Embed Size (px)
Citation preview

Eur J Med Chem (1992) 27,437442 0 Elsevier, Paris
437
Synthesis and antidepressant activity of 5-(1-aryl-4-piperazino)methyl-2-amino-2-oxazolines
JJ Bose’, C Jan-y I*, A Carpyz, E Panconi3, P Descas3
‘Laboratoire de Chimie Physique; 2URA 605 CNRS, Universite’ de Bordeaux II, 3, place de la Victoire, 33076 Bordeaux Cedex;
3Dkpartement Recherche et De’veloppement, Laboratoires Sarget, 33701 Me’rignac Cedex, France
(Received 23 March 1991; accepted 12 November 1991)
Summary - The synthesis of 20 5-(l-aryl-4-piperazino)methyl-2-amino-2-oxazolines is described. Antidepressant activity was observed in mice using classical screening tests. Structure-activity relationships were studied and correlated with the nature of the aromatic substituent. Preliminary lipophilic and electronic properties of one lead compound (COR 3224) have been described.
2-amino-2-oxazolines I 1-arylpiperazine I antidepressant activity / structure-activity relationships
Introduction
Considerable interest has been paid to compounds containing a 1-arylpiperazine moiety in particular due to their effects on the central nervous system. Among these compounds trazodone and buspirone, 2 agents presently in clinical practice, may produce some of their pharmacological effects via a central serotonin mechanism [ 1,2].
As part of our search for bioactive compounds, we prepared and tested for antidepressant activity a series of 5-( 1 -aryl-4-piperazino)methyl-2-amino-2-oxazo- lines. The preliminary pharmacological results con- cerning the leader compound (COR 3224, Labora- toires &get, Merignac, France, presently in phase II clinical trials) have been recently reported [3, 41. In this work, we present some pharmacological data on the complete series for study of structure-activity relationships. The electronic and lipophilic properties associated with the pharmacological activity of this series were clarified for COR 3224. 4 3
Chemistry
The general methods used for the preparation of 5( l- aryl-4-piperazino)methyl-2-amino-2-oxazolines 4 are shown in scheme 1. In path A the l-( 1-aryl-4- piperazino)-2,3-epoxypropanes 3 were obtained from
*Correspondence and reprints Scheme 1.
- AWL I \ )(,, e 2.PYRIDYL , 2.PYRIHIDYL

438
appropriate piperazines 1 and epichlorhydrin in a one- pot reaction [5, 61. The non-isolated intermediates 1 -chloro-3-( 1 -aryl-4-piperazino)-propan-2-01s 2 were dehydrohalogenated into 3 using sodium hydroxide. In most cases the epoxides 3 were converted without further purification into the corresponding 5-( l-aryl- 4-piperazino)methyl-2-amino-2-oxazolines 4 by con- densation with cyanamide monosodium salt. The opening of the epoxide ring of 3 leads to the pre- ferential formation of the normal isomer 4 [7]. It was not possible to isolate the abnormal isomeric 4-(1- aryl-4-piperazino)methyl-2-amino-2-oxazolines. In path B the I-chloro-3-(1-aryl-4-piperazino)propan-2- 01s 2 were isolated and reacted directly with 2 equi- valents of cyanamide monosodium salt directly forming the 2-amino-2-oxazolines 4. In both ways the reaction with monosodium salt was performed in methanol which permitted the solubilization of all compounds. The amidine group of the 2-amino-2- oxazoline moiety can induce a tautomeric pheno- menon. It has been shown that an amino form is preponderant in a free base, whereas an imino form is preponderant in a salt [8]. In the 1H NMR spectra of compounds 4 as bases, the intensity of the NH, signal was in agreement with an amino form. The oxazoline protons on the C-4 and C-5 positions formed an ABX system, and the C-5 methine proton was found at about 4.8 ppm (table I). The EI mass spectra of compounds 4 have been studied elsewhere [9]. It was noted that the main fragments were derived from the arylpiperazine moiety.
Pharmacological results
All compounds were evaluated for their anti- depressant activity in mice using the following tests: inhibition of reserpine and apomorphine induced hypothermia and potentiation of yohimbine induced sublethality. Acute toxicity was also determined (table II). Imipramine and viloxazine were included in the pharmacological assays as appropriate standards.
The drugs able to reduce the reserpine induced hypothermia could increase postsynaptic nor- epinephrine [lo]. Compounds 4a (COR 3224), 4e, 4i, 4j, 4r and 4s exhibited good activity with an ED,, < 20 mg/kg po. Among the phenyl substituted compounds, the para substituted ones were the most effective. In 4-alkyloxy substituted compounds, the increase in chain length was detrimental to the activity. Compared to the 4-methoxy derivative 4e, the 3,4-dimethoxy 4q and the 2-methylpiperazinyl substituted 4t were less potent. A chloro substitution in any position induced a decrease in activity.
Hypothermia antagonism induced by high doses of apomorphine is also a specific test used to detect potential antidepressant activity [ 11, 121. This effect is
Table I. Physical data of derivatives 4. Recristallization solvent: a: heptane, b: ethanol, c: trichloroethylene, d: tetra- chloroethylene. 1H NMR: s: singlet, m: multiplet, c: center of AA’BB’ system.
N” SUBSTITUENT YIELD F”C RMN ‘H 8ppm CDCf3 I DMSO Dcj *
4a
4b
4h
4i
H
4-a
3.4-diCI
4-CH3
4-OCH3
3-a
4WCHa)z
3.CH3
4-OH
4-OCOCH3 (2 HBr)
4k 4-OCHpCHzCH3
41 3-OCH3
4m 2-a
4n 4-CF3
40 4-OCH(CH& (2 HCI)
4P 4-OCH2CsH5
4q
“4r
3+diOCHS
(HCI) P-Pyridyl
“43 P-Pyrimidyf
“4l 4-OCH3
32 174 d
37 195
c 14 196
a 32 162
a 30 177
d
26 198
21 173 c
17 177
a
20 231 d
12 160
b 25 162
c 26 170
d
22 145
20 160
c 10 192
b
35 193
27 160
b 34 156
c
29 179 a
IO 130
4.13 4.9-4.7 3 m
* 5.7 4.6-4.4
3 m 4.9-4.4
m
5-4.2
m 5.1-4.4
m
* 5.6 3 4.9-4.3
m
5.1-4.5 m
5.1-4.5 m
‘7.6-4.9 4.9-4.3
m m ‘11.2-8.9 6-5.5
m m
5.1-4.5 m
5-4.4
m
5.2-4.5 m
* 5.6 3 4.9-4.3 m
‘12-6.4 6-5.5 m m
5.24.3 m
‘12-6 5.6-5.1
m m
* 5.83 4.94.3
m
‘5.7 3 4.9-4.3
m
‘6.4-5 4.94.3
3.9-2.5 m
3.6-2.2
m 4-2.3
m
4-2.1
m 4.1-2.3
m 4-2.3
m 4-2.3
m 4-2.2
m
3.9-2.3
m 4.2-3
m
4.2-2.3 m
4-2.3
m 4.1-2.3
m
3.9-2.2 m
4.7-2.9
m 4.2~2.3
m 4.2-2.6
m 3.9-2.2
m
4-2.2
m 4-2.1
7.3-6.6 m
7c
7.3-6.5
m
6.9 c
6.9 c
7.4-6.6
m
6.8 c
7.3-6.5 m
7.64.9 m
7c
6.9 c
7.3-6.3
m 7.5-6.6
m
7.2 c
6.9 c
7.7-6.8
m 7-6.2
m 6.2-6.4
m
6.5-6.4
m 6.6 c
CH3 piper a m m m
+*4r: Aryl = 2-pyridyl; **4s: Aryl = 2-pyrimidyl; **4t: R, = 4-OCH,, R2 = CH,.
usually considered as related to the stimulation of dopaminergic receptors; it also implicates a /3-adre- nergic system [ 131. Except 4f and 40, all compounds active in the previous test were active in this one. Any substitution on the phenyl ring gave no significant improvement of the activity.
Most of the tested compounds presented a low toxicity after single oral administration since the LD,,
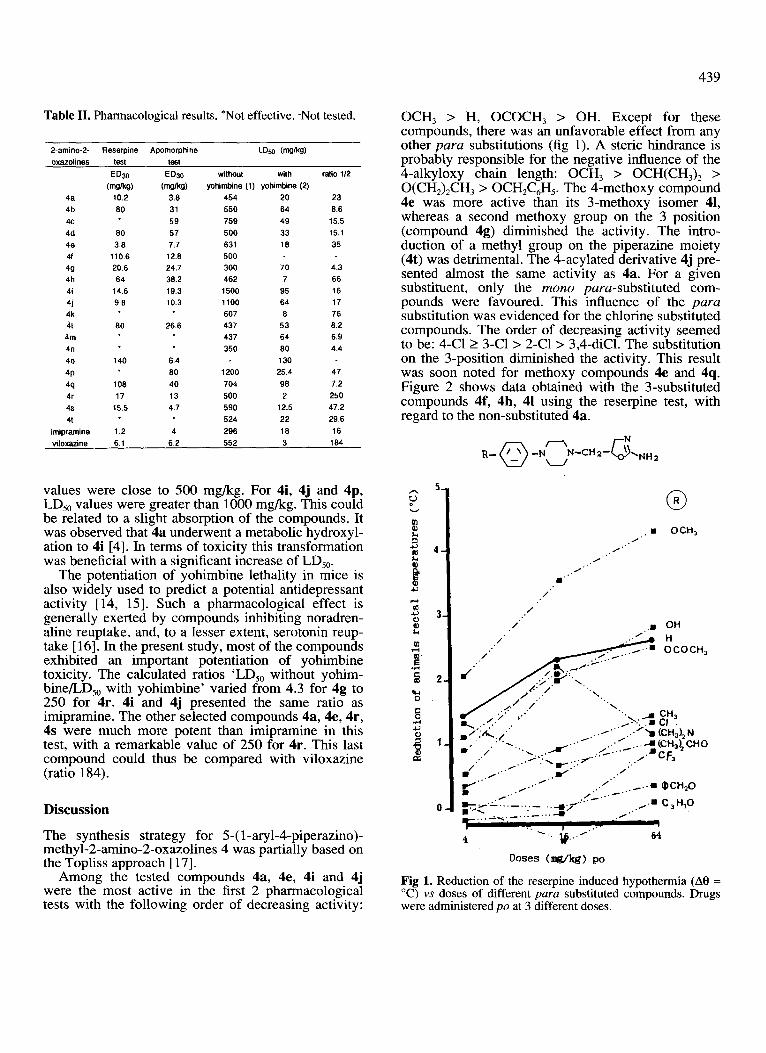
Table II. Pharmacological results. *Not effective. -Not tested.
2.amino-2. Reserpine Apomorphine LQXI (w/kg) oxazolines test test
4a 4b 4c 4d 40 41
49 4h 4i 41 4k 41
4m 4n 40
4P
‘w 4r 4s 4t
imipramine
WO mM4)
10.2 60 1
60 3.6
110.6 20.6
64 14.6 9.6
80 26.6
140
106 17
15.5
1.2
ED30 WWW
3.6 31 59 57 7.7
12.6 24.7 36.2 19.3 10.3
6.4 60 40 13 4.7
4
without with yohimbine (1) yohtmbine (2)
454 20 550 64 759 49 500 33 631 16 500 300 70 462 7
1500 95 1100 64 607 6 437 53 437 64 350 80
130 1200 25.4 704 96
500 2 590 12.5 524 22 296 16
ratio 112
23 6.6
15.5 15.1 35
4.3 66 16 17 76 6.2 6.9 4.4
47 7.2
250 47.2
29.6 16
viloxazine 6.1 6.2 552 3 184
values were close to 500 mg/kg. For 4i, 4j and 4p, LD,, values were greater than 1000 mg/kg. This could be related to a slight absorption of the compounds. It was observed that 4a underwent a metabolic hydroxyl- ation to 4i [4]. In terms of toxicity this transformation was beneficial with a significant increase of LD,,.
The potentiation of yohimbine lethality in mice is also widely used to predict a potential antidepressant activity [14, 151. Such a pharmacological effect is generally exerted by compounds inhibiting noradren- aline reuptake, and, to a lesser extent, serotonin reup- take [ 161. In the present study, most of the compounds exhibited an important potentiation of yohimbine toxicity. The calculated ratios ‘LD,, without yohim- bine/L,D,, with yohimbine’ varied from 4.3 for 4g to 250 for 4r. 4i and 4j presented the same ratio as imipramine. The other selected compounds 4a, 4e, 4r, 4s were much more potent than imipramine in this test, with a remarkable value of 250 for 4r. This last compound could thus be compared with viloxazine (ratio 184).
Discussion
The synthesis strategy for 5-( 1-aryl-4-piperazino)- methyl-2-amino-2-oxazolines 4 was partially based on the Topliss approach [ 171. Doses (wUke> PO
Among the tested compounds 4a, 4e, 4i and 4j were the most active in the first 2 pharmacological
Fig 1. Reduction of the reserpine induced hypothermia (A0 =
tests with the following order of decreasing activity: “C) vs doses of different paru substituted compounds. Drugs were administered po at 3 different doses.
439
OCH, > H, OCOCH, > OH. Except for these compounds, there was an unfavorable effect from any other para substitutions (fig 1). A steric hindrance is probably responsible for the negative influence of the 4-alkyloxy chain length: OCH, > OCH(CH,), > O(CH,),CH3 > OCH,C,H,. The 4-methoxy compound 4e was more active than its 3-methoxy isomer 41, whereas a second methoxy group on the 3 position (compound 4g) diminished the activity. The intro- duction of a methyl group on the piperazine moiety (4t) was detrimental. The 4-acylated derivative 4j pre- sented almost the same activity as 4a. For a given substituent, only the mono para-substituted com- pounds were favoured. This influence of the para substitution was evidenced for the chlorine substituted compounds. The order of decreasing activity seemed to be: 4-Cl2 3-Cl > 2-Cl> 3,4-diC1. The substitution on the 3-position diminished the activity. This result was soon noted for methoxy compounds 4e and 4q. Figure 2 shows data obtained with the 3-substituted compounds 4f, 4h, 41 using the reserpine test, with regard to the non-substituted 4a.
R- ‘-’ -N OC N-Cl-l?- ‘NH.2
0 R ./..
n ocn, ,/.’
,/.’ ,/.’
m” /
/’ /.‘ .8 OH
.,~ -. -:‘%H,)?N
_.’ /’ :’ __. A cti,r CHO
440
5-
I-
3-
2-
l-
0,
'NH2
0 R
o”’ r 1 4 4 16 64
Doses (&kg) PO
Fig 2. Reduction of the reserpine induced hypothermia (At3 = “C) for the 3-substituted 4f, 4h, 41 with regard to 4a. Drugs were administered po at 3 different doses.
The replacement of the phenyl ring of 4a by a pyridyl or a pyrimidyl ring resulted in slighty less potent (4r) or equipotent (4s) compounds. Results on acute toxicity showed that toxicity was 2-fold lower in compounds 4i and 4j than in 4a and 4e. Knowing that the metabolic pathway of 4a led to 4j and combining activity and toxicity results, it seems that the best results are obtained for the pm-u hydroxylated com- pound 4i or for compounds which might be hydroxyl- ated after metabolism ie, 4a, or after enzymatic hydrolysis ie, 4e and 4j. This could also explain the lower activity of compounds 4k, 40 and 4p which are resistant to metabolism, or of compounds 4b, 4d, 4g and 4n for which metabolic hydroxylation is imposs- ible. The most toxic compound was 4g, which con- tains an anilino moiety. Finally, the 2 pyridyl and pyrimidyl compounds 4r, 4s were found to be slighty less toxic than 4a itself.
The mechanism of 5-( I-aryl-4-piperazino)methyl- 2-amino-2-oxazolines action not being completely elucitated, we can only obtain an insight into the main molecular properties related to the activity. An account of the X-ray crystal structure of 4a as a di- hydrochloride dihydrate of the racemic form has been
published elsewhere, together with a conformational analysis [8]. The aromatic binding site must be deep enough to allow pm-u substituents such as OH or OCH,, and narrow enough to avoid ortho and meta substituents.
Figure 3 represents the 3-D MEP (molecular electrostatic potential) map obtained on the neutral amino form of 4a. Only the negative areas (-10 kcahmol-1) generated by the lone pairs of the heteroatoms have been depicted. One should note that the exocyclic amino group does not generate a negative potential as the negative charge borne by the nitrogen is compensated by the positive charges borne by the 2 hydrogens. It can also be mentioned that the areas of negative potential correspond to the hydrogen bonds which occur in the crystals.
Figure 4 represents the 3-D MLP (molecular lipo- philicity potential) map obtained on the amino form of 4a. The vertical barrier divides the molecule in 2 parts. On the left side is the lipophilic zone generated by the phenyl ring. On the right side, there are 2 hydrophilic zones. The first one is due to the basic nitrogen of the piperazine ring, and the second to the amidine moiety of the 2-amino-2-oxazoline. If we compare this map with the MEP one, we can see some similarities and some differences. The positive potential generated by the phenyl ring corresponds to the lipophilic zone. The negative potentials generated by the non-basic nitrogen of the piperazine and by the 5-membered ring do not correspond to hydrophilic areas. An account of molecular modelling applied to the entire series will be published in a specialized journal.
Fig 3. 3-D-MEP (calculated by the VSS semi-empirical method) of 4a in the crystal conformation. Only isoenergy areas (-10 kcal mol-*) have been displayed.
441
Fig. 4. 3-D-MLP (calculated with the algorithm from [21]) of 4a in the crystal conformation. On the left is the lipophilic zone (PL = 60). On the right are 2 hydrophilic zones (PL = 40). The vertical barrier (PL = 53) divides the molecule into 2 parts which display opposite properties.
Experimental protocols
Chemistry
Satisfactory elemental analysis + 0.4% of calculated values were obtained for all new compounds. Melting points determined with a Kofler hot-stage were uncorrected. IR spectra were recorded on a Beckman Acculab spectrometer as KBr discs. tH and 13C NMR spectra were recorded on Bruker AM-80 and Bruker AC-200 instruments with tetramethylsilane as internal standard. The purity of the synthesized compounds was checked by thin-layer chromatography on silica gel plates Kieselgel 60 F 254 (Merck). HPLC analysis was performed on a Hypersil C-8 column (5 pm, 25 cm) using a Pet&n-Elmer chromatograph equipped with a UV Perkin-Elmer LC 95 absorbance detector (h = 235 nm).
1-Phenylpiperazine hydrochloride was supplied by Hexa- chimie (France) whereas epichlorhydrin and cyanamide mono- sodium salt were obtained from Fluka (Switzerland).
Typical procedure for the preparation of 5-(1 -phenyl-4-pipera- zino)methyl2-amino-2-oxazoline 4a Path A. 18.5 g (0.2 mol) of epichlorhydrin was added dropwise at 40°C to a stirred mixture of 32.4 g (0.2 mol) of l- phenylpiperazine in 200 ml of ethanol. The reaction mixture was stirred for 6 h at room temperature. After addition of 8 g (0.2 mol) of sodium hydroxide in ethanol the mixture was stirred during 15 h at room temperature and filtered. After ethanol evaporation the residue was dissolved in ether and
filtered. The ether phase was concentrated and the crude l-(l- phenyl-4-piperazino)-2,3-epoxypropane 3a obtained as an oil was washed 3 times with hot heptane. 32.7 g (0.15 mol) of 3a diluted in 50 ml of methanol was added dropwise at 20°C to a stirred solution of cyanamide monosodium salt (9.6 g, 0.15 mol) in 200 ml of methanol. Stirring was maintained for 24 h. Then the mixture was concent&ed under reduced pressure. The residue was extracted with ether. After filtration, the organic phase was removed. The crude 2-amino-2- oxazoline was obtained as an oil and crystallized from tetra- chloroethylene to yield 12.5 g (32%) of 4a. mp 174°C; IR (KBr) 3410, 3290, 1685 cm-l; iH NMR 200 MHz (CDCl,) 6 ppm: 7.3-6.8 (m, 5H, phenyl); 4.8 (m, lH, C5-H); 4.13 (s, 2H, NH, exchange with D,O); 3.9, 3.4 (2dd, 2H, C-4-H, J = 13.5, 8.3, 3.5 Hz); 2.75, 2.5 (2dd, 2H, NW&HO, J = 12.2, 8.9, 7.5 Hz); 3.2-2.7 (m, 8H, CH,:piperazine). t3C NMR DMSO d6 6 ppm: C-2 160; C-4 56.6; C-5 76.4; NCH&-5 62.2. Path B. In a 6-l reactor were successively added under stirring 1.8 1 of 5 N NaOH 1200 g of I-phenylpiperazine hydrochloride (6.05 mol) and 1.8 1 of chloroform. The mixture was stirred for 1 h and the aqueous layer was separated. Then 1.8 1 of 2.5 N NaOH was added to the organic laver and biuhasic mixture was stirred for 1 h. The water rayer was separated and the organic layer washed with 1.8 1 of water and then concentrated, leading to an oil (975 g). The l-phenylpiperazine la was used as a crude oil. Dropwise addition of epichlorhydrin (615 g, 6.65 mol) on a solution of la (975 g, 6.02 mol) in 3 1 of ethanol was performed in 1 h. Then the mixture was stirred at room temperature for 24 h. The white solid part was filtered, washed with cold ethanol (0.3 1) and then dried under vacuum at 45°C for 8 h leading to 900 g of 1-chloro 3-(1-phenyl4-piperazino)- propan-2-01 2a (yield 59%); mp 89-90°C (ethanol); IR (KBr) 1580, 1240, 920 cm-t: tH NMR (CDCl,) 6 ppm: 7.5-6.7 (m, 5H, phenyl); 4 (m, lH, CHOH); 3.8-3.4 (m, 2H, CH,CI); 3.3-2.4 (m, 1 lH, NCH, and OH). Monosodium cyanamide salt (465 g, 6.84 mol), dry methanol (6.75 1) and 2a (900 g, 3.5 mol) were introduced in a reactor and stirred for 12 h at room temperature. The mineral salts were filtered off with Celite 535 (the filtering agent was first washed with 6 N HCl and then with water). The organic layer was concentrated and poured into ether (5 1) under stirring. The mixture was stirred for 1 h at room temperature. The solid phase was filtered and washed with ether (0.5 1). These ether layers were kept for further processing. The solid was dissolved in chloroform, the organic layer washed with water (0.5 1), dried and concentrat- ed. The crude mixture was then stirred in heptane (3 1) at room temperature for 1 h. The solid obtained was washed with heptane and dried under vacuum leading to a first crop of 4a. A second crop was obtained by filtration of the ether layers after their concentration (final yield 30%). Other title compounds were similarly prepared, analyzed and characterized by their elemental and spectral data (table I).
Pharmacology
The products were administered orally as suspensions in a 5% Tween 80 solution in distilled water. Experiments were carried out on male OFA Swiss mice weighing 20 to 26 g. The animals were kept at constant environmental temperature of 21 + 1°C and had free access to food and water. Acute toxicity was evaluated after oral administration (10 animals per dose) and LD,, value calculated.
Reserpine hypothermia interaction Before each experiment animal rectal temperature was recorded with a probe thermometer at a constant depth. The

442
References mice were divided into groups of 6 animals in such a way than the mean rectal temperature was the same in each group. According to Bourin et al [18], the hypothermia induced by reserpine (2.5 mg/kg ip) was noted 4 h after administration, The tested products were then administered and their effects assessed 60 min later.
Apomorphine hypothermia interaction Groups of 6 mice were placed in small individual boxes. Control and treated animals were administered 30 min before apomorphine (16 mg/kg, SC) and mice were isolated in boxes. To determine the percentage of hypothermic inhibition, rectal temperature was measured for each animal with a thermistor before all injections, and at the end of the experiment, ie, 30 min after apomorphine injection [ 131.
Interaction with yohimbine As in the schedule described by Quinton [ 141, yohiibine (30 mg/kg SC) induced on average 10% mortality in mice. Potentiation of this lethality was investigated by administration of tested compounds 30 min before yohimbine hydrochloride injection. The percentage of mortality was observed after 24 h and compared with controls.
Molecular modelling
MEP calculations and display Charge calculations were performed with the CNINDO/D program [19]. MEP calculations were performed with the VSS program [20] available in the CHEM-X package (developed and distributed by Chemical Design Ltd, Oxford, UK).
MLP calculations and display Calculations were performed with the algorithm nrouosed bv Audry et al [21]. The X values were taken from-B<oto et al [22]. The program was initially written by Croizet [23] and imnlemented in CHEM-X bv Tinland and Barbanton (un- published results).
.z
1
;
4
5
6
7 8
9
10
11 12
13
14 15 16
:i
19
;7
22
23
Dommisse C. De Vane CL (1985) Drug Int Clin Pharm 19,624628
~ , ”
Fuller RW (1986) Psychopharmacol Bull 22,825-828 Bourin M. Creuzet MH. Jarrv C. Collombel C (1988) Arzneim Forsch 38,666-668 d ’
. ,
Damaj MI, Trouvin JH, Lambrey B, Jacquot C (1990) J Pharm Sci 79,6,5 16-5 18 Jarry C, Golse R, Panconi E, Creuzet MH (1986) Eur J Med Chem 21,138-142 Luu-Due C, Beney C, Jarry C, Feniou C (1986) J Label Camp Radiopharm 24,1001-1003 Parker HE, fraacs NS (1959) Chem Rev 59,737-799 Jarrv C. Bose JJ. Ouhabi J. Carov A (1990) Arch Pharm 323: 157-161 Bourgeois G, Jarry C, Bose JJ, Deleris G, Pays E (1991) Orp Mass Suectr 26. 109-l 12 S&er IHI ‘ Rathbmr RC, Kattau R (1979) J Pharm Pharmacol25,108-110 Bourin M (1990) Fundam Clin Pharmacol4,49-64 Petit S. Nallet JP. Guillard M. Dreux J. Chermat R. Poncelet M, Bulacd C, Simon P (1990) Eur J Med Chem 25641-652 Puech AJ, Chermat R, Poncelet M, Doare L, Simon P (198 1) Psychopharmacology 75,84-91 Quinton RM (1963) Br J Pharmacol21,51-56 Simon P, Boissier JR (1975) Znt JMed Res 3, 14-17 Malick JB (1983) Drug Dev Res 3,357-363 Topliss JG (1972) JMed Chem 15,10061011 Bourin M, Poncelet M, Chermat R, Simon P (1982) J Pharmacol18,621-625 Chung-Phillips A (1989) J Comput Chem 10, 17-34 Giessner-Prettre C ( 1974) OCPE 11.249-26 1 Audry E, Dubost JP, Cdll~ter JC, Dallet P (1986) Eur J Med Chem 21,71-72 Broto P, Moreau G, Vandyke C (1984) Eur J Med Chem 19,79-84 Croizet F (1990) These de doctorat en Pharmacie, Universite de Bordeaux II






![Section 1. Identification de la substance/ du mélange et de ... · diéthyle 2-[[5- (3-éthoxy-1-éthoxycarbonyl-3-oxo-propyènel)amino]-1,3,3-triméthyle-cyclohexyle]méthylamino]butanedioate](https://img.pdfslide.fr/doc/110x75/5e47ef5fa5876c78da23daf7/section-1-identiication-de-la-substance-du-mlange-et-de-dithyle-2-5-.jpg)






















![Final version 3 - u-bordeaux1.frgrenet.drimm.u-bordeaux1.fr/pdf/2006/HUANG_CHI-HAO_2006.pdf2 nickel acetylacetonate P3HT poly(3-hexylthiophene) PCBM [6,6]-phenyl C61-butyric acid methyl](https://img.pdfslide.fr/doc/110x75/5e66813a46681f71863cc0b2/final-version-3-u-2-nickel-acetylacetonate-p3ht-poly3-hexylthiophene-pcbm-66-phenyl.jpg)