2)
(5)
(a)
(b)
100
87
100
(80)
100100
Z
Y
FEMS Microbiol Ecol 65 (2008) 180–192 c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
187Bacterial diversity associated with mycorrhizal roots

Rubrivivax subgroup and Firmicutes among those of the 373
tested isolates was not in accordance with hybridization of
the AM396929-probe with reference strains belonging to
these groups.
Phylogenetic diversity of partial 16S rRNAgene--IGS sequences from root isolates
The 173 partial 16S rRNA gene–IGS sequences of the isolates
with a high level of similarity to Oxalobacteraceae (7) and
Comamonadaceae (166) plus the most closely related se-
quences identified by BLAST analysis were analysed through
phylogenetic inference using the neighbour-joining method.
Phylogenetic analyses were performed separately on the
corresponding partial 16S rRNA gene (Fig. 3a) and IGS
(Fig. 3b) sequences.
The partial 16S rRNA gene sequences analysed were
distributed into two monophyletic branches, supported by
a significant bootstrap value (100), corresponding to the
Comamonadaceae and the Oxalobacteraceae families (Fig.
3a). The Comamonadaceae branch encompassed all the
sequences (166) showing 97% similarity to M. petroleiphi-
lum (gi: 6063088), 57 from M. truncatula J5 and 109 from
TRV25 (Fig. 3a). Within this branch, partial 16S rRNA gene
sequences of the 166 isolates were distributed into the same
subbranch, supported by a significant bootstrap (87), in-
cluding Variovorax paradoxus (gi: 82504534), Variovorax
ginsengisoli (gi: 84453033) and Variovorax dokdonensis (gi:
74423019). The Oxalobacteraceae branch encompassed all
the sequences (7) showing 99% similarity to C. fungivorans
(gi: 33145982), five from J5 and two from TRV25 (Fig. 3a).
IGS sequences were distributed into two branches (Y and
Z) supported by a high bootstrap value (100) (Fig. 3b). All
the sequences (166) showing 97% similarity to M. petrolei-
philum (gi: 6063088), 57 from M. truncatula J5 and 109
from TRV25, were included in branch Y (Fig. 3b). All the
sequences (7) showing 99% similarity to C. fungivorans (gi:
33145982), five from M. truncatula J5 and two from TRV25,
were included in branch Z (Fig. 3b). IGS branches Y and Z
included sequences corresponding to partial 16S rRNA gene
sequences belonging to Comamonadaceae and Oxalobacter-
aceae, respectively (Fig. 3a).
Genetic diversity of root isolates based onBOX-PCR
The genetic diversity of the 173 root isolates was further
analysed by BOX-PCR (Fig. 4). Amplification products
yielded complex genomic fingerprints consisting of
fragments ranging in size from 506 to 4072 bp (data not
shown).
Twenty-five BOX types (indicated by letters A–Y), en-
compassing the majority (74.1%) of the isolates, were
delineated. All isolates distributed into BOX types belonged
to Comamonadaceae and to IGS Y branches (Fig. 3), whereas
the remaining isolates belonged to the Comamonadaceae
and Oxalobacteraceae branches and to the IGS Y and Z
branches (Fig. 3). The distributions of the isolates from
M. truncatula J5 and from TRV25 in the BOX types differed
significantly (Po 0.0001), this difference being significantly
explained by types (M, T, U and V) that included only
isolates from M. truncatula J5. Other types (E, G, H, O and
X) only included isolates from TRV25 (Fig. 4).
The partial 16S rRNA gene sequences of a representative
isolate per BOX-type were further subjected to a phyloge-
netic analysis (data not shown). The corresponding
results indicated that isolates belonging to BOX types C, G,
I, K, L, M, N, O, P, Q, R and T and those belonging to
BOX types A, B, D, E, F, H, J, S, U, V, W, X and Y
were grouped into the branches defined in Fig. 3 that
include V. ginsengisoli (gi: 84453033) and V. paradoxus
(gi: 82504534), respectively.
Discussion
The diversity of bacteria from mycorrhizal roots of M.
truncatula wild-type and from roots of a mutant impaired
in the ability to establish AM was compared, both plant
genotypes being cultivated in a fallow soil. This comparison
relied on two complementary strategies based on the diver-
sity analysis of DNA extracted from roots and from bacterial
isolates. The first consisted of phylogenetic analysis of
sequences of partial 16S rRNA gene–IGS, amplified and
cloned from DNA extracted from roots, belonging to OTUs
preferentially associated with mycorrhizal roots (Offre et al.,
2007). Phylogenetic analysis of partial 16S rRNA gene
sequences of AM396923, AM396926 and AM396929 types
indicated that all were affiliated to Burkholderiales
Num
ber
of is
olat
es
0A
5
10
15
20
25
BOX types
YB C D E F G H I J K L M N O P Q R S T U V W X
M. truncatula J5 (Myc / Nod )
M. truncatula TRV25 (Myc /Nod )
Fig. 4. Frequency distributions of bacterial isolates from Medicago
truncatula J5 and TRV25 in the different BOX types. The two distributions
are significantly different (w2, exact P-valueo 0.0001). This distribution
difference is explained by underlined BOX types as identified by analysis
of contingency-table cell contribution to the w2 value.
FEMS Microbiol Ecol 65 (2008) 180–192c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
188 P. Offre et al.

(Betaproteobacteria) (Fig. 1a). They were grouped into three
clusters belonging to the Comamonadaceae, Oxalobactera-
ceae and Rubrivivax subgroup, respectively (Fig. 1a). Dis-
crimination of the sequences from M. truncatula J5 and
from the mutant TRV25 showed that most of the sequences of
the Comamonadaceae (eight out of nine), Oxalobacteraceae
(85 out of 119) and Rubrivivax subgroup (32 out of 36) were
associated with the mycorrhizal roots. These data support our
previous conclusions establishing that these groups explained
variations in genetic structure of the bacterial communities
associated with mycorrhizal and nonmycorrhizal roots (Offre
et al., 2007). To our knowledge, this is the first report
describing bacteria from the Comamonadaceae, Oxalobacter-
aceae and Rubrivivax subgroup as being preferentially asso-
ciated with AM. The phylogeny of IGS from AM396923,
AM396926 and AM396929–type sequences was fully congru-
ent with that of partial 16S rRNA genes for Comamonadaceae
and Oxalobacteraceae (Fig. 1a and b), whereas IGS sequences
belonging to the Rubrivivax subgroup were clustered into
two branches, one including only three sequences. This is
consistent with the fact that IGS sequences are known to
be more variable than 16S rRNA gene sequences (Gurtler &
Stanisich, 1996).
The second strategy was based on characterization of
Burkholderiales isolates from a large collection of isolates
(2412) that gave a positive hybridization with the polynu-
cleotide AM396929-probe. We chose to make a polynucleo-
tide rather than an oligonucleotide probe to avoid
false negatives (Amann & Ludwig, 2000). The difficulty
experienced in raising AM396923-probes targeting the
Comamonadaceae and Rubrivivax subgroup may be related
to the larger size of the corresponding sequence types
(1170 bp) than that of the AM396929-probe targeting
Oxalobacteraceae (980 bp). The specificity test indicated
that, unexpectedly, the AM396929-probe not only targeted
Oxalobacteraceae but also the Comamonadaceae, Alphapro-
teobacteria and, to a lesser extent, the Rubrivivax subgroup,
Gammaproteobacteria and Firmicutes (Fig. 2). This could be
related to the low specificity of the AM396929-probe.
Results of the specificity test of the AM396929-probe are in
agreement with the assignment of the 373 strains, showing a
positive signal with the AM396929-probe, made on the basis
of their partial 16S rRNA gene sequences to the following
groups: Oxalobacteraceae (seven isolates), Comamonadaceae
(166 isolates), Alphaproteobacteria (135 isolates), Betapro-
teobacteria (32 isolates) (other than Oxalobacteraceae and
Comamonadaceae) and Gammaproteobacteria (33 isolates).
However, the absence of sequences belonging to the Rubri-
vivax subgroup and Firmicutes among those of the 373
isolates was not in accordance with positive hybridization
of a reference strain of the Rubrivivax subgroup and one of
Firmicutes. This discrepancy could be related to the fact that
we had access only to culturable bacteria and to a limited
number of isolates. Only the sequences shown to have a high
level of similarity to those of Oxalobacteraceae or Comamo-
nadaceae were subjected to the phylogenetic analysis. Phy-
logenetic analyses of the corresponding sequences
confirmed that 166 of the isolates belonged to Comamona-
daceae (Fig. 3a) and seven to Oxalobacteraceae (Fig. 3a). The
low abundance of bacteria belonging to Oxalobacteraceae
compared with the high frequency of the corresponding
sequence in DNA directly extracted from roots might be
related to the difficulty in growing the corresponding
bacteria. Indeed, among this family, C. fungivorans is known
to be chitinolytic (de Boer et al., 2004) and therefore a
growth medium including chitin would have increased the
probability of isolating such bacteria. Despite their small
number, Oxalobacteraceae isolates were more abundant in
mycorrhizal (five) than in nonmycorrhizal (two) roots,
supporting conclusions made from analysis of DNA directly
extracted from roots showing a higher frequency of
AM396929 (65 out of 119) and AM396926-type (20 out of
119) sequences in mycorrhizal than in nonmycorrhizal roots
(30 and 4 out of 119, respectively) (Fig. 1). Conversely,
bacteria belonging to Comamonadaceae were mostly iso-
lated from nonmycorrhizal (109) than from mycorrhizal
roots (57), contrasting with data obtained from DNA
directly extracted from roots, which showed a higher
frequency of AM396923-type sequences in mycorrhizal
(eight out of nine) than in nonmycorrhizal (one out of
nine) roots (Fig. 1). This discrepancy could be explained by
assessment of the 16S rRNA gene–IGS sequence-type fre-
quency, which was based on similar amounts of PCR
products for A-RISA analysis (Ranjard et al., 2003) for both
plant genotypes, whereas bacterial abundance based on
enumeration of total bacterial cells with orange acridine
(Hobbie et al., 1977) was higher in nonmycorrhizal than in
mycorrhizal roots (data not shown), in agreement with the
higher release of photosynthates in nonmycorrhizal roots
(Bago et al., 2000; Johnson et al., 2002).
The genetic diversity of Comamonadaceae isolates asso-
ciated with mycorrhizal and nonmycorrhizal roots differed
significantly as indicated by their different distribution in
the BOX types (Fig. 4). This difference was explained by
BOX types only including isolates from mycorrhizal roots,
suggesting that the corresponding populations were prefer-
entially associated with AM. Root colonization by fungi,
including AM, has been described as providing ecological
niches favourable for specific bacterial populations
(Andrade et al., 1997; Johansson et al., 2004; de Boer et al.,
2005; Toljander et al., 2007). The preferential association
between Comamonadaceae and Oxalobacteraceae and my-
corrhizal roots has already been described for ectomycorrhi-
zas of Scots pine: Suillus flavilus (Izumi et al., 2006),
Tomentella (Khetmalas et al., 2002) and Tuber borchii (Bar-
bieri et al., 2005) ectomycorrhizal fungi. In addition,
FEMS Microbiol Ecol 65 (2008) 180–192 c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
189Bacterial diversity associated with mycorrhizal roots

Collimonas (Oxalobacteraceae), previously shown to grow at
the expense of living fungi (de Boer et al., 2004, 2005), was
found to be associated with lichen symbiosis in forest soils
(Mannisto & Haggblom, 2006) and with the oak-Scleroderma
citrinum ectomycorrhizosphere (Uroz et al., 2007).
The preferential association of Comamonadaceae and Ox-
alobacteraceae with AM suggests that AM fungi are favourable
for the survival of these bacteria and reciprocally that, from an
evolution point of view, these bacteria are beneficial to
mycorrhization. This hypothesis is supported by the demon-
stration made by Frey et al. (1997) of the promoting effect on
ectomycorrhization of a bacterial isolate representative of the
bacterial community associated with the mycorrhizosphere of
Laccaria bicolor. Intimate associations between Glomerybacter
bacteria, belonging to Burkholderiales, and various AM fungi
(Gigaspora margarita, Scutellospora persica and Scutellospora
castanea) were also demonstrated, these bacteria being con-
sidered as endosymbionts of AM fungi (Bianciotto et al.,
2003). However, one may not exclude the possibility that some
bacterial populations preferentially associated with mycorrhi-
zal roots may also live at the expense of AM fungi. Among
Oxalobacteraceae, C. fungivorans has been shown to grow at
the expense of soil fungi (de Boer et al., 2004) and Janthino-
bacterium agaricidamnosus to be pathogenic of mushroom
(Lincoln et al., 1999).
The present study has advanced our knowledge of the
diversity of Burkholderiales associated with endomycorrhizal
roots by following an approach based jointly on the diversity
analysis of DNA extracted from roots and from bacterial
isolates. Analysis of root DNA confirmed the preferential
association of Burkholderiales bacteria with mycorrhizal
roots reported previously (Offre et al., 2007). We have
further specified that they belong to the Comamonadaceae
and Oxalobacteraceae families and to the Rubrivivax sub-
group. The results obtained by the culturable approach
suggest that the Oxalobacteraceae are preferentially asso-
ciated with endomycorrhizal roots. This could be further
supported by the RNA-stable isotope probing approach
applied by Vandenkoornhuyse et al. (2007), allowing the
identification of bacterial populations utilizing carbon sub-
strates released by mycorrhizal plant roots.
A discrepancy was found between the molecular and
cultivation-based approaches for Comamonadaceae and
could possibly be related to the different quantitative types
of information yielded. However, BOX-PCR analysis of
these isolates, which provided higher resolution analysis of
the diversity of bacterial isolates than that based on partial
16S rRNA gene sequences, indicated that isolates from
mycorrhizal and nonmycorrhizal roots differed. Comamo-
nadaceae isolates explaining these differences and Oxalobac-
teraceae isolates from mycorrhizal roots are currently being
tested for their possible beneficial effect on mycorrhizal
colonization of roots.
Acknowledgements
This study was supported by programs funded by INRA
Sante des Plantes et Environnement (Rhizosphere Ecology
of Annual Medics program) and the Burgundy regional
project (02511CPO2S188) and a doctoral fellowship to P.O.
B.P. was supported by a doctoral fellowship from Italian
MIUR and a Vinci project grant. The authors are grateful to
D. Pouhair and S. Leclercq for technical assistance, to G. Duc
(UMR LEG, Dijon, France) for providing seeds of M.
truncatula and to W. de Boer (NIOO-KNAW, Centre for
Terrestrial Ecology; Heteren, the Netherlands) and M.
Devers (UMR MSE, Dijon, France) for providing C. fungi-
vorans Ter331 and Variovorax sp. MD1 strains, respectively.
Authors’contribution
P.O. and B.P. contributed equally to this study.
References
Altschul SF, Gish W, Miller W, Myers EW & Lipman DJ (1990)
Basic local alignment search tool. J Mol Biol 215: 403–410.
Amann R & Ludwig W (2000) Ribosomal RNA-targeted nucleic
acid probes for studies in microbial ecology. FEMS Microbiol
Rev 24: 555–565.
Andrade G, Mihara KL, Linderman RG & Bethlenfalvay GJ
(1997) Bacteria from rhizosphere and hyphosphere soils of
different arbuscular-mycorrhizal fungi. Plant Soil 192: 71–79.
Artursson V, Finlay RD & Jansson JK (2005) Combined
bromodeoxyuridine immunocapture and terminal-restriction
fragment length polymorphism analysis highlights differences
in the active soil bacterial metagenome due to Glomus mosseae
inoculation or plant species. Environ Microbiol 7: 1952–1966.
Bago B, Pfeffer PE & Sachar-Hill Y (2000) Carbon metabolism
and transport in arbuscular mycorrhizas. Plant Physiol 124:
949–957.
Barbieri E, Bertini L, Rossi I, Ceccaroli P, Saltarelli R, Guidi C,
Zambonelli A & Stocchi V (2005) New evidence for bacterial
diversity in the ascoma of the ectomycorrhizal fungus Tuber
borchii Vittad. FEMS Microbiol Lett 247: 23–35.
Bever JD, Pringle A & Schultz PA (2002) Dynamics within the
plant-arbuscular mycorrhizal fungal mutualism: testing the
nature of community feedback. Mycorrhizal Ecology
(van der Heijden MGA & Sanders IE, eds), pp. 267–292.
Springer-Verlag, Berlin.
Bianciotto V, Lumini E, Bonfante P & Vandamme P (2003)
‘Candidatus Glomeribacter gigasporarum’ gen. nov., sp. nov.,
an endosymbiont of arbuscular mycorrhizal fungi. Int J Syst
Evol Micr 53: 121–124.
Brundrett MC (2002) Coevolution of roots and mycorrhizas of
land plants. New Phytol 154: 275–304.
Budi SW, van Tuinen D, Martinotti MG & Gianinazzi S (1999)
Isolation from the Sorghum bicolour mycorrhizosphere of a
bacterium compatible with arbuscular mycorrhiza
FEMS Microbiol Ecol 65 (2008) 180–192c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
190 P. Offre et al.

development and antagonistic towards soilborne fungal
pathogens. Appl Environ Microbiol 65: 5148–5150.
de Boer W, Leveau JHJ, Kowalchuk GA, Klein Gunnewiek PJA,
Abeln ECA, Figge MJ, Sjollema K, Janse JD & van Veen JA
(2004) Collimonas fungivorans gen. nov., sp. nov., a chitinolytic
soil bacterium with the ability to grow on living fungal hyphae.
Int J Syst Evol Microbiol 54: 857–864.
de Boer W, Folman LB, Summerbell RC & Boddy L (2005) Living
in a fungal world: impact of fungi on soil bacterial niche
development. FEMS Microbiol Rev 29: 795–811.
Devers M, Henry S, Hartmann A & Martin-Laurent F (2005)
Horizontal gene transfer of atrazine-degrading genes (atz)
from Agrobacterium tumefaciens St96-4 pADP1: Tn5 to
bacteria of maize cultivated soil. Pest Manag Sci 61: 870–880.
Eparvier A, Lemanceau P & Alabouvette C (1991) Population
dynamics of non-pathogenic Fusarium and fluorescent
Pseudomonas strains in rockwool, a substratum for soilless
culture. FEMS Microbiol Ecol 86: 177–184.
Forbes BA, Sahm DE & Weissfeld AS (1998) Bailey & Scott’s
Diagnostic Microbiology, 10th edn. Mosby Inc., St Louis.
Frey P, Frey-Klett P, Garbaye J, Berge O & Heulin T (1997)
Metabolic and genotypic fingerprinting of fluorescent
pseudomonads associated with Douglas fir Laccaria bicolor
mycorrhizosphere. Appl Environ Microbiol 63: 1852–1860.
Galibert F, Finan TM, Long SR et al. (2001) The composite
genome of the legume symbiont Sinorhizobium meliloti.
Science 293: 668–672.
Galtier N, Gouy M & Gautier C (1996) SeaView and Phylo_win,
two graphic tools for sequence alignment and molecular
phylogeny. Comput Appl Biosci 12: 543–548.
Gill SR, Fouts DE, Archer GL et al. (2005) Insights on evolution
of virulence and resistance from the complete genome analysis
of an early methicillin-resistant Staphylococcus aureus strain
and a biofilm-producing methicillin-resistant Staphylococcus
epidermidis strain. J Bacteriol 187: 2426–2438.
Goodner B, Hinkle G, Gattung S et al. (2001) Genome sequence
of the plant pathogen and biotechnology agent Agrobacterium
tumefaciens C58. Science 294: 2323–2328.
Gurtler V & Stanisich VA (1996) New approaches to typing and
identification of bacteria using the 16S–23S rDNA spacer
region. Microbiology 142: 3–16.
Hobbie JE, Daley RJ & Jasper S (1977) Use of nucleopore filters
for counting bacteria by epifluorescence microscopy. Appl
Environ Microbiol 33: 1225–1228.
Izumi H, Anderson IC, Alexander IJ, Killham K & Moore ERB
(2006) Endobacteria in some endomycorrhiza of Scots pine
(Pinus sylvestris). FEMS Microbiol Ecol 56: 34–43.
Johansson JF, Paul LR & Finlay RD (2004) Microbial interactions
in the mycorrhizosphere and their significance for sustainable
agriculture. FEMS Microbiol Ecol 48: 1–13.
Johnson D, Leake JR, Ostle N, Ineson P & Read DJ (2002) In situ
(CO2)-13C pulse labelling of upland grassland demonstrates a
rapid pathway of carbon flux from arbuscular mycorrhizal
mycelia to the soil. New Phytol 153: 327–334.
Kaneko T, Nakamura Y, Sato S et al. (2002) Complete genomic
sequence of nitrogen-fixing symbiotic bacterium
Bradyrhizobium japonicum USDA110. DNA Res 9: 189–197.
Khetmalas MB, Egger KN, Massicotte HB, Tackaberry LE &
Clapperton MJ (2002) Bacterial diversity associated with
subalpine fir (Abies lasiocarpa) ectomycorrhizae following
wildfire and salvage-logging in central British Columbia. Can J
Microbiol 48: 611–625.
Kimura M (1980) A simple model for estimating evolutionary
rates of base substitutions through comparative studies of
nucleotide sequences. J Mol Evol 16: 111–120.
King EO, Ward MK & Raney DE (1954) Two simple media for the
demonstration of pyocyanin and fluorescein. J Lab Clin Med
44: 301–307.
Klironomos JN, McCune J, Hart M & Neville J (2000) The
influence of arbuscular mycorrhizae on the relationship
between plant diversity and productivity. Ecol Lett 3: 137–141.
Lincoln SP, Fermor TR & Tindall BJ (1999) Janthinobacterium
agaricidamnosum sp. nov., a soft rot pathogen of Agaricus
bisporus. Int J Syst Evol Microbiol 49: 1577–1589.
Mannisto MK & Haggblom MM (2006) Characterization of
psychrotolerant heterotrophic bacteria from Finnish Lapland.
Syst Appl Microbiol 29: 229–243.
Marschner P, Crowley DE & Lieberei R (2001) Arbuscular
mycorrhizal infection changes the bacterial 16S rDNA
community composition in the rhizosphere of maize.
Mycorrhiza 11: 297–302.
Mazurier S, Lemunier M, Hartmann A, Siblot S & Lemanceau P
(2006) Conservation of type III secretion system genes in
Bradyrhizobium isolated from soybean. FEMS Microbiol Lett
259: 317–325.
Meyer JR & Linderman RG (1986) Selective influence on
populations of rhizosphere or rhizoplane bacteria and
actinomycetes by mycorrhizas formed by Glomus fascicolatum.
Soil Biol Biochem 18: 191–196.
Mougel C, Offre P, Ranjard L, Corberand T, Gamalero E, Robin C
& Lemanceau P (2006) Dynamic of the genetic structure of
bacterial and fungal communities at different development
stages of Medicago truncatula Gaertn. cv. Jemalong line J5. New
Phytol 170: 165–175.
Nei M & Li W (1979) Mathematical model for studying genetic
variation in terms of restriction endonucleases. Proc Natl Acad
Sci USA 76: 5269–5273.
Normand P, Ponsonnet C, Nesme X, Neyra M & Simonet P
(1996) ITS analysis of prokaryotes. Molecular Microbial
Ecology Manual (Akkermans DL, van Elsas JD &
de Bruijn EI, eds), pp. 1–12. Kluwer Academic Publishers,
Amsterdam.
Offre P, Pivato B, Siblot S, Gamalero E, Corberand T, Lemanceau
P & Mougel C (2007) Identification of bacterial groups
preferentially associated with mycorrhizal roots of Medicago
truncatula. Appl Environ Microbiol 73: 913–921.
Perriere G & Gouy M (1996) WWW-Query: an on-line retrieval
system for biological sequence banks. Biochimie 78: 364–369.
FEMS Microbiol Ecol 65 (2008) 180–192 c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
191Bacterial diversity associated with mycorrhizal roots

Pivato B, Mazurier S, Lemanceau P, Siblot S, Berta G, Mougel C &
van Tuinen D (2007) Medicago species affect the community
composition of arbuscular mycorrhizal fungi associated with
roots. New Phytol 176: 197–210.
Ranjard L, Lejon DPH, Mougel C, Schehrer L, Merdinoglu D &
Chaussod R (2003) Sampling strategy in molecular microbial
ecology: influence of soil sample size on DNA fingerprinting
analysis of fungal and bacterial communities. Environ
Microbiol 5: 1111–1120.
Remy W, Taylor TN, Hass H & Kerp H (1994) Four hundred-
million-year-old vesicular arbuscular mycorrhizae. Proc Natl
Acad Sci USA 91: 11841–11843.
Roccourt J, Grimont F, Grimont PAD & Seeliger HPR (1982)
DNA relatedness among serovars of Listeria monocytogenes
sensu lato. Curr Microbiol 7: 383–388.
Sagan M, Morandi E, Tarenghi E & Duc G (1995) Selection of
nodulation and mycorrhizal mutants in the model plant
Medicago truncatula (Gaertn.) after g-ray mutagenesis. Plant Sci
111: 63–71.
Saitou N & Nei M (1987) The neighbor-joining method: a new
method for reconstructing phylogenetic trees. Mol Biol Evol 4:
406–425.
Schußler A, Schwarzott D & Walker C (2001) A new fungal
phylum, the Glomeromycota: phylogeny and evolution. Mycol
Res 105: 1413–1421.
Simon R, Prifer U & Puhler A (1983) A broad range mobilization
system for in vivo genetic engineering: transposon mutagenesis
in Gram-negative bacteria. Biotechnology 1: 784–791.
Smith SE & Read DJ (1997) Mycorrhizal Symbiosis. Academic
Press, London.
Stall RE, Loschke DC & Jones JB (1986) Linkage of copper
resistance and avirulence loco on a self-transmissible plasmid
in Xanthomonas camprestris pv. vesicatoria. Phytopathology 76:
240–243.
Streitwolf-Engel R, Boller T, Wiemken A & Sanders IR (1997)
Clonal growth traits of two Prunella species are determined by
co-occurring arbuscular mycorrhizal fungi from a calcareous
grassland. J Ecol 85: 181–191.
Thompson JD, Higgins DG & Gibson TJ (1994) CLUSTAL W:
improving the sensitivity of progressive multiple sequence
alignment through sequence weighting, positions-specific gap
penalties and weight matrix choice. Nucleic Acids Res 22:
4673–4680.
Toljander JF, Bjorn DL, Paul LR, Elfstrand M & Finlay RD (2007)
Influence of arbuscular mycorrhizal mycelial exudates on soil
bacterial growth and community structure. FEMS Microbiol
Ecol 61: 295–304.
Uroz S, Calvaruso C, Turpauld MP, Pierrat JC, Mustin C
& Frey-Klett P (2007) Effect of the mycorrhizosphere
on the genotypic and metabolic diversity of the
bacterial communities involved in mineral
weathering in a forest soil. Appl Environ Microbiol 73:
3019–3027.
Vandenkoornhuyse P, Ridgway P, Watson IJ, Fitter AH & Young
JPW (2003) Co-existing grass species have distinctive
arbuscular mycorrhizal communities. Mol Ecol 12:
3085–3095.
Vandenkoornhuyse P, Mahe S, Ineson P, Staddon P, Ostle N,
Cliquet J-B, Francez A-J, Fitter AH & Youg JPW (2007) Active
root-inhabiting microbes identified by rapid incorporation of
plant-derived carbon into RNA. Proc Natl Acad Sci USA 104:
16970–16975.
van der Heijden MGA, Klironomos JN, Ursic M, Moutioglis P,
Streitwolf-Engel R, Boller T, Wiemken A & Sanders IR (1998)
Mycorrhizal fungal diversity determines plant biodiversity,
ecosystem variability and productivity. Nature 396:
69–72.
Versalovic J, Schneider M, de Bruijn FJ & Lupski JR (1994)
Genomic fingerprinting of bacteria using repetitive sequence-
based polymerase chain reaction. Meth Mol Cell Biol 5:
25–40.
Wang B & Qiu YL (2006) Phylogenetic distribution and evolution
of mycorrhizas in land plants. Mycorrhiza 16: 299–363.
FEMS Microbiol Ecol 65 (2008) 180–192c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
192 P. Offre et al.





























