Embed Size (px)
Citation preview

Mise au point
Prise en charge des états de mal épileptiques de l’enfant épileptique
Treatment of status epilepticus in children with epilepsy
C. Chiron a, N. Bahi-Buisson a,*, P. Plouin b
a Service de neuropédiatrie et maladies métaboliques, hôpital Necker–Enfants-Malades, AP-HP, 149, rue de Sèvres, 75015 Paris,France et Inserm U29, Marseille, France
b Service d’explorations fonctionnelles, hôpital Necker–Enfants-Malades, AP-HP, 149, rue de Sèvres, 75015 Paris, France
Reçu le 5 décembre 2003 ; accepté le 25 mars 2004
Disponible sur internet le le 18 mai 2004
Résumé
Chez l’enfant épileptique, la prise en charge d’un état de mal (EM) dépend du type de syndrome épileptique, principalement pour éviterl’utilisation de médicaments aggravants, comme les barbituriques par voie intraveineuse (i.v.) dans l’épilepsie myoclonique sévère dunourrisson (EMSN) (syndrome de Dravet) ou les benzodiazépines i.v. dans un EM tonique du syndrome de Lennox-Gastaut. Le recours à laréanimation ne doit pas être systématique en cas d’EM convulsif (EMC) et est inutile en cas d’EM non convulsif (EMNC). Les EMCgénéralisés sont rencontrés dans les EMSN avant trois ans, les épilepsies généralisées symptomatiques et les épilepsies partielles lésionnelles.Ils requièrent en urgence du diazépam intrarectal à domicile, puis, en hospitalisation, du diazépam et du clonazépam i.v. voire de la phénytoïnei.v. en surveillant les taux plasmatiques. Les EMC partiels des épilepsies partielles lésionnelles font courir le risque de déficit focal etrequièrent le même traitement. Les EMNC généralisés sont des états d’absence ou myocloniques, souvent longtemps méconnus jusqu’audiagnostic électroencéphalographique. Fréquents dans les épilepsies myocloniques, lésionnelles ou non, ils répondent aux benzodiazépinesi.v. La fréquence des EMNC partiels est sûrement sous-estimée surtout chez le nourrisson où la vidéo-électroencéphalographie est le moyendiagnostique de choix et le traitement le même que celui des EMC partiels.© 2004 Elsevier SAS. Tous droits réservés.
Abstract
The treatment of status epilepticus (SE) in children with epilepsy depends on the epilepsy syndrome, in order to avoid worsening drugs suchas IV barbiturates in severe myoclonic epilepsy in infancy (SMEI) (Dravet’s syndrome) or IV benzodiazepam in tonic SE of Lennox-Gastautsyndrome. Intensive care procedures should not be systematical in convulsive SE (CSE) and are not indicated in non-convulsive SE (NCSE).Generalized CSE mostly involve SMEI before 3 years of age, symptomatic generalized epilepsy and partial lesional epilepsy. Treatment is anemergency and relies on IV benzodiazepines and, if necessary, IV phenytoine using plasmatic concentrations for an optimal management. Thepartial CSE of partial lesional epilepsy can result in focal deficit and need the same treatment as generalized CSE. NCSE consist in absenceand/or myoclonic SE and are often unrecognised during a long time until EEG is performed. They mostly involve myoclonic epilepsies and canbe controlled by IV benzodiazepines. The frequency of partial NCSE is underestimated, particularly in infants. Diagnosis relies on video EEGand treatment is the same as that used in partial CSE.© 2004 Elsevier SAS. Tous droits réservés.
Mots clés : État de mal épileptique ; Épilepsie ; Électroencéphalogramme
Keywords: Status epilepticus; Epilepsy; Electroencephalography
* Auteur Correspondant.Adresse e-mail : [email protected] (N. Bahi-Buisson).
Archives de pédiatrie 11 (2004) 1217–1224
www.elsevier.com/locate/arcped
© 2004 Elsevier SAS. Tous droits réservés.doi:10.1016/j.arcped.2004.03.127

Un état de mal est classiquement défini, chez l’enfantcomme chez l’adulte, comme une crise prolongée plus de30 minutes ou des crises successives sans reprise de cons-cience entre chaque crise. Cette définition est en fait l’objetde critiques car elle ne prend pas en compte la gravité decertaines crises pourtant plus courtes alors que d’autres situa-tions pourtant plus prolongées font moins courir le risque deséquelles [1]. Il est admis par tous qu’un traitement anticon-vulsivant doit être instauré devant toute crise convulsive quise prolonge ou se répète sans attendre cette durée fatidique de30 minutes [2].
1. Différents types d’états de mal épileptiques
Nous nous limiterons ici aux états de mal chez l’enfantépileptique (nouveau-né exclu), qu’ils soient révélateurs del’épilepsie ou que l’épilepsie soit au contraire déjà connue.L’épilepsie est en effet pourvoyeuse de près de la moitié descas d’état de mal et 15 % des enfants épileptiques présente-ront dans leur vie au moins un état de mal [3]. Il ne fauttoutefois pas oublier que les causes occasionnelles sont bienplus fréquentes avant l’âge de deux ans (80 %) qu’après(46 %) [4] ; parmi celles qui requièrent un traitement spéci-fique et urgent, les états de mal partiels fébriles du nourrissonnon épileptique doivent faire penser à l’encéphalite herpéti-que, les non fébriles à l’hématome sous-dural.
Comme les adultes, les enfants peuvent présenter diffé-rents types d’états de mal épileptiques : schématiquement desétats de mal convulsifs, généralisés ou partiels, ou des étatsde mal non convulsifs, également généralisés ou partiels. Cesderniers sont réputés plus rares que chez l’adulte et corres-pondent à des crises partielles complexes prolongées, tandisque les états de mal non convulsifs généralisés sont aucontraire plus fréquents correspondant à des états d’absenceou myocloniques. Dans le cas des états de mal convulsifs, lediagnostic différentiel pose peu de problème, à l’inverse desétats de mal non convulsifs qui nécessitent un enregistrementélectroencéphalographique pour affirmer l’origine épilepti-que des troubles de conscience. La symptomatologie peut eneffet se réduire à une obnubilation, voire à de minimessecousses erratiques et le diagnostic d’état de mal peut êtreméconnu longtemps, tout particulièrement chez le très jeuneenfant. En revanche, il existe des situations « limites d’étatsde mal », spécifiques de l’enfant, rencontrées dans des syn-dromes que l’on ne voit pas chez l’adulte, et qui requièrent untraitement particulier comme l’épilepsie du nourrisson aveccrises partielles migrantes ou les pointes ondes continues dusommeil.
Tous les syndromes épileptiques de l’enfant sont suscep-tibles de donner des états de mal, toutefois les épilepsiesidiopathiques en sont des causes bien plus rares que lesépilepsies symptomatiques ou cryptogéniques [5–7]. Le syn-drome épileptique conditionne le type d’état de mal, sa fré-quence et sa prise en charge thérapeutique. Ainsi, le risque derécurrence des états de mal est de 13 % toutes épilepsies
confondues tandis qu’il atteint 44 % dans les épilepsiessymptomatiques [8]. Par ailleurs, certains antiépileptiquesont une efficacité particulière dans certains syndromes, surles états de mal comme sur les crises, tandis que d’autresantiépileptiques sont potentiellement aggravants égalementdans certains syndromes. En plus du diagnostic d’état de mal,le diagnostic du syndrome épileptique est un élément impor-tant d’une bonne prise en charge thérapeutique. Lorsquel’épilepsie commence par un état de mal (un tiers environ desétats de mal symptomatiques sont suivis d’une épilepsie), laprise en charge repose principalement sur le type de criseobservé. Un consensus a été établi en 1995 qui prend encompte quelques particularités des états de mal chez l’enfantmais qui s’applique avant tout aux états de mal convulsifs [2].
L’urgence de la prise en charge dans les états de malconvulsifs est justifiée par la gravité de la situation. Non tantpar le risque vital qui semble moins élevé chez les épilepti-ques (4 % sur une série hospitalière de 50 enfants [9]) quedans les états de mal occasionnels symptomatiques (jusqu’à15 % [3]), que par la morbidité qui est très élevée chezl’épileptique, particulièrement chez l’enfant (79 % de sé-quelles) [10]. Dans la série de Pinard, 60 % des patientsprésentaient une aggravation après un état de mal, soit de leurépilepsie (42 %), soit cognitive ou comportementale (36 %),soit motrice (38 %) [9]. Le risque d’aggravation était plusdurable dans les états de mal convulsifs et plus élevé danscertains syndromes comme les épilepsies partielles lésion-nelles ou l’épilepsie myoclonique sévère du nourrisson, tan-dis qu’il est au contraire nul en cas d’épilepsie idiopathique[10], situation rare bien que certains syndromes commel’épilepsie partielle occipitale à début précoce, épilepsie par-tielle bénigne récemment individualisée, présentent un fortrisque d’états de mal partiels (44 %) [11].
Expérimentalement, des crises prolongées, surtout de typeclonique ou tonique, sont source de mort neuronale parnécrose ou par apoptose [12]. Cependant, si la souffrancecellulaire, l’œdème régional et les lésions de la barrièrehématoencéphalique au moment de l’état de mal sont biendémontrés par l’IRM de diffusion [13], l’atrophie corticalesecondaire est inconstante [14]. La particulière gravité descrises prolongées chez l’enfant est liée à la survenue deséquelles fonctionnelles parfois majeures sur un cerveau endéveloppement, en raison de la modification des réseauxneuronaux induite par les crises [15]. Par ailleurs, l’existenced’une anomalie corticale préexistante est un facteur nonseulement favorisant mais aussi aggravant des états de malchez l’enfant, comme c’est le cas de l’épilepsie temporalemésiale avec sclérose hippocampique faisant suite à une crisefébrile prolongée dans la première année de vie. Dans ce cas,l’état de mal convulsif entraînerait des remaniements fonc-tionnels de l’hippocampe qui s’exprimeraient par une épilep-sie temporale à l’âge adulte, au moment où les connexions decette structure avec le cortex deviennent fonctionnelles. Unemalformation développementale de l’hippocampe pourraitaussi être la cause de la crise longue initiale [16].
Enfin, d’autres facteurs de risques d’état de mal ont étéidentifiés : la fièvre et une modification de traitement avaient
1218 C. Chiron et al. / Archives de pédiatrie 11 (2004) 1217–1224

précédé l’état de mal chez respectivement 20 et 25 % despatients de la série de Pinard [9]. Dans les épilepsies symp-tomatiques, l’existence d’un foyer à l’élec-troencéphalogramme (EEG), de crises partielles avec géné-ralisation secondaire, d’un état de mal inaugural et de lésionsdiffuses sur l’IRM sont autant de facteurs de risques d’état demal [17]. Enfin, un traitement inadapté par carbamazépine oupar phénytoine peut être responsable de l’apparition d’étatsde mal convulsifs ou d’états d’absence dans les épilepsiesgénéralisées idiopathiques [18].
À partir de ces notions générales, il apparaît clairementque c’est le syndrome épileptique qui conditionne avant toutla diversité de sémiologie, de gravité et donc la prise encharge des états de mal des épilepsies de l’enfant. Nousverrons donc ces différents aspects dans les syndromes épi-leptiques les plus fréquemment en cause.
2. États de mal convulsifs
2.1. États de mal convulsifs généralisés
On distingue les états de mal cloniques et tonicocloniques,et les états de mal toniques.
Les états de mal (tonico) cloniques émaillent l’évolutionde trois grands types de syndromes épileptiques de l’enfant :l’épilepsie myoclonique sévère du nourrisson (EMSN) (syn-drome de Dravet), les épilepsies partielles (EP) non idiopa-thiques, surtout celles avec une lésion identifiée, et les épi-lepsies généralisées symptomatiques (EGS), c’est-à-dire desépilepsies qui ont en général à la fois une composante par-
tielle et une composante généralisée. Ces trois syndromesreprésentent respectivement 24, 17 et 12 % des états de malchez les 50 patients étudiés par Pinard [9]. Dans ces troissituations, la crise peut se prolonger de façon généraliséeaprès avoir commencé soit de façon généralisée ou hémicor-porelle dans l’EMSN ou de façon focale même si cela a pupasser inaperçu dans les EP. Nous décrirons en détail le casde l’EMSN car les états de mal y sont souvent inauguraux etleur prise en charge conditionne en partie le pronostic ulté-rieur de l’enfant.
Dans l’EMSN, les états de mal cloniques sont presqueconstants (90 % des patients), souvent fébriles, souvent hé-micorporels ou à prédominance unilatérale, alternants d’uncôté ou de l’autre, avec un déficit moteur postcritique dequelques minutes à quelques heures (Fig. 1). Ils apparaissenttoujours avant l’âge d’un an et se répètent à une fréquencevariable pendant les deux à trois premières années. Le dia-gnostic de ce syndrome est simple. Il repose sur le début dansla première année, les crises éventuellement fébriles, ce typed’état de mal survenant chez un enfant sans antécédents, avecun examen normal, un EEG et une IRM normaux [19].Jusqu’à un ou deux ans, l’enfant a un développement normalpuis ce dernier s’infléchit et à terme 100 % des patients ont unretard mental. La responsabilité des états de mal précocesdans cette évolution toujours défavorable est largement sus-pectée. Certains antiépileptiques sont susceptibles d’aug-menter la fréquence de ces états de mal comme la carbama-zépine, le phénobarbital, le vigabatrin, la lamotrigine [20].L’association du stiripentol, inhibiteur du cytochrome P450,avec le clonazépam semble au contraire supprimer les états
Fig. 1. Épilepsie myoclonique sévère du nourrisson.Crise clonique bilatérale (clonies enregistrées sur l’EMG, 2 deltoïdes). Sur l’EEG, pointes rythmiques généralisées synchrones des clonies et dépressionpostcritique prolongée.
1219C. Chiron et al. / Archives de pédiatrie 11 (2004) 1217–1224

de mal [21]. La fièvre, qui est un facteur déclenchant habi-tuel, doit être systématiquement traitée.
La prise en charge thérapeutique des états de mal (tonico)cloniques généralisés ne diffère pas fondamentalement chezl’enfant et l’adulte, à l’exception des doses et des particula-rités de l’EMSN dans laquelle l’utilisation des barbituriquesest contre-indiquée. Dans notre expérience, on a recours audiazepam intrarectal dès le début de la crise (0,5 mg/kg), audomicile, puis au clonazepam i.v. (0,1 mg/kg/6 heures), enfinà la phénytoïne i.v. Il est exceptionnel d’avoir à indiquer uneintubation trachéale chez un enfant avec ce schéma thérapeu-tique. Pour la phénytoïne, nous injectons une dose de chargede 15 mg/kg, puis nous réinjectons 5 mg/kg quatre heuresplus tard si le taux plasmatique de phénytoïne deux heuresaprès la dose de charge est inférieur à 20 mg/l. Enfin, nousadaptons les doses toutes les huit heures jusqu’à 36 heures enutilisant des dosages plasmatiques de phénytoïne et le tauxplasmatique extrapolé (à partir d’un modèle mathématique)que nous obtiendrions si nous injections la dose théoriqued’entretien de 5 mg/kg. En pratique, nous avons à injecter desdoses bien inférieures, ce qui permet d’éviter le surdosageprogressif en phénytoïne, principale cause de résistance del’état de mal, par effet paradoxal [22]. D’autres moléculesont aussi été proposées dont l’usage varie selon les équipes etles pays : le valproate i.v. [23], le midazolam i.v. [24] oumême buccal en première intention [25].
Les états de mal toniques sont beaucoup plus rares que lesétats de mal (tonico)cloniques. Principalement rencontrésdans le syndrome de Lennox-Gastaut, ils peuvent être aggra-vés voire déclenchés par les Benzodiazépines i.v. tandisqu’ils répondent en règle à la phénytoïne.
2.2. États de mal convulsifs partiels
On les rencontre avant tout dans les épilepsies partiellessymptomatiques, au premier rang desquelles deux syndro-mes neurocutanés (l’angiomatose de Sturge-Weber et la sclé-rose tubéreuse de Bourneville), mais aussi dans des malfor-mations corticales (dysplasie corticale focale, hémi-mégalencéphalie, agénésie du corps calleux) et dans desséquelles anoxo-ischémiques (hémiplégie cérébrale infan-tile). Ainsi, dans la maladie de Sturge-Weber, les états de malmoteurs sont fréquemment inauguraux, dès la première an-née de vie et responsables d’un déficit moteur définitif avecretard mental, si bien que certains prônent l’instauration d’untraitement antiépileptique avant même l’apparition des pre-mières crises. Dans toute épilepsie partielle symptomatique,une augmentation récente et non contrôlable de la fréquencedes crises avec un retentissement fonctionnel fait craindrel’apparition d’un état de mal convulsif : le régime cétogène apermis d’arrêter les crises dans 90 % des cas sur une sériepersonnelle récente de dix enfants.
Le traitement médical initial de ces états de mal partielsrepose sur les mêmes principes que celui des états de malgénéralisés, l’urgence étant ici moins liée à la peur de trou-bles végétatifs qu’à celle d’une hémiplégie ou d’une hémia-
nopsie séquellaires. Lorsque l’épilepsie est pharmacorésis-tante et les états de mal récurrents, situation de loin la plusfréquente dans ces épilepsies partielles lésionnelles, la chi-rurgie d’exérèse du foyer épileptogène est à discuter.
À la « limite des états de mal », le syndrome récemmentdécrit d’épilepsie du nourrisson avec crises partielles mi-grantes [26] débute dans les premiers mois de vie par descrises partielles qui sont multifocales, extrêmement fréquen-tes et s’intriquent les unes dans les autres de façon prolongée,l’une débutant dans une autre région du cerveau avant que laprécédente ne soit terminée (Fig. 2), induisant donc unesémiologie des crises très polymorphes, avec ou sans phéno-mènes moteurs. La majorité des décharges sont infraclini-ques. Ces nourrissons, qui n’ont pourtant aucune lésion céré-brale et sont exempts de toute maladie métabolique connue,ont un arrêt de leur développement psychomoteur et de leurcroissance cérébrale et deviennent de grands encéphalopa-thes. Les crises sont extrêmement réfractaires à tous médica-ments connus ; seul le clonazepam injectable à fortes dosesadministré par la sonde gastrique a semblé en diminuer lafréquence [26].
L’encéphalite de Rasmussen représente une autre situa-tion « à la limite des états de mal » par le biais de l’épilepsiepartielle continue qui lui est associée dans la moitié des cas.Débutant le plus souvent avant dix ans, c’est une épilepsieprogressive de la région rolandique qui associe des crisesmotrices unilatérales, des myoclonies parcellaires du bras, dela jambe, ou de la face du même côté, qui sont des myoclo-nies corticales bien qu’ayant rarement une corrélation visiblesur l’EEG car elles sont trop parcellaires, mais qui devien-nent permanentes et s’accompagnent d’une hémiplégie ho-molatérale, d’une atrophie centrale controlatérale et d’unedégradation cognitive. La pharmacorésistance est extrême.La corticothérapie prolongée à fortes doses a pu retarderl’apparition du déficit moteur mais en définitive elle ne sem-ble pas constituer une alternative à l’hémisphérectomie oul’hémisphérotomie, seules capables de stopper l’épilepsie[27,28].
3. États de mal non convulsifs
Les états de mal non convulsifs généralisés se manifestentpar un état d’obnubilation prolongé pendant plusieurs heuresou plusieurs jours, souvent associé à des myoclonies errati-ques et parfois à d’autres types de crises convulsives généra-lisées. L’EEG est absolument nécessaire pour faire le dia-gnostic de ces états de mal sur une désorganisation du tracésans rythme physiologique décelable, avec des ondes lentesdiffuses irrégulièrement encochées de pointes, voire desbouffées diffuses de pointes, polypointes et pointes ondeslentes qui peuvent être corrélées à des myoclonies segmen-taires (Fig. 3). Ces états de mal sont bien souvent méconnuset, du fait de leur prolongation sur une longue période et deleur haut risque de récurrence, peuvent s’accompagner d’unedétérioration psychomotrice (28 patients sur 50 dans la série
1220 C. Chiron et al. / Archives de pédiatrie 11 (2004) 1217–1224
de Stores et al. [29]) au point parfois de faire suspecter uneencéphalopathie progressive [30].
La prise en charge médicamenteuse des états de mal nonconvulsifs ne présente pas le même caractère d’urgence quecelle des états de mal convulsifs. Les benzodiazépines i.v.
constituent le traitement de référence, tandis que le phény-toïne et le phénobarbital i.v. ont plutôt un effet aggravant.
Les états de mal non convulsifs surviennent dans lesépilepsies myocloniques associées à des maladies métaboli-ques (pyridoxino-dépendance, céroïdolipofuchinose, ou mi-
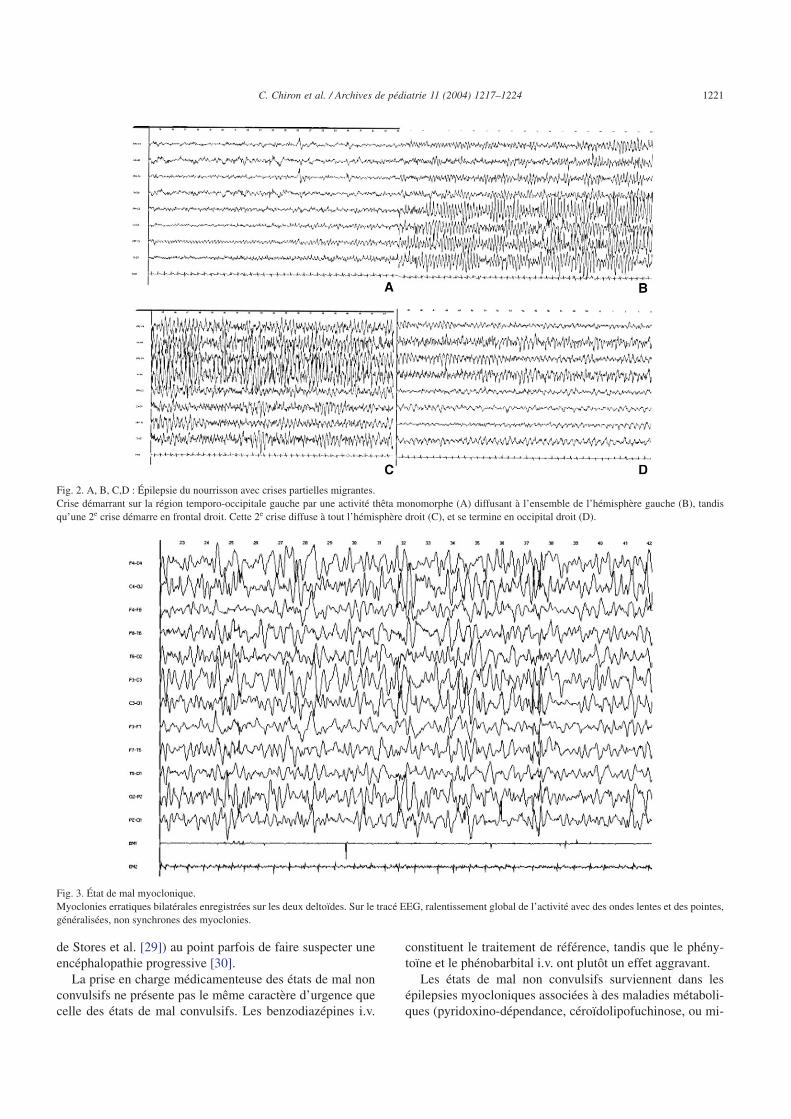
Fig. 2. A, B, C,D : Épilepsie du nourrisson avec crises partielles migrantes.Crise démarrant sur la région temporo-occipitale gauche par une activité thêta monomorphe (A) diffusant à l’ensemble de l’hémisphère gauche (B), tandisqu’une 2e crise démarre en frontal droit. Cette 2e crise diffuse à tout l’hémisphère droit (C), et se termine en occipital droit (D).
Fig. 3. État de mal myoclonique.Myoclonies erratiques bilatérales enregistrées sur les deux deltoïdes. Sur le tracé EEG, ralentissement global de l’activité avec des ondes lentes et des pointes,généralisées, non synchrones des myoclonies.
1221C. Chiron et al. / Archives de pédiatrie 11 (2004) 1217–1224

tochondriopathie), dans le cadre des épilepsies myocloniquessur encéphalopathie non progressive (ENP), ou encore dansdes épilepsies myocloniques sans lésion cérébrale commel’épilepsie myoclonique sévère du nourrisson ou l’épilepsiemyoclono-astatique.
L’épilepsie myoclono-astatique (EMA ou syndrome deDoose) débute après deux ans par des crises généraliséescloniques chez un enfant antérieurement normal, puis appa-raissent des myoclonies parcellaires ou massives entraînantdes chutes, de fréquents états de mal myocloniques. L’évo-lution est dans deux tiers des cas, une épilepsie résistanteavec crises toniques nocturnes et un retard intellectuel, etdans un tiers des cas une guérison sans séquelles [31].
Dans les épilepsies myocloniques sur encéphalopathienon progressive au premier rang desquelles figure le syn-drome d’Angelman, l’épilepsie myoclonique débute plus tôt,à trois mois en moyenne, que chez des patients avec unelésion cérébrale et un retard préexistants. Par ailleurs, cesenfants présentent des états de mal myocloniques précoces etprolongés dont le diagnostic est souvent longtemps méconnu[32].
Dans le syndrome de Lennox-Gastaut, les états de mal nonconvulsifs sont le plus souvent des états d’absence, corres-pondant à des absences atypiques subcontinues, associées àdes bouffées prolongées de pointes ondes lentes bifrontalessur l’EEG. Quelques formes myocloniques de syndrome deLennox-Gastaut ont été rapportées, qui, en l’absence de lé-sion cérébrale, posent des problèmes nosologiques délicatsavec les formes d’évolution défavorable des EMA [31].
Dans les épilepsies idiopathiques, les états de mal nonconvulsifs sont très rares chez l’enfant et toujours d’évolu-tion bénigne. Ils peuvent être déclenchés par des médica-ments, comme la carbamazépine, le phénytoïne, le phénobar-bital et le vigabatrin, molécules qui expérimentalement
aggravent les absences dans le modèle du rat GAERS. L’arrêtdu produit suffit à stopper l’état de mal [33].
« À la limite des états de mal », les pointes ondes conti-nues du sommeil (POCS) représentent un syndrome épilep-tique spécifique de l’enfant caractérisé par une activité depointes ondes lentes généralisées quasi continues pendant lesommeil lent (Fig. 4), source de régression cognitive. Cephénomène réalise un état épileptique non convulsif infracli-nique. Il s’agit d’un phénomène de généralisation lié à l’âge,qui débute entre deux et huit ans. Un caractère focal est trèssouvent associé. Les causes de cette épilepsie sont multiples,lésionnelles ou non. En effet, d’authentiques épilepsies par-tielles bénignes peuvent s’accompagner de POCS à un mo-ment de leur évolution. Les lésions observées sont surtout denature anoxo-ischémique (porencéphalies ou polymicrogy-ries unilatérales ou bioperculaires) et peuvent être source decrises partielles et de difficultés cognitives associées, sélecti-ves des régions fonctionnelles préférentiellement concer-nées. Les patients présentent en général peu de crises, sauf encas d’implication de la région rolandique où l’on observe desmyoclonies négatives à l’origine de chutes [34]. Cet aspectEEG est particulièrement pharmacorésistant et susceptibled’être aggravé par la carbamazépine, la phénytoïne, le phé-nobarbital qui sont à proscrire chez ces patients [35]. Letraitement repose sur les benzodiazépines orales et l’étho-suximide et rapidement, en cas d’échec, la corticothérapie àfortes doses et prolongée au moins un an pour éviter le risquede rechutes et permettre la récupération des fonctions cogni-tives à l’état antérieur [36].
À la différence des adultes, les états de mal non convulsifspartiels sont plus rares chez l’enfant. Ils sont certainementsous-estimés et il convient de les suspecter devant une obnu-bilation ou des troubles confusionnels. En effet, la pratiqueextensive des enregistrements EEG de longue durée a montré
Fig. 4. Pointes ondes continues du sommeil (POCS).Pointes ondes lentes continues généralisées amples de fréquence 2,5 Hz pendant plus de 85 % du temps de sommeil total.
1222 C. Chiron et al. / Archives de pédiatrie 11 (2004) 1217–1224

que plus de la moitié des crises passaient cliniquement ina-perçues chez le nourrisson, se manifestant par un simple arrêtd’activité ou de la gesticulation. Par ailleurs, l’identificationrécente de l’épilepsie particulière du syndrome du chromo-some 20 en anneau est une démonstration de l’intérêt del’EEG vidéo pour limiter la sous-évaluation de ces états demal souvent prolongés pendant plusieurs heures sous formed’un état confusionnel ou d’obnubilation associé à des ondeslentes rythmiques bifrontales [37].
4. Conclusion
La sémiologie et la prise en charge thérapeutique des étatsde mal de l’enfant épileptique dépend du type de syndromeépileptique. Certains médicaments sont susceptibles d’ag-graver certains états de mal, comme les barbituriques i.v.dans les états de mal convulsifs généralisés de l’EMSN et lesétats de mal généralisés non convulsifs des épilepsies myo-cloniques, ou les benzodiazépines i.v. dans les états de maltoniques du syndrome de Lennox-Gastaut. Le recours à laréanimation est inutile en cas d’état de mal non convulsif etne doit pas être systématique en cas d’état de mal convulsif :l’expérience des services de neuropédiatrie montre qu’unetrès faible minorité d’enfants épileptiques a besoin d’uneintubation trachéale au cours d’un état de mal convulsif pourdes motifs autres que les effets secondaires des médicamentsinjectés.
Les états de mal convulsifs généralisés sont principale-ment le fait de l’épilepsie myoclonique sévère du nourrisson,des épilepsies généralisées symptomatiques et des épilepsiespartielles lésionnelles. Ils requièrent en urgence du diazépamintrarectal à domicile, puis en hospitalisation du clonazépami.v. voire de la phénytoïne i.v. en surveillant les taux plasma-tiques. Les états de mal convulsifs partiels des épilepsiespartielles lésionnelles font courir le risque de déficit focal etrequièrent le même traitement. Les états de mal non convul-sifs généralisés sont des états d’absence ou myocloniques,souvent longtemps méconnus jusqu’au diagnostic élec-troencéphalographique. Fréquents dans les épilepsies myo-cloniques, lésionnelles ou non, ils répondent aux benzodia-zépines i.v. La fréquence des états de mal non convulsifspartiels est sûrement sous-estimée surtout chez le nourrissonoù la vidéo-EEG est le moyen diagnostique de choix : letraitement est le même que celui des états de mal convulsifspartiels.
Références
[1] Coeytaux A, Jallon P. Des difficultés de définir et de classifier l’état demal épileptique. Neurophysiol Clin 2000;30:133–8.
[2] XIVe conférence de consensus de réanimation et de médecined’urgence. Ann Fr Anesth Reanim, 15. 1996. p. 106–9 Prise en chargede l’état de mal épileptique (Enfants–Adultes). 23 June 1995, LeKremlin-Bicêtre.
[3] Fountain N. Status epilepticus: risk factors and complications. Epilep-sia 2000;Suppl 2:S23–30.
[4] Shinnar S, Pellock JM, Moshe SL, Maytal J, O’Dell C,Driscoll SM, et al. In whom does status epilepticus occur: age-relateddifferences in children. Epilepsia 1997;38:907–14.
[5] Berg AT, Shinnar S, Levy SR, Testa FM. Status epilepticus in childrenwith newly diagnosed epilepsy. Ann Neurol 1999;45:618–23.
[6] Coeytaux A, Jallon P, Galobardes B, Morabia A. Incidence of statusepilepticus in French-speaking Switzerland: (EPISTAR). Neurology2000;55:693–7.
[7] Shinnar S, O’Dell C, Berg AT. Distribution of epilepsy syndromes in acohort of children prospectively monitored from the time of their firstunprovoked seizure. Epilepsia 1999;40:1378–83.
[8] Shinnar S, Maytal J, Krasnoff L, Moshe S. Recurrent status epilepti-cus in children. Ann Neurol 1992;31:598–604.
[9] Pinard J. Étude des états de mal dans les épilepsies de l’enfant [Thèse].Université Pierre et Marie Curie; 1988.
[10] Barnard C, Wirell E. Does status epilepticus in children cause devel-opmental deterioration and exacerbation of epilepsy? J Child Neurol1999;14:787–94.
[11] Ferrie C, Beaumanoir A, Guerrini R, Kivity S, Vigevano F, Takai-shi Y, et al. Early-onset benign occipital seizure susceptibility syn-drome. Epilepsia 1997;38:285–93.
[12] Coulter D, DeLorenzo R. Basic mechanisms of status epilepticus.AdvNeurol 1999;79:725–33.
[13] Lansberg MG, O’Brien MW, Norbash AM, Moseley ME, Morell M,Albers GW, et al. MRI abnormalities associated with partial statusepilepticus. Neurology 1999;52:1021–7.
[14] Salmenpera T, Kalviainen R, Partanen K, Mervaala E, Pitkanen A.MRI volumetry of the hippocampus, amygdala, entorhinal cortex, andperirhinal cortex after status epilepticus. Epilepsy Res 2000;40:155–70.
[15] Engert F, Bonhoeffer T. Dendritic spine changes associated withhippocampal long-term synaptic plasticity. Nature 1999;399:66–70.
[16] Baulac M, De Grissac N, Hasboun D, Oppenheim C, Adam C, Arzi-manoglou A, et al. Hippocampal developmental changes in patientswith partial epilepsy: magnetic resonance imaging and clinicalaspects. Ann Neurol 1998;44:223–33.
[17] Novak G, Maytal J, Alshansky A, Ascher C. Risk factors for statusepilepticus in children with symptomatic epilepsy. Neurology 1997;49:533–7.
[18] Osorio I, Reed RC, Peltzer JN. Refractory idiopathic absence statusepilepticus: A probable paradoxical effect of phenytoin and carbam-azepine. Epilepsia 2000;41:887–94.
[19] Dravet C. Severe myoclonic epilepsy in infants and its related syn-dromes. Epilepsia 2000;41(Suppl 9):7.
[20] Guerrini R, Dravet C, Genton P, Belmonte A, Kaminska A, Dulac O.Lamotrigine and seizure aggravation in severe myoclonic epilepsy.Epilepsia 1998;39:508–12.
[21] Chiron C, Marchand MC, Tran A, Rey E, d’Athis P, Vincent J, et al.Stiripentol in severe myoclonic epilepsy in infancy: a randomisedplacebo-controlled syndrome-dedicated trial. The Lancet 2000;356:1638–42 STICLO study group.
[22] Richard MO, Chiron C, d’Athis P, Rey E, Aubourg P, Dulac O, et al.Phenytoin monitoring in status epilepticus in infants and children.Epilepsia 1993;34:144–50.
[23] Uberall MA, Trollmann R, Wunsiedler U, Wenzel D. Intravenousvalproate in pediatric epilepsy patients with refractory status epilepti-cus. Neurology 2000;54:2188–9.
[24] Yoshikawa H, Yamazaki S, Abe T, Oda Y. Midazolam as a first-lineagent for status epilepticus in children. Brain Dev 2000;22:239–42.
[25] Scott RC, Besag FM, Neville BG. Buccal midazolam and rectaldiazepam for treatment of prolonged seizures in childhood andadolescence: a randomised trial. Lancet 1999;353:623–6.
[26] Coppola G, Plouin P, Chiron C, Robain O, Dulac O. Migrating partialseizures in infancy: a malignant disorder with developmental arrest.Epilepsia 1995;36:1017–24.
1223C. Chiron et al. / Archives de pédiatrie 11 (2004) 1217–1224

[27] Vining EP, Freeman JM, Pillas DJ, Uematsu S, Carson BS,Brandt J, et al. Why would you remove half a brain? The outcome of58 children after hemispherectomy-the Johns Hopkins experience:1968 to 1996. Pediatrics 1997;100:163–71.
[28] Villemure JG, Rasmussen T. Functional hemispherectomy in chil-dren. Neuropediatrics 1993;24:53–5.
[29] Stores G, Zaiwalla Z, Styles E, Hoshika A. Non-convulsive statusepilepticus. Arch Dis Child 1995;73:106–11.
[30] Tassinari CA, Michelucci R, Forti A, Salvi F, Plasmati R, Rub-boli G, et al. The electrical status epilepticus syndrome. Epilepsy ResSuppl 1992;6:111–5.
[31] Kaminska A, Ickowicz A, Plouin P, Bru MF, Dellatolas G, Dulac O.Delineation of cryptogenic Lennox-Gastaut syndrome and myoclonicastatic epilepsy using multiple correspondence analysis. Epilepsy Res1999;36:15–29.
[32] Viani F, RomeoA, Viri M, Mastrangelo M, Lalatta F, SelicorniA, et al.Seizure and EEG patterns in Angelman’s syndrome. J Child Neurol1995;10:467–71.
[33] Marescaux C, Vergnes M. Genetic Absence Epilepsy in Rats fromStrasbourg (GAERS). Ital J Neurol Sci 1995;16:113–8.
[34] Guerrini R, Genton P, Bureau M, Parmeggiani A, Salas-Puig X,Santucci M, et al. Multilobar polymicrogyria, intractable drop attackseizures, and sleep- related electrical status epilepticus. Neurology1998;51:504–12.
[35] Veggiotti P, Beccaria F, Guerrini R, Capovilla G, Lanzi G. Continuousspike-and-wave activity during slow-wave sleep: syndrome or EEGpattern? Epilepsia 1999;40:1593–601.
[36] Yasuhara A, Yoshida H, Hatanaka T, Sugimoto T, Kobayashi Y,Dyken E. Epilepsy with continuous spike-waves during slow sleepand its treatment. Epilepsia 1991;32:59–62.
[37] Inoue Y, Fujiwara T, Matsuda K, Kubota H, Tanaka M, Yagi K, et al.Ring chromosome 20 and nonconvulsive status epilepticus. A newepileptic syndrome. Brain 1997;120:939–53.
1224 C. Chiron et al. / Archives de pédiatrie 11 (2004) 1217–1224