Embed Size (px)
Citation preview

Résumé : Le séquençage du gé-nome humain est à l’origine de ladécouverte de millions de varia-tions de séquence dans le génomehumain. Ces variations de sé-quence d’ADN sont dans leurimmense majorité limitées à despolymorphismes de nucléotideunique (Single Nucléotide Polymor-phisms ou SNP) ; cette forme cou-rante de polymorphisme se ren-contre environ toutes les 1 000 basesdans le génome humain et 1,8 mil-lion de SNP sont actuellementrépertoriés. L’accès à ces SNP dansles banques de données publiquesa ouvert la possibilité d’étudierl’influence de ces polymorphismesrépertoriés sur la prédisposition àcertaines maladies génétiques ainsique sur la réponse aux médica-ments. L’identification de ces nom-breux SNP tient beaucoup aux pro-grès des techniques d’analyse del’ADN et nous décrivons, dans cetterevue, différentes méthodes pou-vant être utilisées pour l’étude depolymorphismes. Les techniquesd’étude des SNP reposent sur deuxprincipes fondamentaux : d’unepart la création ou l’élimination parle SNP d’un site de coupure pourune enzyme de restriction donnée,et d’autre part la formation d’unmésappariement due à la présenced’une séquence variante lors d’unehybridation. La technique originelled’étude des SNP est le Southernblot couplé à une digestion enzy-matique permettant d’étudier lespolymorphismes de taille de frag-ments de restriction (RFLP). Ce pro-
cédé s’est largement simplifié parl’arrivée de la technique de réactionen chaîne par polymerase (PCR).D’autres approches telles que lachromatographie à haute perfor-mance en conditions dénaturantes(dHPLC) ou la PCR en temps réelpermettent une discrimination desallèles sauvages et variants. Enrevanche, le séquençage reste laseule technique permettant dedéterminer la nature et la positiondes SNP connus et inconnus. Enfin,l’apparition des techniques à hautdébit permet de prévoir des chan-gements d’échelle considérablesdans la recherche des SNP.
Mots clés : Polymorphismes d’unnucléotide (SNP) – Génotypage –RFLP – Discrimination allèle-spéci-fique – Séquençage
Techniques for SNP detection
Abstract: Sequencing the humangenome has allowed the discoveryof millions of DNA sequencevariants. Sequence variations inhuman DNA are mainly present asSingle Nucleotide Polymorphisms(SNPs); this common form of varia-tion is found about once every1,000 bases in the human genomeand 1.8 million SNPs have nowbeen identified and located. Theaccessibility of databases of SNPsopens the possibility of studyingthe influence of these polymor-phisms on disease risks as wellas on drug responses. Numerous
approaches have been set up forthe identification of SNPs. In thisreview we describe the main tech-niques used for the identification ofthese polymorphisms. They relyon two major consequences ofsequence variations: the apparitionor the disappearance of restrictionenzyme sites or the alteration ofDNA strand hybridization due to thepresence of a mismatch. Southernblotting and restriction endonu-cleases have allowed the develop-ment of the technique of restrictionfragment length polymorphisms(RFLPs), now performed on PCR pro-ducts. Several other approaches suchas denaturing high-performanceliquid chromatography or real-timePCR can detect allele differencesupon re-hybridization and hetero-duplex formation. However, DNAsequencing remains the obligatestep for the positive identification ofknown or unknown SNPs. At last,the development of high-throughputmethods allows a large increase inthe rate of discovery of SNPs likely.
Keywords: Single Nucleotide Poly-morphisms (SNPs) – Genotyping –RFLPs – Allele discrimination –Sequencing
Introduction
Il existe d’un individu à l’autre, dansles populations humaines, desvariations de séquence de l’ADNappelées polymorphismes qui peu-vent ne se traduire par aucune
OR
IG
IN
AL
7
Techniques de recherche des polymorphismes génétiques
V. Le Morvan1, J.-L. Formento2, G. Milano2, J. Bonnet1, J. Robert1
1 Laboratoire de Pharmacologie des Agents Anticancéreux, CNRS FRE 2618, Université Victor Segalen Bordeaux 2 et Institut Bergonié, 229, cours de l’Argonne, F-33076 Bordeaux Cedex, France2 Laboratoire d’Oncopharmacologie, Centre Antoine-Lacassagne, 33, avenue de Valombrose, F-06189 Nice Cedex 2,France
Correspondance : Dr V. Le Morvan, e-mail : [email protected]
Oncologie (2005) 7: 7-16© Springer 2005DOI 10.1007/s10269-005-0146-8

conséquence pathologique. Biensouvent ces variations n’entraînentaucune modification qualitative ouquantitative du produit du gène(quand, par exemple ces polymor-phismes impliquent des substitu-tions nucléotidiques affectant latroisième base d’un codon ou lesrégions non codantes de l’ADN).Dans d’autres cas, impliquant desvariations de la séquence codanteou des régions régulatrices, ellespeuvent entraîner une variationphénotypique, par exemple desmodifications de l’activité d’unenzyme ou la couleur des yeux.Cette variation naturelle de laséquence d’ADN, propriété fonda-mentale à la base de l’évolution desgénomes, est par ailleurs respon-sable des différences individuellesde prédisposition à certaines ma-ladies [7], ou de réponses indivi-duelles à des médicaments [21].
Les différents types
de polymorphisme
Les translocations, inversions,
délétions, insertions
Les premières observations de mo-dification de l’ADN ont été mises enévidence par des études cytogéné-tiques de cas pathologiques qui ontrévélé l’existence de remaniementschromosomiques plus ou moins im-portants. Au sein de certaines popu-lations, certains de ces remaniementschromosomiques interviennent àdes fréquences élevées et sont alorsconsidérés comme des polymor-phismes, mais les translocations, lesdélétions et insertions sont généra-lement la cause d’anomalies phéno-typiques graves [3]. Dans une régioncodante, les insertions ou délétionsd’un nombre de bases non multiplede trois entraînent une modificationdu cadre de lecture, à l’origine d’uneprotéine très altérée ou tronquée[25] ; si le nombre de bases inséréesou délétées est multiple de trois, il yaura addition ou perte d’acides ami-nés : c’est le cas de la délétion ∆F508de la mucoviscidose [20].
Les séquences répétées en chaîne
(en tandem)
Les séquences répétées en chaînesont fréquentes dans le génome ;
elles sont de taille variable, consti-tuées de répétitions d’un motif uni-taire de taille variable. Selon la tailledu motif et le nombre de répéti-tions, on distingue : 1) les satellites,pour lesquels le motif (long jusqu’àquelques centaines de paires debases) est répété un grand nombrede fois, de l’ordre de 100 000 [6] ;2) les minisatellites (ayant des mo-tifs de quelques dizaines de pairesde bases répétés jusqu’à quelquescentaines de fois), très polymor-phes dans leur séquence et leurnombre de répétitions [14] ; 3) lesmicrosatellites (Short TandemRepeats ou STR), comportant desséries de répétitions n’excédantpas le plus souvent quelques di-zaines, d’un motif de base court (de1 à 10 paires de bases) et répartissur l’ensemble du génome [16].
Les mutations ponctuelles
Les mutations ponctuelles sont lasource prépondérante des poly-morphismes dans la population.Un million huit cent mille SingleNucleotide Polymorphisms (SNP)ont été trouvés dans le génomehumain et sont disponibles dansdes banques de données spécia-lisées [27]. La modification la plussimple est le remplacement d’unebase par une autre. Dans le cas duremplacement d’une base puriquepar une autre base purique (A/G) oud’une base pyrimidique par uneautre base pyrimidique (C/T), onparle de transition. Les remplace-ments d’une base purique par unebase pyrimidique, ou réciproque-ment, sont appelés des transver-sions. Dans les régions codantes,ces substitutions peuvent entraînerune modification de la nature del’acide aminé codé en fonction deleur position dans le codon. Lesmutations de la première ou de laseconde base entraînent plus fré-quemment une substitution d’ami-noacide que les mutations de latroisième base en raison de ladégénérescence du code géné-tique. Ainsi, les mutations de la troi-sième base entraînent dans 72 %des cas des mutations silencieuses.
Les nouvelles techniques d’ana-lyse de l’ADN ont permis la mise en
évidence d’une très importante va-riabilité interindividuelle de l’ADNtant au niveau chromosomique(remaniements), qu’au niveau nu-cléotidique (mutations ponctuelles).Notre revue, sans être exhaustive,s’attachera à présenter les tech-niques les plus classiques d’ana-lyses des polymorphismes.
Techniques d’analyse
des polymorphismes
de type SNP
Les techniques d’analyse des SNPsont multiples. Elles sont réaliséessur de l’ADN génomique provenantde sang total (pour l’étude despolymorphismes constitutionnels)ou de tumeur (pour les polymor-phismes somatiques). Il faut diffé-rencier les méthodes d’étude despolymorphismes connus de cellesvisant à identifier des polymor-phismes encore inconnus.
Techniques d’analyse
des SNPs répertoriés
Les polymorphismes de taille de fragments de restriction ou RFLP (Restriction Fragment Length Polymorphism)On appelle polymorphisme de taillede fragments de restriction une va-riation individuelle de la séquencede l’ADN révélée par une modifica-tion de la carte de restriction (appa-rition ou disparition d’un site decoupure pour un enzyme de res-triction spécifique) [15]. Celle-ci estmise en évidence par la méthode deSouthern blot qui montre des diffé-rences individuelles dans la tailledes fragments obtenus après diges-tion par un enzyme de restriction etrévélation par une sonde. Un RFLPest donc défini par un couple enzymede restriction/sonde et correspond àun emplacement strictement définisur le génome : un locus génétique.Les différentes versions correspon-dant à un même emplacement surle génome sont exclusives les unesdes autres sur un même chromo-some, et correspondent à des allèles.Chez un hétérozygote, les deuxallèles sont visibles : les RFLP sontdonc des marqueurs co-dominants.
Cette technique nécessite l’iden-tification d’un site de restriction
ON
CO
LO
GI
E8
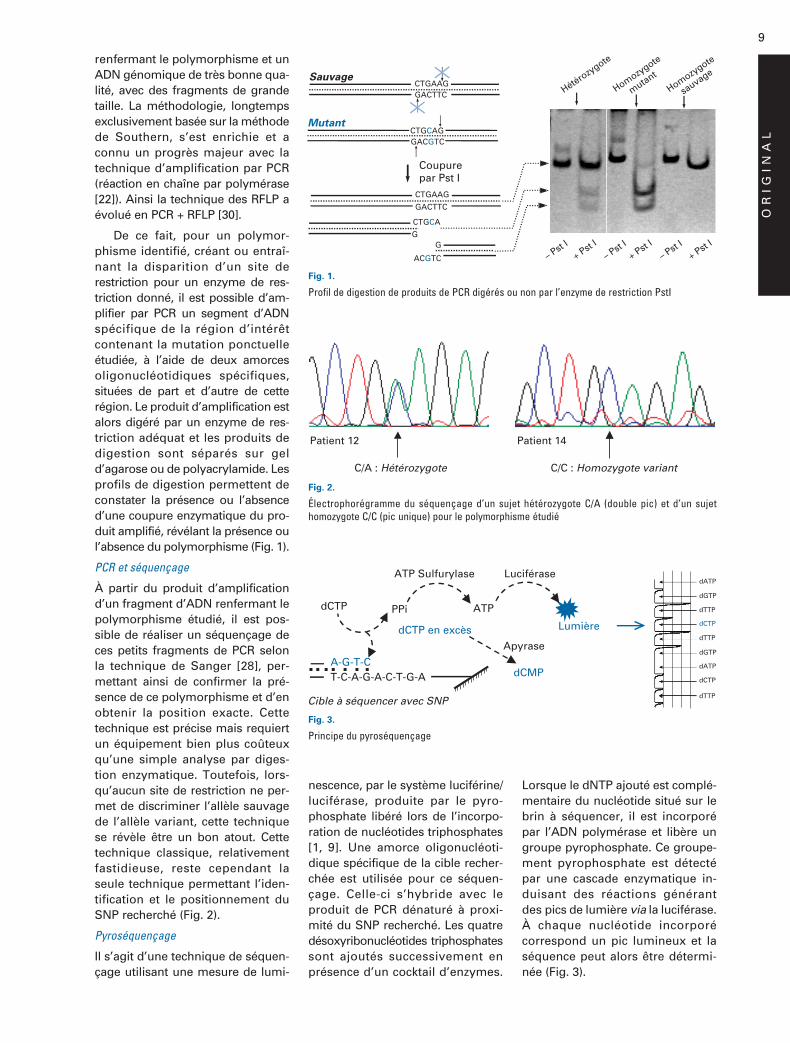
renfermant le polymorphisme et unADN génomique de très bonne qua-lité, avec des fragments de grandetaille. La méthodologie, longtempsexclusivement basée sur la méthodede Southern, s’est enrichie et aconnu un progrès majeur avec latechnique d’amplification par PCR(réaction en chaîne par polymérase[22]). Ainsi la technique des RFLP aévolué en PCR + RFLP [30].
De ce fait, pour un polymor-phisme identifié, créant ou entraî-nant la disparition d’un site derestriction pour un enzyme de res-triction donné, il est possible d’am-plifier par PCR un segment d’ADNspécifique de la région d’intérêtcontenant la mutation ponctuelleétudiée, à l’aide de deux amorcesoligonucléotidiques spécifiques,situées de part et d’autre de cetterégion. Le produit d’amplification estalors digéré par un enzyme de res-triction adéquat et les produits dedigestion sont séparés sur geld’agarose ou de polyacrylamide. Lesprofils de digestion permettent deconstater la présence ou l’absenced’une coupure enzymatique du pro-duit amplifié, révélant la présence oul’absence du polymorphisme (Fig. 1).
PCR et séquençage
À partir du produit d’amplificationd’un fragment d’ADN renfermant lepolymorphisme étudié, il est pos-sible de réaliser un séquençage deces petits fragments de PCR selonla technique de Sanger [28], per-mettant ainsi de confirmer la pré-sence de ce polymorphisme et d’enobtenir la position exacte. Cettetechnique est précise mais requiertun équipement bien plus coûteuxqu’une simple analyse par diges-tion enzymatique. Toutefois, lors-qu’aucun site de restriction ne per-met de discriminer l’allèle sauvagede l’allèle variant, cette techniquese révèle être un bon atout. Cettetechnique classique, relativementfastidieuse, reste cependant laseule technique permettant l’iden-tification et le positionnement duSNP recherché (Fig. 2).
Pyroséquençage
Il s’agit d’une technique de séquen-çage utilisant une mesure de lumi-
nescence, par le système luciférine/luciférase, produite par le pyro-phosphate libéré lors de l’incorpo-ration de nucléotides triphosphates[1, 9]. Une amorce oligonucléoti-dique spécifique de la cible recher-chée est utilisée pour ce séquen-çage. Celle-ci s’hybride avec leproduit de PCR dénaturé à proxi-mité du SNP recherché. Les quatredésoxyribonucléotides triphosphatessont ajoutés successivement enprésence d’un cocktail d’enzymes.
Lorsque le dNTP ajouté est complé-mentaire du nucléotide situé sur lebrin à séquencer, il est incorporépar l’ADN polymérase et libère ungroupe pyrophosphate. Ce groupe-ment pyrophosphate est détectépar une cascade enzymatique in-duisant des réactions générantdes pics de lumière via la luciférase.À chaque nucléotide incorporécorrespond un pic lumineux et laséquence peut alors être détermi-née (Fig. 3).
OR
IG
IN
AL
9
+ Pst I
GG
Hétérozygote
Homozygote
mutant
Homozygote
sauvage
+ Pst I
– Pst
I– P
st I
+ Pst I
– Pst
I
Sauvage
Mutant
Coupure par Pst I
CTGAAGGACTTC
CTGCAGGACGTC
CTGAAG
GACTTC
CTGCA
ACGTC
Fig. 1.
Profil de digestion de produits de PCR digérés ou non par l’enzyme de restriction PstI
Patient 12 Patient 14
C/A : Hétérozygote C/C : Homozygote variant
Fig. 2.
Électrophorégramme du séquençage d’un sujet hétérozygote C/A (double pic) et d’un sujethomozygote C/C (pic unique) pour le polymorphisme étudié
.... . . .
dATP
dGTP
dTTP
dCTP
dTTP
dGTP
dATP
dCTP
dTTP
ATP Sulfurylase Luciférase
dCTP PPi ATP
Apyrase
LumièredCTP en excès
Cible à séquencer avec SNP
dCMP T-C-A-G-A-C-T-G-AA-G-T-C
Fig. 3.
Principe du pyroséquençage

PCR et hybridation spécifique d’allèle
Le principe de cette techniquerepose sur le fait que le mésap-pariement d’une base lors d’unehybridation spécifique est suffisantpour déstabiliser l’hybride forméentre une sonde et une séquence-cible. La réussite de cette opérationrepose sur le choix de la sonde(spécifique de la zone où se trouvele SNP) qui doit permettre unebonne discrimination. Le principeest d’immobiliser la sonde spé-cifique de l’allèle sur un supportsolide ; on réalise ensuite unehybridation avec les échantillonsd’ADN marqués (radioactivité, fluo-rescence), amplifiés par PCR etdénaturés. On peut, inversement,immobiliser les produits de PCRet réaliser l’hybridation avec dessondes marquées. Différents lavagesfont suite et l’hybridation est visua-lisée par détection du marquage.Les lavages permettent d’éliminertout ce qui n’est pas parfaitementhybridé, et seule une hybridationparfaite du SNP recherché donneraun signal.
Différentes techniques ont vu lejour selon que l’on immobilise leproduit de PCR ou la sonde spéci-fique. De plus, les modes de détec-tion des marquages sont multipleset les plus utilisés sont la fluores-cence [11] ou la détection par Fluo-rescence Resonance Energy Trans-fer (FRET) [5]. Il est possible detravailler avec une sonde discrimi-nante (Fig. 4) ou deux sondes dis-criminantes, spécifiques de chacundes deux allèles. Dans ce derniercas, chaque sonde est marquéeavec un fluorochrome différent,permettant ainsi une simple ana-lyse de fluorescence vert-rouge(Fig. 5). Cette technique d’hybrida-tion de sondes discriminantes spé-cifiques d’allèles a été appliquéepour les SNP sur DNA-arrays et per-met un criblage à haut débit [2].
Extension d’amorce
L’extension d’oligonucléotides estune technique robuste de discrimi-nation allélique car une ADN poly-mérase, ne possédant pas d’acti-vité 3’ → 5’ exonucléasique, nepeut allonger un oligonucléotide
que si celui-ci est parfaitementcomplémentaire en 3’ de laséquence cible. Dans ce cas, selonl’amorce oligonucléotidique discri-minante utilisée, il y aura forma-tion ou non d’un produit d’amplifi-cation de l’ADN lors de la PCR [12](Fig. 6).
Mesure du point de fusion par PCR en temps réel (rt-PCR)
La technique de PCR en temps réel(rt-PCR) repose sur la détection et laquantification d’un marqueur fluo-rescent au cours de la réaction. Lesignal fluorescent est directementproportionnel à la quantité de pro-
ON
CO
LO
GI
E10
AG
C
AGC C
+
Révélation du marquage de la sonde (ex : Fluorescence)
Hybridation parfaite
Fig. 4.
Principe de l’hybridation spécifique d’allèle avec une sonde discriminante
G
C
G
C
A
T
A
+
T
Révélation différentielle de chacun des marquages des sondes
Hybridation parfaitede l’allèle sauvage
Hybridation parfaitede l’allèle variant
Fig. 5.
Principe de l’hybridation spécifique d’allèle avec deux sondes discriminantes
AG
C
+
GC C
A
GC C
A
Hybridation parfaite Mésappariement
Obtention d’un produit de PCR Aucun produit de PCR
Pas d’élongation possible
Fig. 6.
Principe de la technique d’extension d’amorces
duits de PCR générés. En mesurantl’intensité de fluorescence émiseà chaque cycle, il est possible desuivre la formation des produitsde PCR pendant la phase exponen-tielle (phase au cours de laquelle laquantité de produits amplifiés esten corrélation directe avec la quan-tité initiale de matrice). Les produitsd’amplification peuvent être détectésselon deux grands principes : parmarquage non spécifique avec desagents se liant à l’ADN double brin(Sybr Green I) et par marquagespécifique à l’aide de sondes fluo-rescentes (Sondes TaqMan oubalises moléculaires).
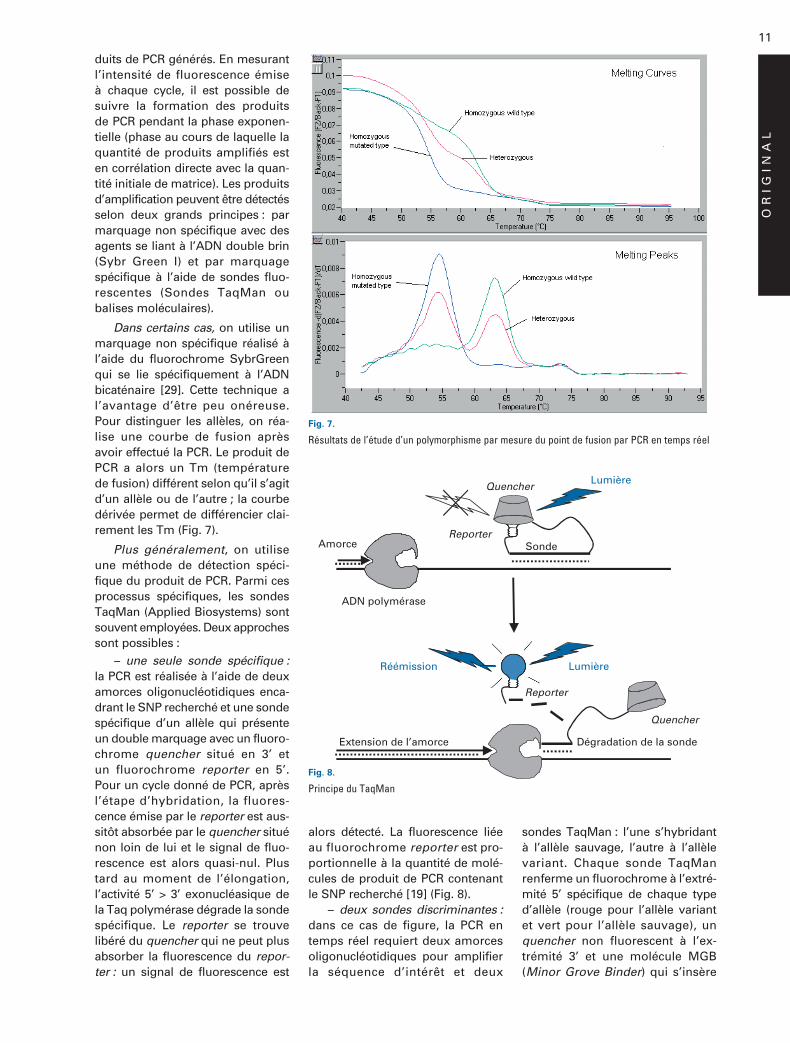
Dans certains cas, on utilise unmarquage non spécifique réalisé àl’aide du fluorochrome SybrGreenqui se lie spécifiquement à l’ADNbicaténaire [29]. Cette technique al’avantage d’être peu onéreuse.Pour distinguer les allèles, on réa-lise une courbe de fusion aprèsavoir effectué la PCR. Le produit dePCR a alors un Tm (températurede fusion) différent selon qu’il s’agitd’un allèle ou de l’autre ; la courbedérivée permet de différencier clai-rement les Tm (Fig. 7).
Plus généralement, on utiliseune méthode de détection spéci-fique du produit de PCR. Parmi cesprocessus spécifiques, les sondesTaqMan (Applied Biosystems) sontsouvent employées. Deux approchessont possibles :
– une seule sonde spécifique :la PCR est réalisée à l’aide de deuxamorces oligonucléotidiques enca-drant le SNP recherché et une sondespécifique d’un allèle qui présenteun double marquage avec un fluoro-chrome quencher situé en 3’ etun fluorochrome reporter en 5’.Pour un cycle donné de PCR, aprèsl’étape d’hybridation, la fluores-cence émise par le reporter est aus-sitôt absorbée par le quencher situénon loin de lui et le signal de fluo-rescence est alors quasi-nul. Plustard au moment de l’élongation,l’activité 5’ > 3’ exonucléasique dela Taq polymérase dégrade la sondespécifique. Le reporter se trouvelibéré du quencher qui ne peut plusabsorber la fluorescence du repor-ter : un signal de fluorescence est
alors détecté. La fluorescence liéeau fluorochrome reporter est pro-portionnelle à la quantité de molé-cules de produit de PCR contenantle SNP recherché [19] (Fig. 8).
– deux sondes discriminantes :dans ce cas de figure, la PCR entemps réel requiert deux amorcesoligonucléotidiques pour amplifierla séquence d’intérêt et deux
sondes TaqMan : l’une s’hybridantà l’allèle sauvage, l’autre à l’allèlevariant. Chaque sonde TaqManrenferme un fluorochrome à l’extré-mité 5’ spécifique de chaque typed’allèle (rouge pour l’allèle variantet vert pour l’allèle sauvage), unquencher non fluorescent à l’ex-trémité 3’ et une molécule MGB(Minor Grove Binder) qui s’insère
OR
IG
IN
AL
11
Fig. 7.
Résultats de l’étude d’un polymorphisme par mesure du point de fusion par PCR en temps réel
QuencherLumière
AmorceReporter
Sonde
ADN polymérase
Réémission Lumière
Reporter
Extension de l’amorce Dégradation de la sonde
Quencher
Fig. 8.
Principe du TaqMan

dans le petit sillon de la doublehélice formée par le duplex ADNcible – sonde spécifique et le stabi-lise. Du fait du rôle stabilisant de lamolécule MGB, les sondes TaqManpeuvent être plus courtes (12 à20 bases contre 15 à 40 bases).Cette stabilisation du complexeADN cible-sonde va de pair avec latempérature de fusion du complexe(Tm). Ainsi, le Tm d’une sonde par-faitement hybridée avec le produitde PCR sera plus élevé que celuid’une sonde s’hybridant imparfaite-ment. Ce phénomène est accentuépar l’utilisation du MGB qui vadéstabiliser un duplex imparfait etdonc faire chuter le Tm.
Au début de l’élongation, lessondes spécifiques d’allèle s’hybri-dent spécifiquement à la matrice,entre les deux amorces nécessairesà l’amplification. La proximité dufluorochrome et du quencher em-pêche la détection de fluorescence.Au cours de l’étape d’élongation dela PCR, la Taq polymérase dégradela sonde hybridée, ce qui entraîneune émission de fluorescence (cf. ci-dessus). La discrimination alléliquesera ensuite réalisée à l’aide d’un pro-gramme qui mesure la fluorescenceà chaque cycle de la PCR et qui per-met la discrimination entre les deuxallèles présents dans les échantil-lons testés. Une augmentation de lafluorescence rouge dans un échan-tillon indiquera une homozygotiepour l’allèle variant ; verte dans unautre échantillon, elle rendra comptede la présence d’un homozygotesauvage ; et à la fois dans le rouge etle vert, marquera la présence d’unindividu hétérozygote présentantchacun des deux allèles recherchés.
Les technologies de Fluores-cence Resonance Energy Transfer(FRET) [5] et des balises moléculai-res (molecular beacons, [33]) sonten pleine expansion. Le FRET reposesur l’utilisation de deux sondes, spé-cifiques du produit de PCR amplifié,qui s’hybrident en tandem sur l’am-plicon pendant l’étape d’hybridationde la PCR. La sonde située en amontdu tandem est marquée en 3’ avecun type de fluorochrome (f1) alorsque la sonde en 5’ est marquée àl’aide d’un fluorochrome reporter
distinct (f2). Ainsi, après l’hybrida-tion des deux sondes adjacentes,l’excitation du fluorochrome f1entraîne un transfert d’énergie aufluorochrome adjacent f2 qui, à sontour, émet un signal fluorescent quisera mesuré. Ce signal est propor-tionnel au nombre de molécules-cibles amplifiées (Fig. 9).
La technique des balises molé-culaires utilise des sondes parti-culières qui renferment trois seg-ments : deux séquences courtescomplémentaires portant respecti-vement un reporter et un quencher
en 5’ et 3’ et encadrant une sé-quence complémentaire du produitde PCR à détecter, renfermant leSNP. En absence de cible, la balisemoléculaire est en configurationfermée en épingle à cheveux, lereporter est proche du quencher,il n’y a pas de signal de fluores-cence. Lorsque la balise moléculaires’associe à sa cible (dans le casd’hybridation spécifique), sa struc-ture s’ouvre, le quencher et lereporter s’éloignent l’un de l’autre,une mesure de fluorescence estalors possible (Fig. 10).
ON
CO
LO
GI
E12
PCR : étape d’hybridation
Réémission Lumière
Matrice
AmorceSonde 1 Sonde 2
f2
Transfert
f1
Fig. 9.
Principe du FRET
Fig. 10.
Principe du fonctionnement des balises moléculaires
+
Séquence cible
Lumière
Quencher
Reporter
Balise moléculaire
Hybridation
LumièreRéémission
Reporter
Quencher

Ces différentes technologiesreposent toutes sur la différence deTm pouvant exister entre un homo-hybride ADN cible-sonde parfaite-ment hybridé et un hétérohybrideoù un mésappariement fait chuterle Tm. Dans la plupart des cas, unsimple mésappariement entre lacible mutée et la sonde cause undéplacement du Tm de fusion del’hybride d’environ 5 à 8 °C. Cepen-dant, ce déplacement dépend de lanature des séquences voisines dela zone mutée. Par exemple, unéchange G > A dans un environne-ment purique conduit à un déplace-ment de seulement 2 °C [26].
Techniques d’analyse
des SNPs inconnus
Ces techniques permettent dedéterminer les ADN présentant despolymorphismes (criblage) mais nedonnent pas de renseignement surla position de ce polymorphisme.Elles doivent toujours être complé-tées par un séquençage.
Le TILLING
Le TILLING (Targeting InducedLocal Lesions in Genomes) est unetechnique de criblage de SNPsinconnus et repose sur l’utilisationde l’endonucléase Cel I, issue ducéleri, qui clive les hétérohybridesd’ADN au niveau d’un mésapparie-ment. Cette approche permet uncriblage rapide car il est possible detravailler sur de longs fragments,ceux-ci pouvant être mélangés. Onpeut faire des mélanges, soit desADN extraits à tester, soit des pro-duits de PCR, amplifiés avec desamorces oligonucléotidiques spéci-fiques du locus contenant le SNP.Dans les deux cas, les échantillonsamplifiés sont dénaturés par chauf-fage puis renaturés pour générerdes hétérohybrides entre les ampli-cons variants (possédant le SNPvariant) d’une part, et les ampliconssauvages d’autre part. Les hétéro-hybrides sont alors digérés en pré-sence de l’endonucléase Cel I et lesproduits obtenus sont visualiséspar électrophorèse en gel dénatu-rant [32]. Les coupures peuventégalement être visualisées sur ungel de séquence si les amorcesde PCR initiales sont marquées par
deux fluorochromes différents [31](Fig. 11).
La PCR-SSCP (Single StrandConformation Polymorphisms)
Dans cette option, une PCR est réa-lisée pour amplifier la région d’inté-rêt. Le produit de PCR est ensuitedénaturé et soumis à une élec-trophorèse dans un gel de poly-acrylamide non dénaturant [24]. Unchangement de séquence corres-pondant à la présence d’une muta-tion ponctuelle, peut entraîner unchangement de la structure tertiairede l’ADN ; il en résulte alors une mo-bilité différente pour les fragmentsportants les différents allèles. Cesmutations sont détectées par l’appa-rition d’une nouvelle bande sur unautoradiogramme (détection radio-active) (Fig. 12). Une variante à cetteapproche est d’utiliser des amorcesfluorescentes lors de la PCR [13, 17],et d’analyser les produits résultantssur un automate de séquençaged’ADN. La détection d’une mutationpar PCR-SSCP est supérieure à80 % pour des fragments amplifiésinférieurs à 300 pb. Pour les frag-ments plus longs, cette techniqueest à éviter [10]. Un autre varianteà la PCR-SSCP a été décrite : la
REF-SSCP (Restriction endonu-clease fingerprinting), qui consisteen l’utilisation d’un mélange d’en-zymes de restriction non spéci-fiques des polymorphismes [18].Une fois ces PCR-SSCP réalisées,les produits de PCR doivent êtreséquencés afin de mettre en évi-dence le polymorphisme identifiéselon des critères de conformationstructurale.
La denaturing High Performance LiquidChromatography (dHPLC)
Le principe de la dHPLC repose surla formation d’hétérohybrides pourdes individus hétérozygotes pourun SNP [23]. Ces individus possè-dent, pour un SNP donné, un rap-port 1 : 1 entre ADN « sauvage » etADN « variant ». Après dénatura-tion des produits issus de la PCR,suivie d’une renaturation à 55 °C, ilse forme un mélange hétérohybride-homohybride. L’analyse de ce mé-lange par dHPLC est réalisée à unetempérature suffisante pour dé-naturer partiellement les hétéro-hybrides, lesquels sont élués plustôt de la colonne que les homo-hybrides, généralement sous laforme d’un double pic (Fig. 13). Afind’identifier le polymorphisme
OR
IG
IN
AL
13
C
AGA
A
G
TC
CT
( )
AT
GC
A GT C
GCT
A
GC TA
GT
GC
CA
TA
ADN sauvage ADN mutant
1. PCR avec oligonucléotides fluorescents
2. Dénaturation et renaturation
3. Coupure avec nucléase
4. Dénaturation
Cel I
5. Séparation sur gel de séquence lecture à deux longueurs d’onde
500 pb
300 pb
200 pb
700 nm 800 nm
homohybrides
hétérohybrides
Fig. 11.
Principe de la technique de TILLING

visualisé par la présence d’un hétéro-hybride, un séquençage est néces-saire. Soulignons que cette tech-nique ne permet pas de distinguerdirectement les sujets homozy-gotes variants des sujets homozy-gotes sauvages.
Le SNP mining
Cette technique repose sur l’utilisa-tion de la protéine mutS de E. coli,marquée par un fluorophore, ca-pable de détecter des mésapparie-ments [2] (brevet déposé par lasociété Nanogen San Diego, CA).Les régions d’ADN sauvage, ampli-fiées par PCR, sont immobiliséessur un support par microélectro-nique. Après dénaturation des pro-duits de PCR, les échantillons àtester sont hybridés sur ces supportsen présence de la protéine mutSqui reconnaît tout mésappariement,témoignant que l’échantillon testéprésente une variation par rapport àl’ADN sauvage (Fig. 14). Le produitde PCR testé est alors séquencépour identifier le nouveau poly-morphisme.
Le séquençage
Le séquençage constitue égale-ment une technique de recherchede nouveaux polymorphismes.Jusqu’à la mise en place de la PCR,ce procédé était très lourd car ilnécessitait un clonage patho-logique. La technique d’amplifica-tion par PCR directement couplée auséquençage constitue une simplifi-cation méthodologique de grandeimportance.
Les polymorphismes
de type microsatellite
(Simple Sequence Length
Polymorphism, SSLP)
Les microsatellites sont des mar-queurs très intéressants dont onpeut caractériser les variations parmesure de la taille après une PCR.Chaque SSLP est en effet carac-térisé par un nombre variable derépétitions de di-, tri- ou tétra-nucléotides à un endroit précis etgénère des fragments de PCR detailles variables lors de l’amplifica-tion par PCR à l’aide de sondes quiencadrent la région variante.
ON
CO
LO
GI
E14
AT
GC
GTC
A
AT C
G
TC
A
AT
GC
Allèle sauvage Allèle mutant
Dénaturation, Renaturation
G
Incubation à 55 °C
Élution à 55 °CSéquençage
Identification du SNP
Fig. 12.
Principe de la PCR-SSCP
Fig. 13.
Principe de la technique de la dHPLC et résultats d’un chromatogramme
Produit de PCRADN sauvage
Produit de PCR ADN variant
1. Dénaturation
2. Refroidissement
3. Gel non dénaturant
Mobilité différente : apparition d’une bande
ADNsauvage
ADN patient 2
ADN patient 1 Protéine SNP mine Cy5
ADN patient 2
Séquençage
Identification du SNPFig. 14.
Principe de la technique de SNP mining
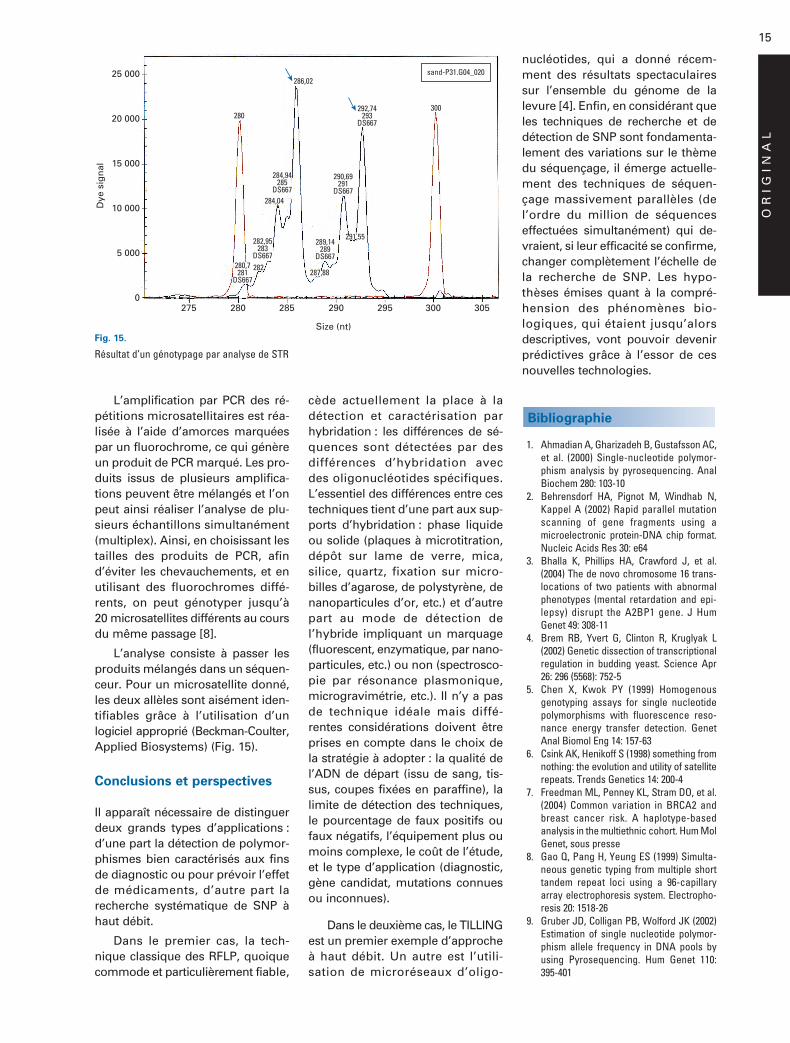
L’amplification par PCR des ré-pétitions microsatellitaires est réa-lisée à l’aide d’amorces marquéespar un fluorochrome, ce qui génèreun produit de PCR marqué. Les pro-duits issus de plusieurs amplifica-tions peuvent être mélangés et l’onpeut ainsi réaliser l’analyse de plu-sieurs échantillons simultanément(multiplex). Ainsi, en choisissant lestailles des produits de PCR, afind’éviter les chevauchements, et enutilisant des fluorochromes diffé-rents, on peut génotyper jusqu’à20 microsatellites différents au coursdu même passage [8].
L’analyse consiste à passer lesproduits mélangés dans un séquen-ceur. Pour un microsatellite donné,les deux allèles sont aisément iden-tifiables grâce à l’utilisation d’unlogiciel approprié (Beckman-Coulter,Applied Biosystems) (Fig. 15).
Conclusions et perspectives
Il apparaît nécessaire de distinguerdeux grands types d’applications :d’une part la détection de polymor-phismes bien caractérisés aux finsde diagnostic ou pour prévoir l’effetde médicaments, d’autre part larecherche systématique de SNP àhaut débit.
Dans le premier cas, la tech-nique classique des RFLP, quoiquecommode et particulièrement fiable,
cède actuellement la place à ladétection et caractérisation parhybridation : les différences de sé-quences sont détectées par desdifférences d’hybridation avecdes oligonucléotides spécifiques.L’essentiel des différences entre cestechniques tient d’une part aux sup-ports d’hybridation : phase liquideou solide (plaques à microtitration,dépôt sur lame de verre, mica,silice, quartz, fixation sur micro-billes d’agarose, de polystyrène, denanoparticules d’or, etc.) et d’autrepart au mode de détection del’hybride impliquant un marquage(fluorescent, enzymatique, par nano-particules, etc.) ou non (spectrosco-pie par résonance plasmonique,microgravimétrie, etc.). Il n’y a pasde technique idéale mais diffé-rentes considérations doivent êtreprises en compte dans le choix dela stratégie à adopter : la qualité del’ADN de départ (issu de sang, tis-sus, coupes fixées en paraffine), lalimite de détection des techniques,le pourcentage de faux positifs oufaux négatifs, l’équipement plus oumoins complexe, le coût de l’étude,et le type d’application (diagnostic,gène candidat, mutations connuesou inconnues).
Dans le deuxième cas, le TILLINGest un premier exemple d’approcheà haut débit. Un autre est l’utili-sation de microréseaux d’oligo-
nucléotides, qui a donné récem-ment des résultats spectaculairessur l’ensemble du génome de lalevure [4]. Enfin, en considérant queles techniques de recherche et dedétection de SNP sont fondamenta-lement des variations sur le thèmedu séquençage, il émerge actuelle-ment des techniques de séquen-çage massivement parallèles (del’ordre du million de séquenceseffectuées simultanément) qui de-vraient, si leur efficacité se confirme,changer complètement l’échelle dela recherche de SNP. Les hypo-thèses émises quant à la compré-hension des phénomènes bio-logiques, qui étaient jusqu’alorsdescriptives, vont pouvoir devenirprédictives grâce à l’essor de cesnouvelles technologies.
1. Ahmadian A, Gharizadeh B, Gustafsson AC,et al. (2000) Single-nucleotide polymor-phism analysis by pyrosequencing. AnalBiochem 280: 103-10
2. Behrensdorf HA, Pignot M, Windhab N,Kappel A (2002) Rapid parallel mutationscanning of gene fragments using amicroelectronic protein-DNA chip format.Nucleic Acids Res 30: e64
3. Bhalla K, Phillips HA, Crawford J, et al.(2004) The de novo chromosome 16 trans-locations of two patients with abnormalphenotypes (mental retardation and epi-lepsy) disrupt the A2BP1 gene. J HumGenet 49: 308-11
4. Brem RB, Yvert G, Clinton R, Kruglyak L(2002) Genetic dissection of transcriptionalregulation in budding yeast. Science Apr26: 296 (5568): 752-5
5. Chen X, Kwok PY (1999) Homogenousgenotyping assays for single nucleotidepolymorphisms with fluorescence reso-nance energy transfer detection. GenetAnal Biomol Eng 14: 157-63
6. Csink AK, Henikoff S (1998) something fromnothing: the evolution and utility of satelliterepeats. Trends Genetics 14: 200-4
7. Freedman ML, Penney KL, Stram DO, et al.(2004) Common variation in BRCA2 andbreast cancer risk. A haplotype-basedanalysis in the multiethnic cohort. Hum MolGenet, sous presse
8. Gao Q, Pang H, Yeung ES (1999) Simulta-neous genetic typing from multiple shorttandem repeat loci using a 96-capillaryarray electrophoresis system. Electropho-resis 20: 1518-26
9. Gruber JD, Colligan PB, Wolford JK (2002)Estimation of single nucleotide polymor-phism allele frequency in DNA pools byusing Pyrosequencing. Hum Genet 110:395-401
Bibliographie
OR
IG
IN
AL
15
25 000
20 000
15 000
10 000
5 000
0
Dye
sig
nal
Size (nt)
275 280 285 290 295 300 305
280300
291,55
280,7 281
DS667
282,95 283
DS667
284,94 285
DS667
292,74 293
DS667
290,69 291
DS667
289,14 289
DS667282
284,04
287,88
286,02sand-P31.G04_020
Fig. 15.
Résultat d’un génotypage par analyse de STR

10. Hayashi K, Yandell DW (1993) How sensi-tive is PCR-SSCP? Hum Mutat 2: 338-46
11. Howell WM, Jobs M, Gyllensten U,Brookes AJ (1999) Dynamic allele-specifichybridization: a new method for scoringsingle nucleotide polymorphisms. Nat Bio-technol 17: 87-8
12. Hsu TM, Kwok PY (2003) Homogeneousprimer extension assay with fluorescencepolarization detection. Methods Mol Biol212: 177-87
13. Iwahana H, Fujimura M, Takahashi Y, et al.(1996) Multiple fluorescence-based PCR-SSCP analysis using internal fluorescentlabelling of PCR products. Biotechniques21: 510-4
14. Jarman AP, Wells RA (1989) Hypervariableminisatellites: recombinaison or innocentbystanders? Trends Genet 5: 367-71
15. Kan YW, Dozy AM (1978) Polymorphism ofDNA sequence adjacent to human beta-globin structural gene: relationship tosickle mutation. Proc Natl Acad Sci USA75: 5631-5
16. Kartin S, Burge C (1995) Dinucleotide rela-tive abundance extremes: a genomicssignature. Trends Genet 11: 283-90
17. Larsen LA, Christiansen M, Vuust J, An-dersen PS (1999) High-throughput single-strand conformation polymorphism analysisby automated capillary electrophoresis:robust multiplex analysis and pattern-based identification of allelic variants. HumMutat 13: 318-27
18. Liu Q, Sommer SS (1995) Restriction endo-nuclease fingerprinting (REF): as sensitivemethod for mutations in long continuoussegments of DNA. Biotechniques 18: 470-7
19. Livak KJ, Flood SJ, Marmaro J, et al. (1995)Oligonucleotides with fluorescent dyes atopposite ends provide a quenched probesystem useful for detecting PCR productand nucleic acid hybridization. PCRMethods Appl 4: 357-62
20. Lucotte G, Hazout S, De Braekeleer M(1995) Complete map of cystic fibrosismutation ∆F508 frequencies in WesternEurope and correlation between mutationfrequencies and incidence of disease.Hum Biol 67: 797-803
21. McLeod HL, Evans WE (2001) Pharmaco-genomics: unlocking the human genomefor better drug therapy. Ann Rev Pharma-col Toxicol 41: 101-21
22. Mullis K, Faloona F, Scharf S, et al. (1986)Specific enzymatic amplification of DNA invitro: the polymerase chain reaction. ColdSpring Harbor Symp Quant Biol 51: 263-73
23. Oefner PJ (2000) Allelic discrimination bydenaturing high-performance liquid chro-matography. J Chromatogr B 739: 345-55
24. Orita M, Suzuki Y, Sekiya T, Hayashi K(1989) Rapid and sensitive detection ofpoint mutations and DNA polymorphismsusing the polymerase chain reaction.Genomics 5: 874-9
25. Puffenberger EG, Hu-Lince D, Parod JM,et al. (2004) Mapping of sudden infantdeath with dysgenesis of the testes syn-drome (SIDDT) by a SNP genome scan andidentification of TSPYL loss of function.Proc Natl Acad Sci USA 101: 11689-94
26. Ririe KM, Rasmussen RP, Wittwer CT(1997) Product differentiation by analysis ofDNA melting curves during the polymerasechain reaction. Anal Biochem 245: 145-60
27. Sachidanandam R, Weissman D, SchmidtSC, et al. (2001) A map of human genomesequence variation containing 1.42 millionsingle nucleotide polymorphisms. Nature409: 928-33
28. Sanger F (1981) Determination of nucleotidesequences in DNA. Science 214: 1205-10
29. Schneeberger C, Speiser P, Kury F, Zeil-linger R (1995) Quantitative detection ofreverse transcriptase-PCR products bymeans of a novel and sensitive DNA stain.PCR Methods Appl 4: 234-8
30. Steinhelper ME, Field LJ (1992) Assignmentof the angiogenin gene to mouse chromo-some 14 using a rapid PCR-RFLP mappingtechnique. Genomics 12: 177-9
31. Stemple DL (2004) TILLING. A high-through put harvest for functional geno-mics. Nat Rev Genet 5: 145-50
32. Till BJ, Burtner C, Comai L, Henikoff S(2004) Mismatch cleavage by single-strandspecific nucleases. Nucleic Acids Res 32:2632-41
33. Tyagi S, Kramer FR (1996) Molecular bea-cons: probes that fluoresce upon hybridi-zation. Nat Biotechnol 14: 303-8
ON
CO
LO
GI
E16





























