Embed Size (px)
Citation preview

notions de base de thermodynamique statistique
Différentes approches (en particulier la mécanique quantique) permettent de modéliser, par exemple à partir de données spectroscopiques, la structure énergétique d'un système chimique donné, et de calculer, par exemple, les différents niveaux d'énergie accessibles (et les niveaux de dégénérescence correspondants) d' une molécule de composition donnée.L'étape suivante consiste à voir comment la connaissance de ces niveaux d'énergie peut être utilisée pour rendre compte des propriétés globales, macroscopiques, du système considéré. C'est l'objet de la thermodynamique statistique: comprendre les relations entre les propriétés microscopiques de la matière et ses propriétés globales (= macroscopiques).On introduit d'abord la distribution de Boltzmann, qui permet de prédire les populations des états. On verra ici comment elle dérive de la distribution des particules entre les états disponibles. Pour sa dérivation, on introduit la fonction de partition, concept mathématique central à ce niveau. On pourra ensuite généraliser au cas de systèmes composés d'assemblages de particules en interaction.
préalable: notion de "populations" d'états d'énergie
Une idée fondamentale pour passer de la mécanique quantique des molécules individuelles à la thermodynamique d'un échantillon réel est de reconnaître que cette dernière concerne le comportement moyen d'un grand nombre de molécules.Par exemple, la pression d'un gaz correspond à la force moyenne exercée par les molécules -> nul besoin de préciser quelle molécule frappe la paroi à un instant donné, pas besoin non plus de considérer les fluctuations de la pression (il est improbable qu'il y ait une soudaine chute ou augmentation du nombre de molécules qui frappent la paroi).Si on considère la température, du fait de l'agitation thermique continue subie par les molécules d'un échantillon à T>0, celles ci se distribuent sur l'ensemble des niveaux d'énergie disponibles. Par exemple, une molécule donnée peut être, à un instant donné, à un niveau d'énergie faible, et se trouver, l'instant d'après, excitée à un niveau d'énergie élevée. Il est matériellement impossible de garder la trace de l'évolution de l'état énergétique d'une molécule donnée, mais on peut s'intéresser au nombre moyen de molécules situées dans un état d'énergie donné: si la température reste constante, le nombre moyen de molécules dans un état d'énergie donné est constant dans le temps; ce nombre est appelé la population de l'état d'énergie considéré.A T=0, seul l'état d'énergie la plus basse est occupé. Quand on augmente la température, une partie des molécules est excitée, c'est à dire portée à des niveaux d'énergie plus élevée, et, à mesure que T augmente, de plus en plus d'états d'énergie deviennent accessibles aux molécules.Mais, quelle que soit la température, il y aura toujours une population plus élevée dans un niveau de basse énergie que dans un de haute énergie. La seule exception à cette règle apparaît quand la température tend vers l' infini: alors tous les états d'énergie ont des populations égales.La distribution de Boltzmann permet de calculer les populations relatives des différents niveaux d'énergie d'un système donné.La "formule de Boltzmann" donne le rapport entre les nombres de particules, respectivement Ni et Nj, occupant les états d'énergie Ei et Ej:
Ni / Nj = exp(-Ei / kT) / exp(-Ej /kT) //k = "constante de Boltzmann"Un point important de la distribution de Boltzmann est qu'elle concerne les populations des états d'énergie, et non des niveaux d'énergie.Comme elle concerne les états, tous les membres d'un ensemble dégénéré d'états "situés au même niveau" d'énergie E auront la même
PhyStatNotes - 1

population, exp(-E / kT). La population d'un état d'énergie E de dégénérescence g sera alors proportionnelle à g.exp(-E / kT).
distribution d'états moléculaires
On considère un système fermé, contenant N Molécules, et d'énergie totale E donné.L'énergie totale du système considéré est constante, mais il est matériellement impossible de préciser comment cette énergie est distribuée entre les molécules: les collisions entre les particule conduisent à une redistribution sans fin de l'énergie, non seulement entre les molécules, mais aussi entre les différents modes de mouvement (translation, vibration, rotation) de chaque molécule.Mais, même si les identités précises des molécules dans chaque état peuvent changer à chaque collision, on peut considérer qu'en moyenne, les populations des états restent constantes. Il est alors possible de décrire la distribution de l'énergie du système, c'est à dire les populations des différents états. On considère qu'il y a, en moyenne, dans chaque état d'énergie ε_i, un nombre spécifique, n_i, de molécules, et on appellera ce nombre "population" de l' état considéré.Dans un premier temps, on s'intéresse au cas où les molécules sont indépendantes, c'est à dire qu'il n'y a entre elles aucune interaction autre que des collisions élastiques. L'énergie totale du système est alors la somme des énergies individuelles des particules. On pose aussi comme principe l'égalité à priori des probabilités: toutes les distributions d'énergie possibles sont également probables.
configuration et poids
Soit une molécule individuelle, qui peut occuper différents niveaux d'énergies ε_0, ε_1, ε_2, ε_3, ... On considère toujours les niveaux d'énergie dans le sens croissant, à partir de ε_0, l'état d'énergie la plus basse, que l'on pose ε_0=0.
<> configuration instantanéeA un instant t, il y a n_0 molécules à l'état ε_0, n_1 molécules à l'état ε_1, ...Alors, la configuration instantanée du système est décrite par la distribution de populations {n_0, n_1, ...}.On peut avoir par exemple la distribution {N,0,0,...}, i.e. toutes les molécules à l'état fondamental. On peut envisager aussi une distribution {N-2,2,0,...}, etc. Cette distribution {N-2,2,0,...} a, "toutes choses égales par ailleurs" [on ne prend pas en compte pour l'instant l'énergie totale, qui est différente entre ces deux cas], un plus haut degré de possibilité que {N,0,0,...}, puisque {N-2,2,0,...} peut être obtenue de plus de façons que {N,0,0,...} En effet, il n'y a qu'une façon d'avoir {N,0,0,...}mais il y a ½.N.(N-1) façons d'avoir {N-2,2,0,...}: on a N choix possibles pour promouvoir une molécule de l'état ε_0 à ε_1, restent alors N-1 choix pour promouvoir une autre molécule parmi les N-1 restantes. Mais on doit diviser ce résultat par 2, puisqu'on ne distingue pas ab de ba (a et b désignant deux molécules) ...Aussi, si, à la suite de collisions entre molécules, le système fluctue entre les distributions d'énergie {N,0,0,...} et {N-2,2,0,...}, on le trouvera dans la majorité des cas dans la seconde configuration, puisque c'est la plus probable. Alors, du point de vue macroscopique, un système libre d' "osciller" entre ces deux configurations aura les propriétés caractéristiques de la seconde configuration.Une configuration générale { n0, n1 ,... } peut être obtenue de N manières différentes , on appelle W le poids de cette configuration.
W = N! / (n0! n1!...)
PhyStatNotes - 2

démonstration - calcul du nombre de manière de distribuer N boules dans différentes cases. La première boule peut être choisie de N différentes manières, la deuxième de N-1 manières parmi les N-1 balles restantes-> N.(N-1).(N-2) ... = N! façons de sélectionner les balles. Mais, s'il y a n0 balles dans la boîte ε0, il y a n0! façons par lesquelles les mêmes balles peuvent avoir été choisies.De même, n1! façons de choisir les n1 balles de la boite ε1, etc.D'où la relation W = N! / (n0! n1!...)
Exempledistribuer 20 objets identiques suivant un arrangement {1,0,3,5,10,1}: W=20! / (1! 0! 3! 5! 10! 1!) = 9,31.108.
Passant aux logarithmes, et utilisant la formule de Stirling, ln(N!) = N.ln(N) - N,
ln(W) = ln(N!) - Σ(ln(n_i!)) = N.lnN - N - Σ(n_i.ln(n_i) - n_i) ->
ln(W) == N.lnN - Σ(n_i.ln(n_i) <> configuration dominante
On a vu que {N-2,2,0} domine {N,0,0}Plus généralement, on démontre qu'il y a une distribution qui a un poids tel qu'elle domine tous les autres, de sorte que le système est "pratiquement toujours" dans cette configuration. Les propriétés macroscopiques du système seront alors celles de cette configuration dominante.Pour trouver cette configuration, on cherche les valeurs de ni qui donne un W maximum. Il s'agit donc de faire varier n_i pour avoir dW=0, et cela sous deux conditions:
Condition 1: le système est fermé: Σ(ni) = NCondition 2: l'énergie du système est donnée: les configurations autorisées sont celles correspondant à une valeur définie de l'énergie totale du système. Cette condition peut s'écrire: Σ(n_i.ε_i) = E-> cette deuxième condition exclut de nombreuses configurations. Par exemple, N,0,0 et N-2,2,0 ont des énergies différentes; elles ne peuvent donc apparaître dans le même système.
<> distribution de Boltzmann La distribution qui maximise W est la "distribution de Boltzmann", décrite par la relation:
n iN=
exp − .i
∑j
exp− . j ; avec = 1
kT
démonstrationIl s'agit de résoudre le système suivant:
PhyStatNotes - 3

d lnW=0 <1>; d ni=0 <2>; i . d ni=0 <3>; avec d lnW=∑i
∂ lnW∂ni
d ni
C'est un problème d'optimisation sous contraintes. La méthode classique de résolution fait appel à la méthode des multiplicateurs de Lagrange: les contraintes <2> et <3>, multipliées par des coefficients ("multiplicateurs de Lagrange"), sont ajoutées à l'équation de variation principale <1>. Les variables sont ensuite traitées comme indépendantes.(1) est remplacé par:
Σ( ∂Log(W)/∂n[i] ).dn[i] + α.Σ dn[i] - β.Σ ε[i].dn[i] = 0 //2ou dln(W) = Σ( ∂Log(W)/∂n[i] + α - β.ε[i] ).dn[i] = 0On traite maintenant les dn[i] comme indépendants; le seul moyen d'avoir dln(W) = 0 est que
∂Log(W)/∂n[i] + α - β.ε[i] = 0 // pour tout iOn a
∂Log(W)/∂n[i] = ∂( N.Log(N) )/∂n[i] - Σj{ ∂(n[j].Log(n[j])) /∂n[i] } dérivation des différents termes:<> ∂( N.Log(N) )/∂n[i] = {∂N/∂n[i]}. Log(N) + N .(1/N) .{∂N/∂n[i]} = Log(N) + 1 //3a<> ∂(n[j].Log(n[j])) /∂n[i] //3b
= (∂n[j]/∂n[i]) . Log(n[j]) + n[j] . (∂Log(n[j])/∂n[i])= (∂n[j]/∂n[i]) . (Log(n[j]) + n[j] / n[j]) //cf 3c= (∂n[j]/∂n[i]) . (Log(n[j]) + 1)
<> ∂(Log(n[j]) /∂n[i] = (1/n[j]) . ∂n[j] /∂n[i] = (1/n[j]) pour j=i; 0 pour j<>i //3c-> ∂Log(W)/∂n[i] //4
= Log(N) + 1 - Σj{ (∂n[j]/∂n[i]) . (Log(n[j]) + 1) }= Log(N) + 1 - (Log(n[i]) + 1)= - Log( n[i] / N)
-> en remplaçant dans∂Log(W)/∂n[i] + α - β.ε[i] = 0,
on a - Log( n[i] / N) + α - β.ε[i] = 0soit,
pour tout i, n[i] / N = exp(α - β.ε[i]) //5par ailleurs, N = Σn[i] = Σ{N.exp(α - β.ε[i])} -> N = N . Σ{exp(α - β.ε[i])} -> 1 = Σ{exp(α - β.ε[i])} = exp(α) . Σ{exp(-β.ε[i])} -> exp(α) = 1 / ( Σ{exp(-β.ε[i])} ) //6<5>+<6> permet d'aboutir à une expression de la distribution de Boltzmann: n[i] / N = exp(-β.ε[i]) / Σ{exp(-β.ε[i])} //7
PhyStatNotes - 4

On verra plus loin comment s'obtient la relation β=1/(k.T), où k est la constante de Boltzmann, équivalente à R/NA (R constante des gaz parfaits, NA nombre d'Avogadro).
fonction de partition moléculaire
Soit pi la proportion de molécules se trouvant à l'état i: pi=ni/NOn appelle q = Σ exp(- β . εi) la fonction de partition moléculaire.Si on a des états dégénérés (= distincts du point de vue quantique, mais possédant la même énergie), on peut les regrouper par niveaux d'énergie:
on a alors : q=niveaux g j . exp − . j , où g j est la dégénérescence du niveau j exemple - écrire une fonction de partition
Pour écrire la fonction de partition d'un système donné, on doit connaître sa structure énergétique, c'est à dire:les énergies des niveaux, leurs niveaux de dégénérescence, le nombre d'états dans chaque niveau.
-> par exemple, si la molécule HCl est traitée comme un rotor rigide, la mécanique quantique conduit aux résultats suivants:énergies des niveaux: hc.B.J.(J+1) , avec J= 0,1,...dégénérescence: chaque niveau contient 2J+1 états
avech constante de Planck,c vitesse de la lumière, B paramètre caractéristique (moment d'inertie) de HCl, J nombre quantique de rotation
-> la fonction de partition est alors: q=∑j=0
∞
2.J1exp −h.c.B.J J1
<> une interprétation de la fonction de partition Pour mieux comprendre la signification de la fonction de partition, examinons comment q varie en fonction de T> Quand T approche 0,
alors β=1/(kT) tend vers l'infini, mais le terme en ε0 = 0 (ou les g0 termes d'énergie ε0 si l'état fondamental est g0-dégénéré) se comporte différemment: en effet, ε0 / kT = 0, quelque soit T-> il reste un seul terme à T=0: -> à T=0, q = g0 . exp(ε0 / kT) = g0 : la fonction de partition est égale à la dégénérescence de l'état fondamental
PhyStatNotes - 5

> Quand T est très élevée, alors, εj / kT tend vers zéro pour tous les j -> q, somme du nombre des états moléculaires, tend vers l'infini.q->∞ quand T->∞
cependant, il existe des cas où le nombre d'états est fini. par exemple, les niveaux de spin d'un radical dans un champ magnétique -> seulement deux états accessiblesq->2 quand T->∞
exemple - évaluer la fonction de partition pour un système doté d'une échelle uniforme de niveaux d'énergieOn considère une molécule avec un nombre infini de niveaux d'énergie non dégénérés régulièrement espacés (0, e, 2e, 3e, ...). C'est le cas des niveaux d'énergies de vibration d'une molécule diatomique suivant l'approximation harmonique (cf. cours Mécanique Quantique).On pourrait s'attendre à ce que la fonction de partition augmente de 1, à T=0, et tende vers l'infini quand T->∞ .On a:
q = Σ exp(- β . εi) = 1 + exp(-β.e) + exp(-2.β.e) + exp(-3.β.e) + ...on démontre que que : 1 + x + x² + ... = 1 /(1-x)en effet, si S = 1 + x + x² +..., alors x.S = x + x² +... = S-1, on en déduit S=1/(1-x)
Appliqué à q : q=1 e−ee−2. e...=1 e−ee− e2 ...= 1 1 −e−e
Si pi = ni / N désigne la proportion de molécules à l'état i, p i=e−e.i
q=1 −e−e .e−. e.i
> quand T est très bas, q est proche de 1: seul l'état de plus basse énergie est significativement peuplé.> quand T augmente, les états de plus haute énergie deviennent progressivement plus peuplés.
Valeurs de q pour différentes valeurs de β.e :β.e = 3 1 0,7 0,3q = 1,05 1,58 1,99 3,86
q s'écarte de 1 à mesure que T augmente; sa valeur donne une indication de l'amplitude du domaine d'états peuplés: plus T augmente, plus on a de chances de trouver des molécules peuplant des niveaux d'énergie élevés.
PhyStatNotes - 6
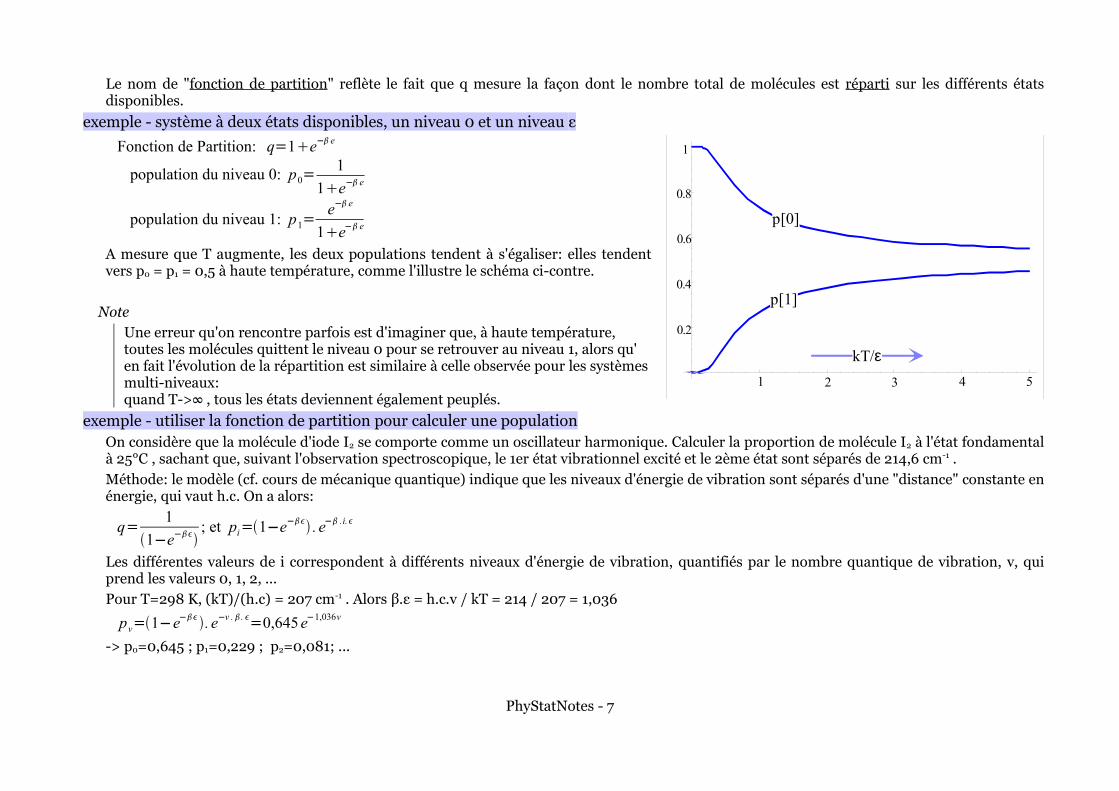
Le nom de "fonction de partition" reflète le fait que q mesure la façon dont le nombre total de molécules est réparti sur les différents états disponibles.
exemple - système à deux états disponibles, un niveau 0 et un niveau ε
Fonction de Partition: q=1e− e
population du niveau 0: p0=1
1e− e
population du niveau 1: p1=e− e
1e− e
A mesure que T augmente, les deux populations tendent à s'égaliser: elles tendent vers p0 = p1 = 0,5 à haute température, comme l'illustre le schéma ci-contre.
NoteUne erreur qu'on rencontre parfois est d'imaginer que, à haute température, toutes les molécules quittent le niveau 0 pour se retrouver au niveau 1, alors qu' en fait l'évolution de la répartition est similaire à celle observée pour les systèmes multi-niveaux: quand T->∞ , tous les états deviennent également peuplés.
exemple - utiliser la fonction de partition pour calculer une populationOn considère que la molécule d'iode I2 se comporte comme un oscillateur harmonique. Calculer la proportion de molécule I2 à l'état fondamental à 25°C , sachant que, suivant l'observation spectroscopique, le 1er état vibrationnel excité et le 2ème état sont séparés de 214,6 cm-1 .Méthode: le modèle (cf. cours de mécanique quantique) indique que les niveaux d'énergie de vibration sont séparés d'une "distance" constante en énergie, qui vaut h.c. On a alors:
q= 11−e−
; et pi=1−e− . e− .i.
Les différentes valeurs de i correspondent à différents niveaux d'énergie de vibration, quantifiés par le nombre quantique de vibration, v, qui prend les valeurs 0, 1, 2, ... Pour T=298 K, (kT)/(h.c) = 207 cm-1 . Alors β.ε = h.c.v / kT = 214 / 207 = 1,036
pv=1−e− . e−v . . =0,645e−1,036v
-> p0=0,645 ; p1=0,229 ; p2=0,081; ...
PhyStatNotes - 7
1 2 3 4 5
0.2
0.4
0.6
0.8
1
p[0]
p[1]
kT/ε

<> fonction de partition moléculaire: approximationsDans de nombreux cas, l'expression analytique exacte de la fonction de partition ne peut pas être calculée. On recourt alors à des approximations.
exemple: particule de masse m libre de se déplacer dans une enceinte de dimension 1 et de longueur XLe seul mode énergétique considéré ici est l'énergie de translation.Le système, à température ambiante, est caractérisé par une très faible séparation entre les niveaux d'énergie et un grand nombre de niveaux accessibles.
Alors, la fonction de partition peut être représentée par q X=2mh2
1/2
. X
Démonstration
<> les niveaux d'énergie d'une particule dans une "boîte 1D" de largeur X sont donnés par En=n2h2
8m X 2 , avec n=1, 2, ...
-> relativement au niveau le plus bas (n=1), posant = h2
8m X 2 , on a εn = (n² - 1) . ε
-> on a alors q X=∑1
∞
e−n2−1
Les niveaux de translation étant très proches les uns des autres, la somme discrète peut être approximée par une intégrale, et on pourra étendre l'intégrale de n=1 à n= 0 et remplacer n²-1 par n². On a alors:
q X ≈∫1
∞
e−n2−1dn ≈∫
0
∞
e−n2 dn = 1
1/2
∫0
∞
e−x2
dx =2mh2
1/2
. X
x2=n2 dn=dx×−1/2
<> factorisationsUn autre aspect des fonctions de partition est de pouvoir être exprimées de manière simple quand l'énergie d'une molécule provient de plusieurs sources indépendantes les unes des autres. En effet, si l'énergie est la somme de contributions indépendantes, la fonction de partition est le produit des fonctions de partition correspondant à ces contributions (ou modes de mouvement).
exemple: "boîte 3D"On considère une molécule libre de se déplacer dans les trois directions, à l'intérieur d'une enceinte de dimensions X-Y-Z.
PhyStatNotes - 8

L'énergie totale, ε, de la molécule est la somme des énergies de translation dans les 3 directions:
n1n2n3=n1
X n2
Y n3
Z
où n1 , n2 , n3 sont les nombres quantiques pour les mouvements dans les directions x, y, z. q=∑tous n
[exp −n1
X −n2
Y −n3
Z ]
q=∑tous n[exp −n1
X ×exp −n2
Y×exp−n3
Z]
q=∑n1exp−n1
X ×∑n2exp−n2
Y ×∑n3exp −n3
Z
q=qX×qY×qZ=2mh2
3/2
. X .Y .Z=2mh2
3/2
.V
ou
q= V3
avec =h
2m
1/2
=h 2mk T −1 /2 = longueur d'onde thermique de la molécule
exemplefonction de partition de translation d'une molécule de H2 dans une enceinte de 100 cm² à 25°C:Λ = 7,12 . 10-11 m = 71,2 pm -> q = 2,77 . 1026 -> 1026 états quantiques thermiquement accessibles
énergie interne et entropie
La fonction de partition moléculaire contient toute l'information nécessaire pour calculer les propriétés thermodynamiques d'un système de particules indépendantes.
-> q joue un rôle, en thermodynamique statistique, similaire au rôle joué par la fonction d'onde en mécanique quantique.-> q est en quelque sorte une "fonction d'onde thermique".
énergie interne
<> relation entre U et q
énergie totale E: E=∑in i .i
Du fait de la domination très forte de la configuration la plus probable, on peut utiliser la distribution de Boltzmann des populations:
PhyStatNotes - 9

sachant que ni=N× pi=Nqe−i ,
on a E= Nq∑i[i e
−i ]
> pour éliminer les ε de cette expression, on observe que dd
e− i=−i e− i
-> E=−N
q∑i[dd
e−i ]=− Nqdd ∑i [e− i]=
−Nqd qd
exemple: fonction de partition pour une molécule "à 2 niveaux"
q=1+exp(-β.ε) -> pour N molécules, l'énergie est E= −N
1e−dd
1e−=N e−
1e− =N
1eE->0 quand T->0 ; E-> ½ N.ε quand T->∞
Comme ε0=0 (on mesure toutes les énergies à partir du niveau accessible le plus bas), E est la valeur de l'énergie par rapport à sa valeur à T=0, U(0). -> l'énergie conventionnelle est U=U(0)+ECependant, considérant que la fonction de partition peut dépendre de variables autres que la température (par exemple le volume), la dérivée d/dβ est en fait une dérivée partielle.
-> l'énergie totale d'un système de N molécules indépendantes s'écrit U = U(0) - (N/q) ( ∂q/∂β )V (ou encore U = U(0) - N ( ∂lnq/∂β )V ) .
-> il suffit de connaître la fonction de partition pour calculer U, par rapport à sa valeur à T=0.
<> où l'on montre que β=1/(kT)On compare l'expression de l'équipartition de l'énergie interne d'un gaz parfait monoatomique,
U = U(0) + 3/2 n R T , où n est le nombre de moles,avec la valeur calculée à partir de la fonction de partition des énergies de translation (ci-après),
U = U(0) + 3/2 N/β , où N est le nombre de molécules.On en déduit que nRT = N/β,
soit, sachant que N=n.NA et R=k.NA (NA: nombre d'Avogadro), β=1/(kT)
PhyStatNotes - 10

démonstration de U = U(0) + 3/2 N/βon a vu sur l'exemple de la "boîte 3D, que q=V/Λ3.
->
∂q∂
V= ∂∂
V3 =V
dd
13=−3 V
4
d d
d d
= dd
[h1 /2
2m1 /2]= 1
21 /2h
2m1/2=
2
∂q∂
V=−3 V
4
2
= −3V23 ; d'où U=U 0−N 3
V−3V2 3 =U 03N
2
entropie statistique
Si la fonction de partition contient toute l'information thermodynamique, elle permet aussi de calculer l'entropie.On sait que d'une part l'entropie est liée à la dispersion de l'énergie, et que, d'autre part, la fonction de partition mesure le nombre d'état thermiquement accessibles. Les deux notions sont donc nécessairement liées.On procède ici en deux étapes:
d'abord démontrer la plus célèbre des relations de la thermodynamique statistique, la formule de Boltzmann,S=k.lnW (où W est le poids de la configuration la plus probable du système)
ensuite, on exprimera W en termes de fonction de partition.
formule de BoltzmannUne variation d'énergie U, où U=U(0) + Σniεi, peut provenir
soit d'une modification des niveaux d'énergie du système: εi -> εi + dεi
soit d'une modification des populations des états: ni -> ni + dni
Autrement dit, dU=dU(0) + Σεi.dni + Σni .dεi <> Quand un système est chauffé à volume constant, les hauteurs des niveaux d'énergie ne varient pas. On a alors, en absence de tout autre changement du système, dU=Σεi.dni . Dans les mêmes conditions, au niveau macroscopique, dU = dqrev = T.dS .dU = T.dS -> dS = dU/T = k β Σεi.dni .Pour des changements de la configuration la plus probable, la seule que l'on a besoin de considérer, on peut écrire:
PhyStatNotes - 11

∂ lnW∂ ni
−i=0
i=∂ lnW∂ ni
dS=k∑ ∂ lnW∂ni
d nik ∑ d ni
et, sachant que, le nombre de molécules étant fixé, Σdni=0,
dS=k∑ ∂ lnW∂ ni
d ni=k d lnW
On obtient ainsi la relation de Boltzmann: S = k lnWQuand on chauffe un système, les valeurs des niveaux des énergies ne varient pas, mais leur peuplement varie (figure ci-contre). Quand on effectue un travail sur le système, les niveaux d'énergie eux-mêmes sont modifiés: on peut se reporter à l'exemple de "la particule dans la boîte" (cf. mécanique quantique): les niveaux dépendent de la taille de l'enceinte, ils s'écartent l'un de l'autre quand la dimension de l'enceinte augmente.L'entropie statistique se comporte de la même façon que l'entropie thermodynamique:
Quand T diminue, la valeur de W (donc de S) diminue: en effet, le nombre de configurations compatibles avec l'énergie totale diminue.A la limite, quand T->0, W->1, et lnW->0: une seule configuration, avec toutes les molécules au niveau d'énergie minimale, est compatible avec E=0.On obtient ainsi une realtion cohérente avec le "troisième principe": S tend vers 0 quand T tend vers 0.
relation entre formule de Boltzmann et fonction de partitionOn met ensuite en relation la formule de Boltzmann avec la fonction de partition.Pour cela, on substitue la relation ln(W) = N.ln(N) - Σni .ln(ni) dans S = k.ln(W), ce qui donne:
S = (U - U(0))/T + N.k.ln(q)démonstration
S=k∑ [ni . ln N −ni . ln ni] =−k∑ [n i . ln niN] =−N k∑ [ pi . ln pi] , avec pi=
niN
pi=e− i
q ln pi=−i−ln q S=−N k [−∑ pii−∑ pi ln q] =k [U−U 0]N k ln q
PhyStatNotes - 12
CHALEUR
TRAVAIL

exemple: calculer l'entropie d'une collection d'oscillateursméthodeon utilise la fonction de partition pour une molécule de type oscillateur harmonique, c'est à dire avec des niveaux énergie de vibration également espacés. on a vu que, dans ce cas, q = 1 /[1 - exp(-βε)]-> l'énergie s'obtient par différenciation: U - U(0) = - (N/q) ( ∂q/∂β )V = N.ε.exp(-βε) /[1 - exp(-βε)] =N.ε/[exp(-βε) - 1]-> S = N.k [ β.ε/[exp(-βε) - 1] - ln[1 - exp(-βε)]]
fonction de partition canonique
Nous allons voir comment généraliser ces résultats à des systèmes comportant des molécules en interaction.
l'ensemble canonique
Pour traiter des systèmes avec particules en interaction, on a besoin d'un nouveau concept essentiel: l'ensemble . Ensemble signifie ici, comme d'habitude, "collection", mais avec dans une acception particulière.
la notion d' "ensemble"Pour construire un ensemble, on prend un système fermé, de volume,composition, et température définis, et on l'imagine répliqué Ň fois.Tous ces Ň systèmes fermés, identiques, sont considérés comme en contact thermique l'un avec l'autre: ils peuvent échanger de l'énergie entre eux. L'énergie totale de l'ensemble de ces systèmes est Ě, et, étant mutuellement en équilibre thermique, ils sont tous à la même température T.Cette collection imaginaire de répliques du système est appelé Ensemble Canonique. La collection étant imaginaire, on est libre de choisir un Ň aussi grand que possible, et on pourra éventuellement faire tendre Ň vers l'infini.Le nombre d'éléments de l'ensemble qui sont à l'énergie Ei est noté ňi, et on peut parler de la configuration de l'ensemble (notion analogue à la configuration du système) et de son poids statistique Ŵ.
configuration dominanteDe même que pour les systèmes envisagés plus haut, certaines configurations de l'ensemble canonique seront beaucoup plus probables que d'autres. Par exemple, il est hautement improbable que l'énergie totale Ě s'accumule dans un seul système.Par analogie avec la discussion précédente, on peut s'attendre à ce qu'il y ait une configuration dominante; on pourra alors évaluer les propriétés thermodynamiques du système en utilisant cette seule configuration la plus probable et en prenant les moyennes sur l'ensemble.A la limite thermodynamique Ň->∞, cette configuration dominante est de loin la plus probable et elle explique de manière dominante les propriétés du système.Le poids statistique d'une configuration {ňi} est Ŵ= Ň! / Π ňi! La configuration de poids statistique maximum, sous contraintes < énergie totale = Ě > et < nombre total de membres = Ň >, est donnée par la
PhyStatNotes - 13

distribution canonique ("canon": qui suit une règle):ňi / Ň = (1/Q) . exp(-β.Ei), avec Q=Σ exp(-β.Ei), appelée Fonction de partition canonique.
Outre le système de type canonique (N, V, T donnés), on définit:Microcanonique: la condition que T est uniforme est remplacée par "tous les systèmes ont la même énergie", autrement dit le système est isoléGrand Canonique: V et T sont les mêmes pour tous les systèmes, mais ils sont ouverts, la matière peut passer de l'un à l'autre; les compositions peuvent varier mais les propriétés chimiques sont uniformes.
Autrement dit:Canonique N ,V ,TMicrocanonique N,V ,EGrand canonique µi, V,T
Nota Bene - Ň ne doit pas être confondu avec N: N est le nombre de molécules du système, Ň est le nombre de répliques imaginaires.
fluctuations à partir de la distribution la plus probable.. /..
l'information thermodynamique contenue dans la fonction de partition
Comme la fonction de partition moléculaire, la fonction de partition porte toute l'information thermodynamique d'un système. Mais Q est plus générale que q, puisqu'elle ne suppose pas que les molécules sont indépendantes. On utilisera donc Q pour les propriétés des phases condensées, ainsi que pour des gaz réels où les interactions intermoléculaires sont importantes.
énergie interneénergie totale de l'ensemble = Ě, nombre de membres = Ň -> énergie moyenne d'un membre = Ě / Ň-> U = U(0) + Ě / Ň, avec Ň->∞fraction pi de membres de l'ensemble à l'état i d'énergie Ei: pi = (1/Q) . exp(-β.Ei)-> U = U(0) + Σ pi . Ei = U(0) + (1/Q) . Σ{ Ei . exp(-β.Ei) }un raisonnement analogue à celui effectué pour q conduit à
U = U(0) - (1/Q) ( ∂Q/∂β )V ,ou U = U(0) - ( ∂lnQ/∂β )V
entropieLe poids total Ŵ d'une configuration de l'ensemble est le produit des poids moyens W de chaque membre de l'ensemble:
PhyStatNotes - 14

Ŵ = W^Ň-> S = k . lnW = k . ln[ Ŵ ^(1/Ň) ] = (k/Ň) . lnŴ -> S = (U - U(0))/T + k . lnQ
molécules indépendantes
Nous allons voir maintenant comment, à partir de l'expression, plus générale, de la fonction de partition canonique, on peut, quand les molécules sont indépendantes, retrouver la fonction de partition moléculaire.Quand les molécules sont indépendantes et discernables (au sens qu'on va voir) on a Q = qN.
démonstrationl'énergie totale d'une collection de N molécules indépendantes est la somme des énergies des molécules.-> l'énergie totale de l'état i du système est Ei = εi(1) + εi(2) + ... + εi(N), où εi(1) est l'énergie de la molécule 1 quand le système est à l'état i, εi(2) pour la molécule 2 quand le système est à l'état i, ..., εi(N) pour la molécule N l'état i.-> la fonction de partition canonique s'écrit alors: Q = Σi{ exp(-β.Ei) } = Σi{ exp(-β.εi(1) -β.εi(2) + ... + -β.εi(N)) } La somme sur tous les états peut être reproduite en faisant en sorte que chaque molécule atteigne tous ses états individuels. Aussi, au lieu de sommer sur tous les états du système, on peut sommer sur tous les états individuels de la molécule 1, puis sur tous les états individuels de la molécule 2, et ainsi de suite jusqu'à N:
Q = [ Σi{ exp(-β.Ei) ](1) .[ Σi{ exp(-β.Ei) ](2) . .... .[ Σi{ exp(-β.Ei) ](N) -> Q = qN
molécules discernables et indiscernablesSi toutes les molécules sont identiques et libres de se déplacer dans tout l'espace, on ne peut les distinguer; alors la relation Q = qN n'est pas valide.Supposons que la molécule 1 est à l'état a, la molécule 2 à l'état b, la 3 à l'état c. Alors, un des membres de l'ensemble a une énergie E=εa + εb + εc, mais ce membre ne peut être distingué d'un autre où 1 est à l'état b, 2 à l'état c, et 3 à l'état a, ou autres permutations entre a,b,c. On dénombre au total 6 permutations pour N=3, et N! dans le cas général.On a donc, dans le cas de molécules indiscernables, compté trop d'états en passant de la somme sur les états du système à la somme effectuée sur les états moléculaires. La discussion est, dans le détail, plus complexe, mais on peut retenir que, sauf à très basse température, le facteur de correction est 1/N!En conséquence,
pour des molécules indépendantes discernables, Q = qN
pour des molécules indépendantes indiscernables, Q = qN/N!Pour que des molécules soient indiscernables, il faut qu'elles soient de même nature (exemple d'un gaz pur), mais ce n'est pas une condition
PhyStatNotes - 15

suffisante. Par exemple, des molécules identiques situées aux noeuds d'un réseau cristallin peuvent être distinguées par leurs coordonnées; on utilisera alors la relation Q=qN pour tous les modes énergétiques considérés comme indépendants de ceux des molécules voisines.
Pour toute collection de molécules dont chacune est dans sa propre "boîte", Q = qN s'appliquera.Par contre, pour des molécules identiques dans un gaz, libres de se déplacer en différents endroits, on ne peut garder la trace de l'identité de chaque molécule. On utilisera alors Q=qN/N!
entropie d'un gaz monoatomiqueéquation de Sackur-Tetrode pour l'entropie de n moles d'un gaz monoatomique, occupant un volume V:
S=n . R . ln e52 .V
n . N A .3 , avec 2= h2
2mk.T 2
pour un gaz parfait, cette relation devient:
S=n . R . ln e52 . k.TP .3
démonstrationgaz de molécules indépendantes -> Q = qN/N! -> S = (U- U(0))/T + N.k.ln(q) - k.ln(N!)le nombre de molécules considéré étant élevé, la formule de Stirling s'applique: S = (U- U(0))/T + N.k.ln(q) - k.( N.ln(N) - N)le seul mode des molécules considéré étant le mouvement de translation des molécules, -> q = V / Λ3, et U-U(0) = 3.N /(2.β) = (3.N)/(2.N) n.R.T = (3/2) n.R.T
>on a alors S = (3/2) n.R + n.R [ ln(V/Λ3) - ln(n.NA) +1 ]= n.R [ ln(e^(3/2)) + ln(V/Λ3) - ln(n.NA) + ln(e) ] -> S = n.R. ln[(e^(5/2) . V) / (n.NA.Λ3 ) ]
Implication: l'expansion d'un gaz monoatomique, d'un volume Vi à un volume Vf, s'accompagne d'une variation d'entropie ΔS = n.R.ln(Vf/Vi) qui est positive (car Vf>Vi); cette augmentation d'entropie apparaît comme une conséquence de l'augmentation, quand le volume de l'enceinte augmente, du nombre d'états translationnels accessibles.En effet, l'expansion du volume disponible induit un "rapprochement" (en 1/L²) des niveaux d'énergie; un plus grand nombre de niveaux deviennent accessible, à une température donnée.
PhyStatNotes - 16
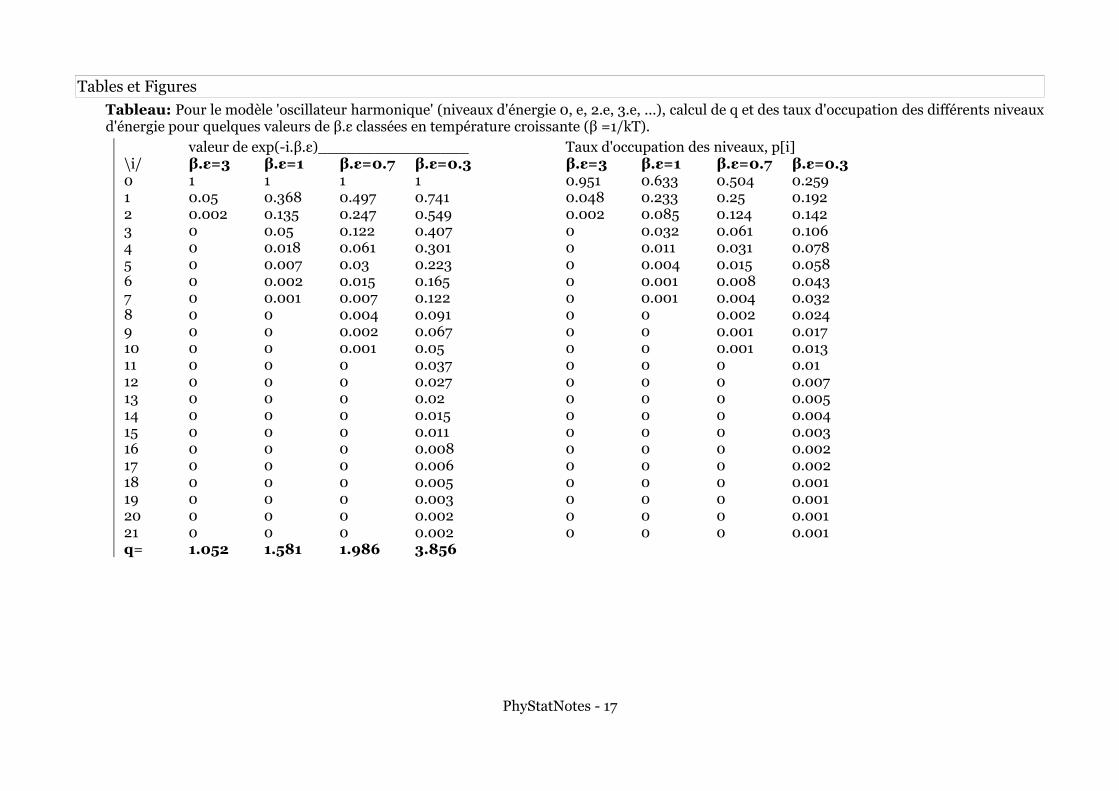
Tables et Figures
Tableau: Pour le modèle 'oscillateur harmonique' (niveaux d'énergie 0, e, 2.e, 3.e, ...), calcul de q et des taux d'occupation des différents niveaux d'énergie pour quelques valeurs de β.ε classées en température croissante (β =1/kT).
valeur de exp(-i.β.ε)________________ Taux d'occupation des niveaux, p[i]\i/ β.ε=3 β.ε=1 β.ε=0.7 β.ε=0.3 β.ε=3 β.ε=1 β.ε=0.7 β.ε=0.30 1 1 1 1 0.951 0.633 0.504 0.2591 0.05 0.368 0.497 0.741 0.048 0.233 0.25 0.1922 0.002 0.135 0.247 0.549 0.002 0.085 0.124 0.1423 0 0.05 0.122 0.407 0 0.032 0.061 0.1064 0 0.018 0.061 0.301 0 0.011 0.031 0.0785 0 0.007 0.03 0.223 0 0.004 0.015 0.0586 0 0.002 0.015 0.165 0 0.001 0.008 0.0437 0 0.001 0.007 0.122 0 0.001 0.004 0.0328 0 0 0.004 0.091 0 0 0.002 0.0249 0 0 0.002 0.067 0 0 0.001 0.01710 0 0 0.001 0.05 0 0 0.001 0.01311 0 0 0 0.037 0 0 0 0.0112 0 0 0 0.027 0 0 0 0.00713 0 0 0 0.02 0 0 0 0.00514 0 0 0 0.015 0 0 0 0.00415 0 0 0 0.011 0 0 0 0.00316 0 0 0 0.008 0 0 0 0.00217 0 0 0 0.006 0 0 0 0.00218 0 0 0 0.005 0 0 0 0.00119 0 0 0 0.003 0 0 0 0.00120 0 0 0 0.002 0 0 0 0.00121 0 0 0 0.002 0 0 0 0.001q= 1.052 1.581 1.986 3.856
PhyStatNotes - 17
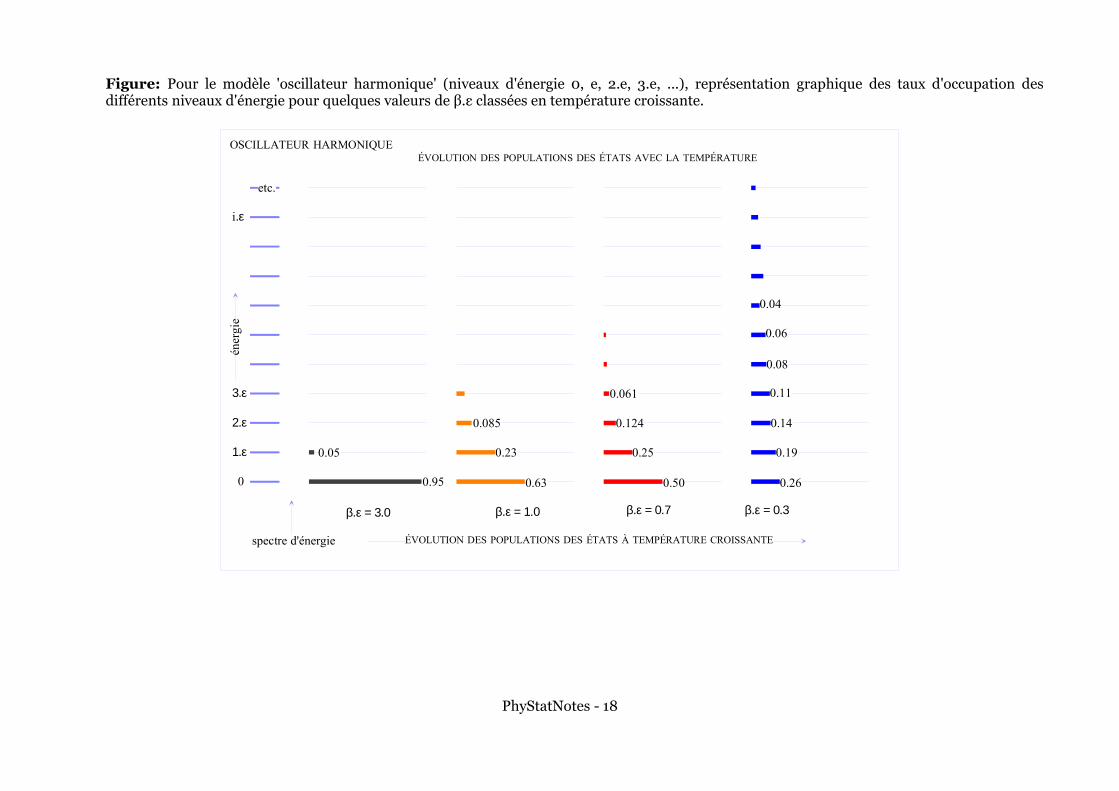
Figure: Pour le modèle 'oscillateur harmonique' (niveaux d'énergie 0, e, 2.e, 3.e, ...), représentation graphique des taux d'occupation des différents niveaux d'énergie pour quelques valeurs de β.ε classées en température croissante.
PhyStatNotes - 18
spectre d'énergie
OSCILLATEUR HARMONIQUE
0
éner
gie
2.ε
3.ε
i.ε
1.ε
β.ε = 3.0 β.ε = 1.0 β.ε = 0.7 β.ε = 0.3
ÉVOLUTION DES POPULATIONS DES ÉTATS AVEC LA TEMPÉRATURE
etc.
ÉVOLUTION DES POPULATIONS DES ÉTATS À TEMPÉRATURE CROISSANTE
0.95
0.05
0.63
0.23
0.085
0.50
0.25
0.124
0.061
0.26
0.19
0.14
0.11
0.08
0.06
0.04

relations générales permutations <> statistiques
On a vu sur quelques exemples détaillés le développement de modèles de systèmes moléculaires suivant une statistique particulière, dite de Boltzmann, ou de Maxwell-Boltzmann-Planck (appelée aussi "distribution canonique"). On généralise ici ce type d'approche à des systèmes de particules obéissant à d'autres règles de distribution.
système considéré dans les trois statistiquesOn considère un système constitué de N objets distincts (particules, molécules) distribués sur un nombre p de "cases" qui correspondent aux différents niveaux d'énergie accessibles par les objets.
n[i] = nombre d'objets sur le niveau i, d'énergie ε[i]On se donne
le nombre total d'objets: N = ∑1..p (n[i]),l'énergie totale du système: U = ∑1..p (n[i].ε[i])
Dans les trois statistiques décrites ci-après, la démarche est la même: <> calculer le poids d'une distribution {n[i]}, c'est à dire le nombre W de configurations possibles d'une distribution {n[i]} des N objets suivant un nombre p de niveaux; <> maximiser W en tenant compte des contraintes sur N et U: pour cela on applique la méthode des multiplicateurs de Lagrange.
rappel: permutations
nombre d'arrangements de n objets distincts
W = n! //2.1<démonstration>
choisir le premier objet: n choixrestent n-1 objets: n-1 choix,restent n_2 objets: n-2... etc. ...-> W = n . (n-1) . (n-2) . ... . 2 . 1 = n!
nombre de manières de distribuer N objets distincts dans p cases distinctesqui contiennent respectivement n[1], n[2], ..., n[p] emplacements
W = N! / (n[1]! . n[2]! ... . n[p]!)(avec n[1]+ n[2] + ... + n[p]= N)W = N! /∏i=1..p(n[i]!) //2.2
PhyStatNotes - 19

exemple:soient N=9 éléments distincts, représentés par les chiffres 1, 2, ..., 9, à répartir en p=3 groupes, respectivement de dimensions n[1]=3, n[2]=2 et n[3]=4
On a alors, au total, N!=362880 arrangements possibles de ces 9 éléments, par exemple |123||45||6789|,mais l'arrangement |321||45||6789| n'est pas différent de l'arrangement |123||45||6789|, puisque les deux arrangements ne diffèrent que par l'ordre des éléments à l'intérieur des groupes, et non par la composition des groupes. Pour chaque groupe i, de dimension n[i], le nombre de permutations de n[i] éléments est n[i]! ->
il n'y a pas 362880 configurations différentes possibles, mais, tenant compte des permutations à l'intérieur de chaque groupe, 362880/(3!2!4!) = 1260 configurations indépendantes.
nombre de manières de distribuer N objets discernables dans p cases distinctes , sans restriction sur les dimensions des casesOn peut mettre "n'importe quel" objet dans "n'importe quelle" case: on a p choix possibles pour le premier objet, on a encore p choix possibles pour le deuxième, ..., et encore p choix possibles pour le Nème. Au total, on a donc N^p choix possibles, quelles que soient les valeurs de N et p.
W = N^p //2.3nombre de manières de distribuer N objets indiscernables dans p cases distinctes , sans restriction sur les dimensions des cases
On suppose maintenant que les objets ne sont pas "discernables" (différentiables). On peut représenter les différents arrangements sous la forme d'une séquence de N+p-1 de longueur, AAAXAAXAAAA, où A représente un objet et X une limite entre deux cases. Ici, les dimensions des séquences AAA... sont libres (pas de restriction sur les dimensions des cases), le nombre de 'X' est fixé, égal à p-1. Il s'agit alors de compter le nombre de façons de choisir N objets parmi un total de N+p-1:
W = (N+p-1)! / [ N! (N+p-1 – N)!]
W = (N+p-1)! / [ N! (p-1)!]
statistique de Maxwell-Boltzmann-(Planck)
C'est la statistique classique développée plus haut pour les modèles moléculaires.N objets discernables, distribués sur un nombre p de "cases" qui correspondent aux différents niveaux d'énergie accessibles par les éléments
soit n[i] le nombre d'objets sur le niveau i d'énergie ε[i]; n[i] peut varier de 0 à Nnombre de manières de distribuer les N molécules sur les différents niveaux,
PhyStatNotes - 20

n[1]au niveau 1, n[2] au niveau 2, ... (cf. relation 2.2 ci-dessus)W = N! / (∏n[i]!)
Mais il faut tenir compte de la dégénérescence des niveaux d'énergie, {g[i]}: sur le niveau i, les n[i] molécules se distribuent suivant g[i] états différents. Dans la statistique Maxwell-Boltzmann-Planck, chaque état d'un niveau i donné peut comporter un nombre quelconque de molécules -> au niveau i on aura g[i]^n[i] configurations possibles (cf. relation 2.3 ci-dessus), ce qui multiplie d'autant le nombre de configurations.-> Nombre total de configurations possibles =
W = N! . (∏i=1..p g[i]^n[i]) / (∏i=1..pn[i]!)Si N est suffisamment grand, l'approximation de Stirling donneLogW = N.logN + ∑[n[i].Log(g[i]] - ∑[n[i].Log(n[i])] - N + ∑(n[i]) = N.logN + ∑( n[i] . Log(g[i]/n[i]) )On cherche ensuite le maximum de logW, sachant que N et (g[i]) sont constants:δLogW = δ[ ∑( n[i] . Log(g[i]/n[i]) ) ] = ∑ [Log(g[i]/n[i])]δn[i] + ∑( n[i] . [ δlog(g[i]) - δlog(n[i]) ] )
= ∑ [Log(g[i]/n[i])]δn[i] - ∑ δn[i]<multiplicateurs de Lagrange> ---->∑ [ Log(g[i]/n[i]) + Logα – β.ε[i] ] . δn[i] = 0 -> Log(g[i]/n[i]) + Logα = β.ε[i]
n[i] = α . g[i] . exp(- β.ε[i])
statistique de Bose-Einstein
<> s'applique aux bosons = particules de spin entier<exemples de bosons> photons, méson π, Hélium He4, ...particules indiscernables, sans limite fixée au nombre de particules sur chacun des étatssur le niveau i, de dégénérescence g[i], n[i] particules sont distribuées suivant g[i] états, chaque état pouvant comporter de 0 à n[i] particulesle nombre de configurations possible pour chaque niveau est alors
W[i] = (n[i] + g[i] – 1)! / [n[i]! . (g[i] – 1)! ]et le nombre total de configurations sur l'ensemble des niveaux du système est
W = ∏i=1..pW[i] = ∏{ (n[i] + g[i] – 1)! / [n[i]! . (g[i] – 1)! ] }supposant n[i] et g[i] >>1, on a alorsLogW = ∑ [ (n[i]+g[i]).Log(n[i]+g[i]) – n[i].Log(n[i]) – g[i].Log(g[i]) ]<multiplicateurs de Lagrange> ...
PhyStatNotes - 21

∑ [ Log(1+g[i]/n[i]) + Logα – β.ε[i] ] . δn[i] = 0-> Log[α .(1+g[i]/n[i])] = β.ε[i]
n[i] = α . g[i] exp(-β.ε[i]) / [1 - α . exp(-β.ε[i])]La distribution des particules dans la statistique de Bose-Einstein diffère de la distribution canonique par le facteur 1/(1 - α . exp(-β.ε[i]))
statistique de Fermi-Dirac
<> s'applique aux fermions = particules de spin n+½<exemples de fermions> électrons, protons, neutron, Hélium He3, ...particules indiscernables, mais obéissant au principe d'exclusion "de Pauli":
un état ne peut être occupé que par une particule au plus, autrement dit, on doit avoir n[i] <= g[i]
le nombre d'arrangements possibles pour un niveau i est alorsW[i] = g[i]! / [n[i]! . (g[i] – n[i])! ] //distribuer n[i] objets dans g[i] cases = C(g[i],n[i])
et le nombre total de configurations sur l'ensemble des niveaux du système est
W = ∏W[i] = ∏{ g[i]! / [n[i]! . (g[i] – n[i])! ] }d'où LogW = ∑ [ g[i].Log(g[i]) – n[i].Log(n[i]) – (g[i] – n[i]).Log(g[i] – n[i]) ] dont la maximisation, par la même démarche que précédemment, ∑ [ Log(g[i]/n[i] - 1) + Logα – β.ε[i] ] . δn[i] = 0conduit à
n[i] = α . g[i] exp(-β.ε[i]) / [1 + α . exp(-β.ε[i])]Cette distribution ne diffère de la précédente que par le changement de signe au dénominateur. De même que précédemment, quand α . exp(-β.ε[i]) devient petit devant 1, on tend vers la distribution de Boltzmann.
récapitulation
Maxwell-Bolzmann-Planckparticules discernables, n[i]=0..N
n[i] = α . g[i] . exp(- β.ε[i])Bose-Einstein
particules indiscernables, n[i]=0..N
PhyStatNotes - 22

n[i] = α . g[i] . exp(-β.ε[i]) / [1 - α . exp(-β.ε[i])]s'applique aux bosons = particules de spin entier, exemples: photons, méson π, Hélium He4, ...
Fermi-Diracparticules indiscernables, n[i]=0..g[i]
n[i] = α . g[i] . exp(-β.ε[i]) / [1 + α . exp(-β.ε[i])]s'applique aux fermions = particules de spin n+½, exemples: électrons, protons, neutron, Hélium He3, ...
PhyStatNotes - 23





























