Embed Size (px)
Citation preview

doi:10.1006/jmbi.2000.3565 available online at http://www.idealibrary.com on J. Mol. Biol. (2000) 297, 481±500
The Crystal Structure of D-Glyceraldehyde-3-phosphate Dehydrogenase from theHyperthermophilic Archaeon Methanothermusfervidus in the Presence of NADP� at 2.1 AÊ Resolution
C. Charron1, F. Talfournier2, M.N. Isupov3, J.A. Littlechild3, G. Branlant2
B. Vitoux1* and A. Aubry1*
1Laboratoire de Cristallographieet ModeÂlisation des MateÂriauxMineÂraux et Biologiques -Groupe Biocristallographie -UPRESA CNRS 7036,Universite Henri PoincareÂNancy I, BP 23954506 Vandoeuvre-leÁs-NancyFrance2Maturation des ARN etEnzymologie MoleÂculaire -UMR CNRS 7567, UniversiteÂHenri PoincareÂ, Nancy I, BP239, 54506, Vandoeuvre-leÁs-Nancy, France3Schools of Chemistry andBiological Sciences, Universityof Exeter, Stocker Road, ExeterEX4 4QD, England
E-mail addresses of the correspo
Abbreviations used: GAPDH, glyrmsd, root-mean-square deviation;
0022-2836/00/010481±20 $35.00/0
The crystal structure of the glyceraldehyde-3-phosphate dehydrogenase(GAPDH) from the archaeon Methanothermus fervidus has been solved inthe holo form at 2.1 AÊ resolution by molecular replacement. Unlike bac-terial and eukaryotic homologous enzymes which are strictly NAD�-dependent, GAPDH from this organism exhibits a dual-cofactor speci-®city, with a marked preference for NADP� over NAD�. The presentstructure is the ®rst archaeal GAPDH crystallized with NADP�. GAPDHfrom M. fervidus adopts a homotetrameric quaternary structure which istopologically similar to that observed for its bacterial and eukaryoticcounterparts. Within the cofactor-binding site, the positively chargedside-chain of Lys33 decisively contributes to NADP� recognition througha tight electrostatic interaction with the adenosine 20-phosphate group.Like other GAPDHs, GAPDH from archaeal sources binds the nicotin-amide moiety of NADP� in a syn conformation with respect to the adja-cent ribose and so belongs to the B-stereospeci®c class ofoxidoreductases. Stabilization of the syn conformation is principallyachieved through hydrogen bonding of the carboxamide group with theside-chain of Asp171, a structural feature clearly different from what isobserved in all presently known GAPDHs from bacteria and eukaryotes.Within the catalytic site, the reported crystal structure de®nitively con-®rms the essential role previously assigned to Cys140 by site-directedmutagenesis studies. In conjunction with new mutation results reportedin this paper, inspection of the crystal structure gives reliable evidencefor the direct implication of the side-chain of His219 in the catalyticmechanism. M. fervidus grows optimally at 84 �C with a maximal growthtemperature of 97 �C. The paper includes a detailed comparison of thepresent structure with four other homologous enzymes extracted frommesophilic as well as thermophilic organisms. Among the variousphenomena related to protein thermostabilization, reinforcement of elec-trostatic and hydrophobic interactions as well as a more ef®cient molecu-lar packing appear to be essentially promoted by the occurrence of twoadditional a-helices in the archaeal GAPDHs. The ®rst one, named a4, islocated in the catalytic domain and participates in the enzyme architec-ture at the quaternary structural level. The second one, named aJ, occursat the C terminus and contributes to the molecular packing within eachmonomer by ®lling a peripherical pocket in the tetrameric assembly.
# 2000 Academic Press
Keywords: glyceraldehyde-3-phosphate dehydrogenase; NADP; crystalstructure; Archaea; Methanothermus fervidus
*Corresponding authorsnding authors: [email protected]; [email protected]
ceraldehyde-3-phosphate dehydrogenase; D-G3P, D-glyceraldehyde-3-phosphate;T�opt, optimal temperature of growth.
# 2000 Academic Press

482 Archaeal GAPDH in Complex with NADP
Introduction
A current view is that the living world is pro-posed to be divided into three domains Archaea,Bacteria, and Eucarya (Woese et al., 1990). Mem-bers of Archaea possess various unusual proper-ties such as the ability to grow under extremeenvironmental conditions of temperature, press-ure, or salinity (Barns et al., 1994; DeLong et al.,1994; Stetter et al., 1993). Archaea are subdividedinto the two kingdoms Euryarchaeota and Cre-narchaeota. The thermoacidophiles, chemo- andorganolithotrophs, and heterotrophs are includedin the Crenarchaeota. The kingdom Euryarchaeo-ta includes the hyperthermophilic Thermococ-cales, the Archaeoglobales, and all of themethanogens and obligate halophiles. Structuralstudies on archaeal enzymes may reveal evol-utionary relationships between proteins from thethree domains, and may advance our under-standing of the molecular basis of the adaptationto extreme environmental conditions.
Methanothermus fervidus is an archaeon which isincluded in the kingdom Euryarchaeota. The opti-mal temperature of growth (T�opt) of this organ-ism is 84 �C, with a maximal growth temperatureof 97 �C (Fabry & Hensel, 1987). Unlike bacterialand eukaryotic glycolytic glyceraldehyde-3-phos-phate dehydrogenases (GAPDH; EC 1.2.1.12)which are strictly NAD�-dependent, GAPDH fromM. fervidus is an enzyme which exhibits a dual-cofactor speci®city, but with a marked preferencefor NADP� over NAD�. This tetrameric protein ofMr 150,000 contains four identical monomers eachcomposed of 337 amino acid residues (Fabry &Hensel, 1988). On the basis of site-directed muta-genesis experiments (Talfournier et al., 1998, 1999),Cys140 from M. fervidus GAPDH has been pro-posed to act as an essential residue during the cata-lytic mechanism by playing a role similar to that ofCys149 in bacterial and eukaryotic GAPDHs.M. fervidus GAPDH shows only 7-16 % sequenceidentity with the GAPDHs from Bacteria andEucarya (Arcari et al., 1993). Deduced from eitherprotein or gene sequencing, nine primary struc-tures of GAPDHs from Archaea are available sofar (Figure 1).
Crystal structures have been solved for GAPDHenzymes isolated from a number of eukaryotic andbacterial species including American lobster(Moras et al., 1975), Chinese lobster (Song et al.,1998), man (Mercer et al., 1976), parasites such asTrypanosoma brucei (Vellieux et al., 1993), Leishmaniamexicana (Kim et al., 1995) and Trypanosoma cruzi(Souza et al., 1998), bacteria such as mesophilicEscherichia coli (DueÂe et al., 1996) and Bacillus coagu-lans (Grif®th et al., 1983), moderately thermophilicBacillus stearothermophilus (Skarzynski et al., 1987),thermophilic Thermus aquaticus (Tanner et al., 1996)and hyperthermophilic Thermotoga maritima(KorndoÈrfer et al., 1995). More recently, preliminarycrystallographic analysis of GAPDHs fromSaccharomyces cerevisiae (Gilboa et al., 1998) and
from spinach chloroplasts (Sabatino et al., 1999)have been reported. Moreover, the three-dimen-sional structure of the archaeal GAPDH from Sulfo-lobus solfataricus, which shows 49 % identity withM. fervidus GAPDH, was recently solved withoutbound cofactor (Fleming et al., 1998; Isupov et al.,1999).
Here we report the detailed crystal structure ofGAPDH from M. fervidus in complex with NADP�
at 2.1 AÊ resolution. As described in a preliminarypaper (Charron et al., 1999), crystals were obtainedunder conditions differing from those previouslyused by Fabry et al. (1988). The GAPDH structurenot only exhibits the classical Rossmann fold, butalso reveals extensive structural homology withthe known GAPDH structures in regions of the cat-alytic domain. More interestingly, the M. fervidusGAPDH shares additional structural propertieswith only the S. solfataricus enzyme, which couldbe relevant to their increased thermostability withrespect to the mesophilic counterparts. In thispaper, the M. fervidus GAPDH is compared withfour other GAPDH enzymes: those from themesophile E. coli (T�opt 37 �C), the moderatethermophile B. stearothermophilus (T�opt 65 �C), thehyperthermophile T. maritima (T�opt 80 �C), andthe hyperthermophile S. solfataricus (T�opt 87 �C).Moreover, structural features responsible for therecognition of NADP� are highlighted and essen-tial residues implicated in the enzymatic activityare tentatively identi®ed.
Results
Residue numbering used here for archaealenzymes corresponds to the M. fervidus GAPDHsequence (Figure 1). The ®nal model of M. fervidusGAPDH is made up of one tetramer in the asym-metric unit. Because of the lack of electron densityfor the C-terminal residue, each monomer containsresidues 1-336, one NADP� and one sulphate ion.These monomers are named O, P, Q and R, follow-ing the naming scheme of the B. stearothermophilusGAPDH model. The asymmetric unit also contains960 water molecules.
Model quality
Although the re®nement process employed nonon-crystallographic symmetry restraints, confor-mations of all four subunits in the ®nal re®ned tet-ramer OPQR are very similar. Using the P subunitas a reference, the root-mean-square deviations(rmsd) of backbone atoms after superimposition ofthe O, Q, and R subunits are 0.280 AÊ , 0.210 AÊ and0.203 AÊ , respectively. Likewise, conformations ofthe bound NADP� molecules are very similar,since superimpositions of the cofactors from the O,Q, and R subunits onto the P subunit NADP�
result in rmsd of 0.133 AÊ , 0.165 AÊ and 0.098 AÊ ,respectively.
The model has tight stereochemical restraintswith rmsd on bond lengths, 0.010 AÊ , and on bond

Figure 1. Primary structure alignment of presently known GAPDH sequences from Archaea based on the 99.1release of the PRODOM database (Sonnhammer & Kahn, 1994) available on http://protein.toulouse.inra.fr/pro-dom.html The S. solfataricus GAPDH sequence published by Arcari et al. (1993) was modi®ed in accordance with sub-sequent data published by Jones et al. (1995). Sequence numbering refers to the M. fervidus enzyme and invariantresidues are shown in bold. The GAPDH primary structure from the halophilic archaeon Haloarcula vallismortis(Brinkmann & Martin, 1996) was not included in the alignment, due to its closer relationship to sequences of bacterialand eukaryotic GAPDHs. Abbreviations: A. f., Archaeoglobus fulgidus; P. w., Pyroccus woesei; P. h., Pyrococcus horikoshii;Mb. b., Methanobacterium bryantii; Mb. f., Methanobacterium formicicum; Mb. t., Methanobacterium thermoautotrophicum;M. f., Methanothermus fervidus; Mc. j., Methanococcus jannashii; S. s., Sulfolobus solfataricus.
Archaeal GAPDH in Complex with NADP 483
angles, 2.342 �. Averaged B-factors are 37.56 AÊ 2 forthe protein chain, 36.09 AÊ 2 for NADP�, 60.65 AÊ 2
for sulphate ions, 51.67 AÊ 2 for water molecules and38.74 AÊ 2 for the whole model. The vast majority ofresidues (91.5 %) lie in the most favoured regionsof the Ramachandran plot (Figure 2), as de®ned byPROCHECK (Laskowski et al., 1993). Only Asp289of each monomer has unfavourable main-chain tor-sion angles as found in the S. solfataricus GAPDH(Isupov et al., 1999). Located in the i � 1 position ofa bII0 turn (Gunasekaran et al., 1996; Hutchinson &Thornton, 1994), that connects two b-strands, thisresidue seems to be characteristic of archaealGAPDHs since it is conserved in enzymes fromMethanobacterium bryantii, Methanobacterium formici-cum, Methanobacterium thermoautotrophicum, andS. solfataricus (Figure 1). As it was observed in theS. solfataricus GAPDH structure, Pro179, Pro187and Pro191 in each subunit adopt the cis confor-mation in the X-Pro preceding bond.
Monomer structure
As in the case of the GAPDH from S. solfataricus(Isupov et al., 1999), the overall structure of themonomer from M. fervidus consists of two foldingdomains and is similar to that previously describedfor bacterial and eukaryotic GAPDHs. Archaealmonomers of GAPDHs from M. fervidus and S. sol-fataricus show an rmsd value of superimposed Ca
positions of 1.043 AÊ (Figure 3).The NAD(P)-binding domain is essentially com-
posed of residues 1 to 138 and has an a/b foldingpattern characteristic of the Rossmann fold (Lesk,1995; Schulz, 1992), with a central parallel b-sheetcomposed of six canonical strands, named bA tobF. This structural core is completed on both edgesby small extra strands named bC0 and bE0 and¯anked on both sides by seven a-helices, namedaA to aG. The cofactor-binding domain is com-pleted by residues 300 to 336, which include twoa-helices named aH and aJ. They connect the

Figure 2. Ramachandran dia-gram of the distribution of dihedralangles phi and psi for the ®nalstructure of M. fervidus GAPDH (Pmonomer). Glycine residues arerepresented by triangles, all otherresidues by squares. Darkenedareas correspond to the energeti-cally most favourable phi,psiangles. The single residue Asp289with disallowed phi,psi angles islabelled. The Figure was drawnwith PROCHECK (Laskowski et al.,1993).
484 Archaeal GAPDH in Complex with NADP
carboxy-terminal end of the monomer to both theamino-terminal and the carboxy-terminal parts ofregion 1-138 via hydrogen bonds and salt bridges(Table 1). In this NAD(P)-binding domain, mosthydrophobic residues are organized into two mainclusters, one on each side of the central b-sheet.The larger hydrophobic cluster includes 29 resi-dues and the other one 13 residues. Moreover, asmall hydrophobic bridge composed of 12 residuesis connected to both clusters mentioned above viaVal72 and Val31.
The catalytic domain comprises a mixed b-sheetof eight strands (b1 to b8) and four a-helices (a1 toa4). Besides hydrogen bond and salt-bridge net-works (Table 1), the catalytic domain contains twohydrophobic clusters. The larger one includes 29residues and is located in the core of the domain,between the large helix a1 and the central b-sheet.The small hydrophobic cluster includes 12 residuesand lies on the other side of the b-sheet.
Unlike the structures of known GAPDHs frombacterial and eukaryotic origins, salt bridges occurbetween residues from the catalytic domain andresidues from the NAD(P)-binding domain(Table 1).
Quaternary structure
The M. fervidus GAPDH is a tetramer withapproximate 222 symmetry. The solvent-accessiblesurface of the tetramer is 44,000 AÊ 2 whereas thetheoretical solvent-accessible area of a single mono-mer would be 14,340 AÊ 2. So, about 24 % of the sub-
unit surface is buried within the core of thetetramer.
The P/O (Q/R) interface is the largest one witha surface area of 1650 AÊ 2. Fifteen hydrogen bondsare formed across this interface, including ®ve saltbridges (Table 2). Besides hydrogen bonds and saltbridges, this interface contains a large hydrophobiccluster where nine residues (Val164, Val166, Ile183,Leu190, Met209, Val211, Val213, Met224 andMet295) of one subunit interact with their counter-parts in the other subunit. These residues arelocated in strands b2, b3, b4 and b8, and in theS-loop.
The P/R (O/Q) interface area is 1320 AÊ 2. Thir-teen hydrogen bonds are formed across this inter-face, including nine salt bridges (Table 3), whichform a network between both subunits. This net-work involves three residues from helices aA andaB of P subunit and ®ve residues from R subunitwhich are located in a fragment centred on thehelix a4. A few hydrophobic contacts are alsoformed between Phe38 and Met42 from the Psubunit and Leu255, Ala259, and Leu268 from Rsubunit. Equivalent contacts from the R subunit tothe P subunit are also made within this interface.
The P/Q (O/R) interface is the smallest one,490 AÊ 2. Only one hydrogen bond is formed acrossthis interface: P178GlyN . . . Q176SerO. Both resi-dues are located in the S-loop. Other residueswithin the S-loops of both subunits also contributeto this interface through hydrophobic interactions:Ala170, Val175, Ile180, Ala182, and Ile184.

Figure 3. Stereo plot of Ca superimposition of the GAPDH monomers from M. fervidus (red) and S. solfataricus(green) using LSQKAB program (Kabsch, 1976).
Archaeal GAPDH in Complex with NADP 485
NADP� conformation and binding site
NADP� is bound to the enzyme in an extendedconformation, with a distance of 8.55 AÊ betweenthe C01 carbon atoms of both riboses (Figure 4).Both furanose rings are in the C(20)-endo confor-mation. The adenine ring adopts an anti confor-mation, while that of nicotinamide is syn.
In the carboxamide plane of the nicotinamidemoiety, the O7N atom is trans with respect to theC2N atom of the pyridinium ring. Within NADP�,an internal hydrogen bond N7N . . . O1N occurs inall subunits with an average distance of 2.65 AÊ
between both atoms. Furthermore, the syn confor-mation of nicotinamide is stabilized by a hydrogenbond between the carboxamide N7 atom and theside-chain of Asp171. In the nicotinamide mononu-cleotide moiety, the 20-hydroxyl group of the riboseforms a hydrogen bond with the main chain ofGly110. Another hydrogen bond is formedbetween one 50-phosphate oxygen atom of thenicotinamide mononucleotide and the main-chainamide group of residue 12.
Within the adenosine mononucleotide moiety,one adenosine 50-phosphate oxygen atom interactsthrough hydrogen bonding with main-chain NHand side-chain OH of Thr11. All of the three 20-phosphate oxygen atoms of the adenosine ribosenot involved in the phosphomonoester bond are
hydrogen-bonded to protein residues: the ®rst onesolely interacts with the side-chain of Lys33; thesecond one is also connected to the side-chain ofLys33 and interacts with main-chain amide groupsof residues 34 and 35; the last one acts as a hydro-gen acceptor with respect to the side-chain ofThr34. Finally, the adenine ring of NADP� isstabilized by hydrophobic interactions with Thr34and Ala53 on one side and Thr86, Pro87 on theother side.
Sulphate-binding site
One sulphate ion was built into each of the sub-units before ®nal model re®nement. The averagedistance between the sulphur atom of the sulphateion and the sulphur atom of catalytic Cys140 is3.93 AÊ . In all subunits, the tetrahedral sulphategroup adopts an orientation where one of its fouroxygen atoms probably accepts the thiol group ofCys140 through a hydrogen bond. Apart from thisinteraction, an extensive hydrogen bond networkis formed between the sulphate ion and its bindingsite: all of the four sulphate oxygen atoms arehydrogen-bonded to side-chains involving Ser139,Asn141, Arg167, His193 and His194 (Figure 5). Allof these residues are strictly conserved in archaealsequences known to date (Figure 1).
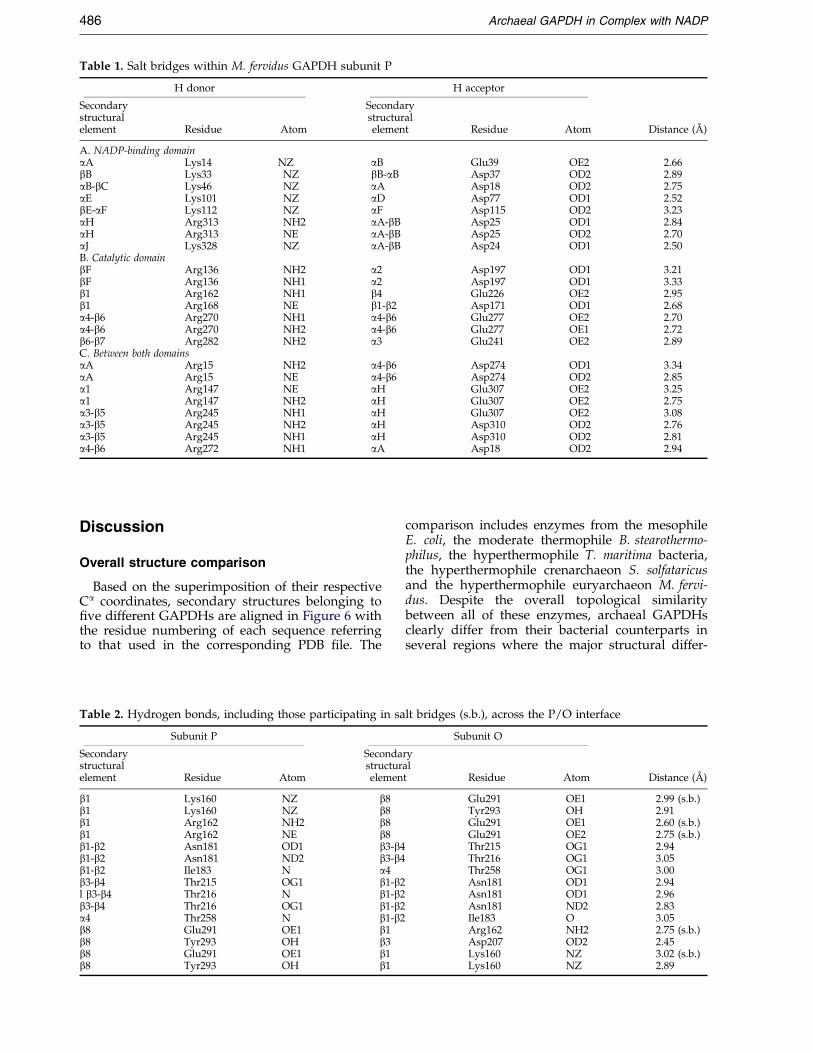
Table 1. Salt bridges within M. fervidus GAPDH subunit P
H donor H acceptor
Secondary Secondarystructural structuralelement Residue Atom element Residue Atom Distance (AÊ )
A. NADP-binding domainaA Lys14 NZ aB Glu39 OE2 2.66bB Lys33 NZ bB-aB Asp37 OD2 2.89aB-bC Lys46 NZ aA Asp18 OD2 2.75aE Lys101 NZ aD Asp77 OD1 2.52bE-aF Lys112 NZ aF Asp115 OD2 3.23aH Arg313 NH2 aA-bB Asp25 OD1 2.84aH Arg313 NE aA-bB Asp25 OD2 2.70aJ Lys328 NZ aA-bB Asp24 OD1 2.50B. Catalytic domainbF Arg136 NH2 a2 Asp197 OD1 3.21bF Arg136 NH1 a2 Asp197 OD1 3.33b1 Arg162 NH1 b4 Glu226 OE2 2.95b1 Arg168 NE b1-b2 Asp171 OD1 2.68a4-b6 Arg270 NH1 a4-b6 Glu277 OE2 2.70a4-b6 Arg270 NH2 a4-b6 Glu277 OE1 2.72b6-b7 Arg282 NH2 a3 Glu241 OE2 2.89C. Between both domainsaA Arg15 NH2 a4-b6 Asp274 OD1 3.34aA Arg15 NE a4-b6 Asp274 OD2 2.85a1 Arg147 NE aH Glu307 OE2 3.25a1 Arg147 NH2 aH Glu307 OE2 2.75a3-b5 Arg245 NH1 aH Glu307 OE2 3.08a3-b5 Arg245 NH2 aH Asp310 OD2 2.76a3-b5 Arg245 NH1 aH Asp310 OD2 2.81a4-b6 Arg272 NH1 aA Asp18 OD2 2.94
486 Archaeal GAPDH in Complex with NADP
Discussion
Overall structure comparison
Based on the superimposition of their respectiveCa coordinates, secondary structures belonging to®ve different GAPDHs are aligned in Figure 6 withthe residue numbering of each sequence referringto that used in the corresponding PDB ®le. The
Table 2. Hydrogen bonds, including those participating in s
Subunit P
Secondary Secondastructural structurelement Residue Atom elemen
b1 Lys160 NZ b8b1 Lys160 NZ b8b1 Arg162 NH2 b8b1 Arg162 NE b8b1-b2 Asn181 OD1 b3-bb1-b2 Asn181 ND2 b3-bb1-b2 Ile183 N a4b3-b4 Thr215 OG1 b1-bl b3-b4 Thr216 N b1-bb3-b4 Thr216 OG1 b1-ba4 Thr258 N b1-bb8 Glu291 OE1 b1b8 Tyr293 OH b3b8 Glu291 OE1 b1b8 Tyr293 OH b1
comparison includes enzymes from the mesophileE. coli, the moderate thermophile B. stearothermo-philus, the hyperthermophile T. maritima bacteria,the hyperthermophile crenarchaeon S. solfataricusand the hyperthermophile euryarchaeon M. fervi-dus. Despite the overall topological similaritybetween all of these enzymes, archaeal GAPDHsclearly differ from their bacterial counterparts inseveral regions where the major structural differ-
alt bridges (s.b.), across the P/O interface
Subunit O
ryalt Residue Atom Distance (AÊ )
Glu291 OE1 2.99 (s.b.)Tyr293 OH 2.91Glu291 OE1 2.60 (s.b.)Glu291 OE2 2.75 (s.b.)
4 Thr215 OG1 2.944 Thr216 OG1 3.05
Thr258 OG1 3.002 Asn181 OD1 2.942 Asn181 OD1 2.962 Asn181 ND2 2.832 Ile183 O 3.05
Arg162 NH2 2.75 (s.b.)Asp207 OD2 2.45Lys160 NZ 3.02 (s.b.)Lys160 NZ 2.89

Figure 4. Hydrogen bond network (broken lines)between NADP� and protein atoms. NADP� bonds aredrawn in white while protein bonds are in yellow. TheFigure was produced with MOLSCRIPT (Kraulis, 1991).
Figure 5. Ribbon diagram of the sulphate binding site.The Figure was produced with MOLSCRIPT (Kraulis,1991). The sulphate ion and side-chains of Ser139,Cys140, Asn141, Arg167, His193 and His194 involved insulphate binding are shown in ball-and-stick represen-tation. His219, whose side-chain is also shown inball-and-stick representation, is likely implicated in thecatalytic mechanism.
Archaeal GAPDH in Complex with NADP 487
ences consist of (i) the deletion of two short b-strands, (ii) the shortening of a few loops includingthe S-loop and (iii) the presence of ®ve additionala-helices.
(i) A region at the N terminus (residues 46-66in the bacterial enzymes), which generally involvestwo b-strands (four in B. stearothermophilusGAPDH), is lost in M. fervidus and S. solfataricusGAPDHs. These b-strands are located at one edgeof the central b-sheet in the cofactor domain ofbacterial GAPDHs (Figure 6).
(ii) Comparisons between archaeal and bac-terial enzymes show that four loops are shorter inboth archaeal GAPDHs than their counterparts inbacterial enzymes. In particular, the S-loopbetween strands b1 and b2 in the M. fervidussequence is 8-11 residues shorter than the corre-sponding ones in the bacterial sequences. AlthoughS-loops from M. fervidus and B. stearothermophiluspoint in opposite directions, they remain similarlyconnected in both enzymes via the P/Q and O/Rsubunit interfaces. In the same manner, the loopbetween helix a3 and strand b5 in M. fervidusGAPDH is four residues shorter than that in the
T. maritima enzyme and three residues shorter thanthose in the B. stearothermophilus and E. colienzymes. Finally, two other M. fervidus loops,a2-b3 and b7-b8, are one residue shorter than theT. maritima and B. stearothermophilus correspondingloops. On the contrary, the M. fervidus loopbetween strand b2 and helix a2 is two residueslonger than the B. stearothermophilus loop and even®ve residues longer than the T. maritima loop. InM. fervidus and S. solfataricus GAPDHs, the loopb2-a2 contributes to the interface between P and Osubunits via van der Waals contacts with the loopb6-b7. Met209 and Met295 also appear to be inclose vicinity of the loop b2-a2. Area calculationsshow that 21 % of Met209 accessible surface and9 % of Met295 accessible surface are buried by resi-dues 188 to 191 from the loop b2-a2. In particular,the Leu190 side-chain points toward Met209 andMet295 and speci®cally increases hydrophobic con-tacts across the P/O interface. Thus, the larger sizeof loop b2-a2 probably contributes to the stabiliz-ation of the M. fervidus quarternary structure. So,the present results do not clearly support someproposals, suggesting that a decrease in the lengthsof the loops connecting secondary structureelements would be important for thermostabiliza-tion (Vieille & Zeikus, 1996).
(iii) Besides loop deletions or insertions,Figure 6 shows the presence of ®ve additional a-helices in archaeal enzymes, among which the twolargest ones are helices a4 and aJ.
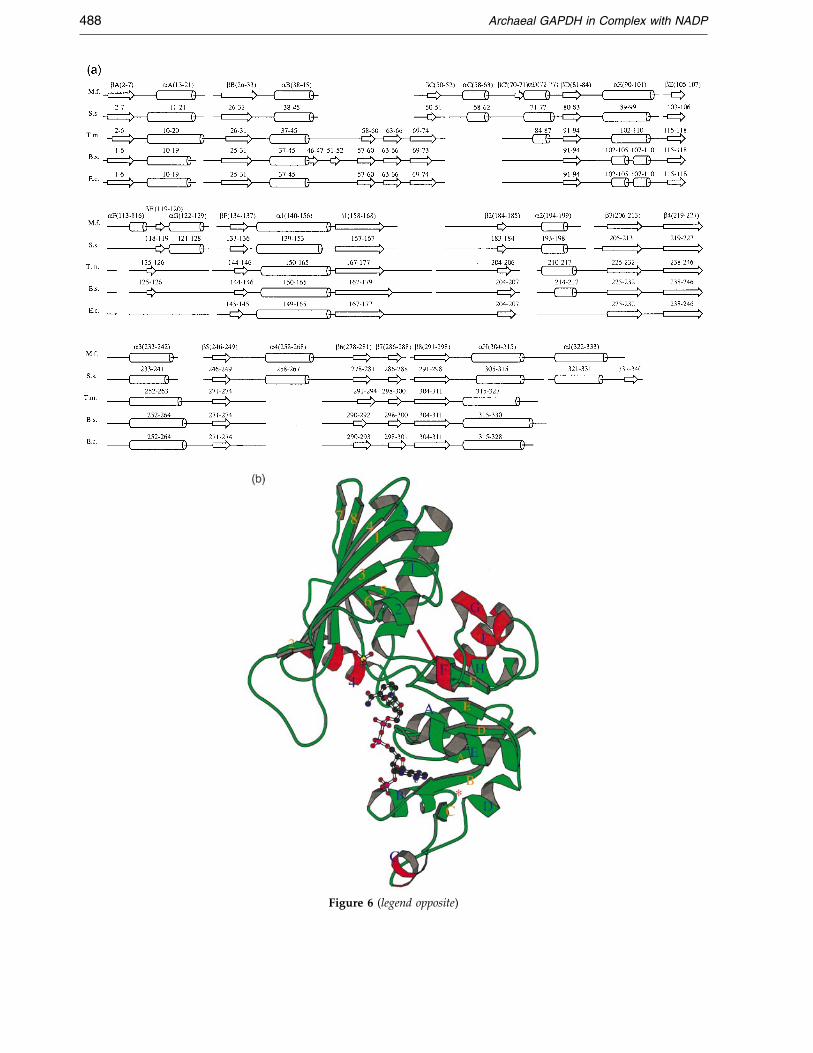
Figure 6 (legend opposite)
488 Archaeal GAPDH in Complex with NADP
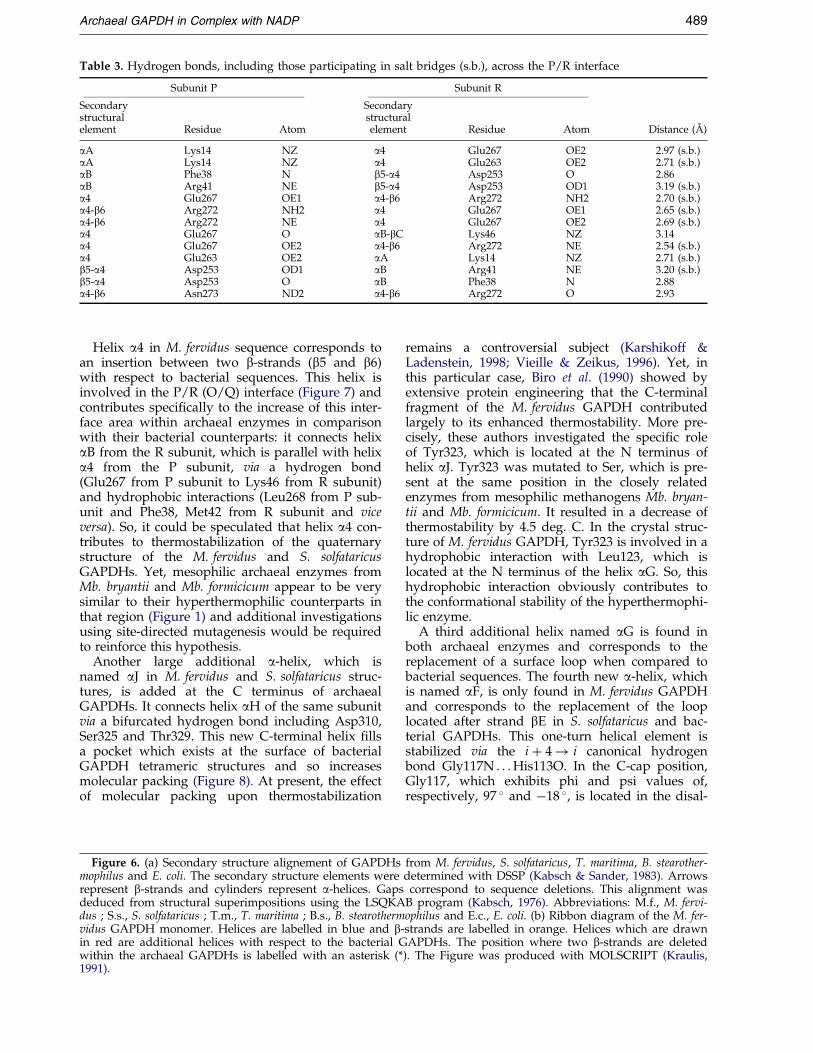
Table 3. Hydrogen bonds, including those participating in salt bridges (s.b.), across the P/R interface
Subunit P Subunit R
Secondary Secondarystructural structuralelement Residue Atom element Residue Atom Distance (AÊ )
aA Lys14 NZ a4 Glu267 OE2 2.97 (s.b.)aA Lys14 NZ a4 Glu263 OE2 2.71 (s.b.)aB Phe38 N b5-a4 Asp253 O 2.86aB Arg41 NE b5-a4 Asp253 OD1 3.19 (s.b.)a4 Glu267 OE1 a4-b6 Arg272 NH2 2.70 (s.b.)a4-b6 Arg272 NH2 a4 Glu267 OE1 2.65 (s.b.)a4-b6 Arg272 NE a4 Glu267 OE2 2.69 (s.b.)a4 Glu267 O aB-bC Lys46 NZ 3.14a4 Glu267 OE2 a4-b6 Arg272 NE 2.54 (s.b.)a4 Glu263 OE2 aA Lys14 NZ 2.71 (s.b.)b5-a4 Asp253 OD1 aB Arg41 NE 3.20 (s.b.)b5-a4 Asp253 O aB Phe38 N 2.88a4-b6 Asn273 ND2 a4-b6 Arg272 O 2.93
Archaeal GAPDH in Complex with NADP 489
Helix a4 in M. fervidus sequence corresponds toan insertion between two b-strands (b5 and b6)with respect to bacterial sequences. This helix isinvolved in the P/R (O/Q) interface (Figure 7) andcontributes speci®cally to the increase of this inter-face area within archaeal enzymes in comparisonwith their bacterial counterparts: it connects helixaB from the R subunit, which is parallel with helixa4 from the P subunit, via a hydrogen bond(Glu267 from P subunit to Lys46 from R subunit)and hydrophobic interactions (Leu268 from P sub-unit and Phe38, Met42 from R subunit and viceversa). So, it could be speculated that helix a4 con-tributes to thermostabilization of the quaternarystructure of the M. fervidus and S. solfataricusGAPDHs. Yet, mesophilic archaeal enzymes fromMb. bryantii and Mb. formicicum appear to be verysimilar to their hyperthermophilic counterparts inthat region (Figure 1) and additional investigationsusing site-directed mutagenesis would be requiredto reinforce this hypothesis.
Another large additional a-helix, which isnamed aJ in M. fervidus and S. solfataricus struc-tures, is added at the C terminus of archaealGAPDHs. It connects helix aH of the same subunitvia a bifurcated hydrogen bond including Asp310,Ser325 and Thr329. This new C-terminal helix ®llsa pocket which exists at the surface of bacterialGAPDH tetrameric structures and so increasesmolecular packing (Figure 8). At present, the effectof molecular packing upon thermostabilization
Figure 6. (a) Secondary structure alignement of GAPDHsmophilus and E. coli. The secondary structure elements wererepresent b-strands and cylinders represent a-helices. Gapsdeduced from structural superimpositions using the LSQKAdus ; S.s., S. solfataricus ; T.m., T. maritima ; B.s., B. stearothermvidus GAPDH monomer. Helices are labelled in blue and bin red are additional helices with respect to the bacterial Gwithin the archaeal GAPDHs is labelled with an asterisk (*1991).
remains a controversial subject (Karshikoff &Ladenstein, 1998; Vieille & Zeikus, 1996). Yet, inthis particular case, Biro et al. (1990) showed byextensive protein engineering that the C-terminalfragment of the M. fervidus GAPDH contributedlargely to its enhanced thermostability. More pre-cisely, these authors investigated the speci®c roleof Tyr323, which is located at the N terminus ofhelix aJ. Tyr323 was mutated to Ser, which is pre-sent at the same position in the closely relatedenzymes from mesophilic methanogens Mb. bryan-tii and Mb. formicicum. It resulted in a decrease ofthermostability by 4.5 deg. C. In the crystal struc-ture of M. fervidus GAPDH, Tyr323 is involved in ahydrophobic interaction with Leu123, which islocated at the N terminus of the helix aG. So, thishydrophobic interaction obviously contributes tothe conformational stability of the hyperthermophi-lic enzyme.
A third additional helix named aG is found inboth archaeal enzymes and corresponds to thereplacement of a surface loop when compared tobacterial sequences. The fourth new a-helix, whichis named aF, is only found in M. fervidus GAPDHand corresponds to the replacement of the looplocated after strand bE in S. solfataricus and bac-terial GAPDHs. This one-turn helical element isstabilized via the i � 4! i canonical hydrogenbond Gly117N . . . His113O. In the C-cap position,Gly117, which exhibits phi and psi values of,respectively, 97 � and ÿ18 �, is located in the disal-
from M. fervidus, S. solfataricus, T. maritima, B. stearother-determined with DSSP (Kabsch & Sander, 1983). Arrows
correspond to sequence deletions. This alignment wasB program (Kabsch, 1976). Abbreviations: M.f., M. fervi-ophilus and E.c., E. coli. (b) Ribbon diagram of the M. fer-
-strands are labelled in orange. Helices which are drawnAPDHs. The position where two b-strands are deleted
). The Figure was produced with MOLSCRIPT (Kraulis,
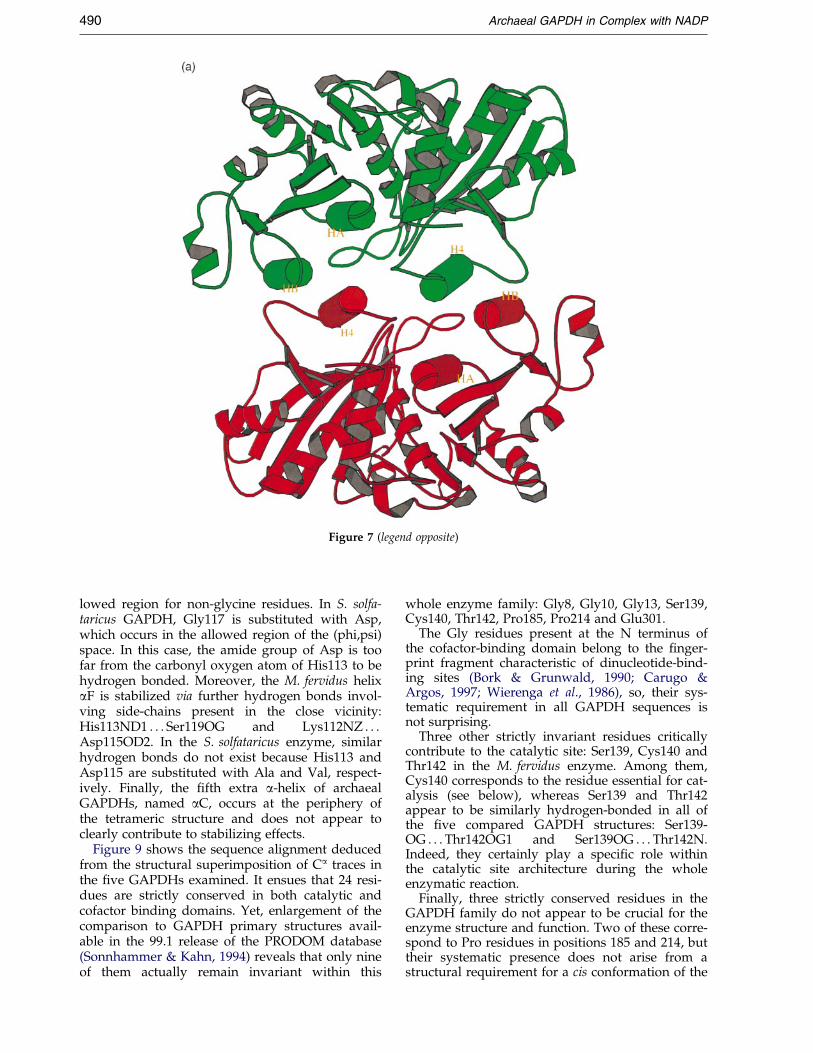
Figure 7 (legend opposite)
490 Archaeal GAPDH in Complex with NADP
lowed region for non-glycine residues. In S. solfa-taricus GAPDH, Gly117 is substituted with Asp,which occurs in the allowed region of the (phi,psi)space. In this case, the amide group of Asp is toofar from the carbonyl oxygen atom of His113 to behydrogen bonded. Moreover, the M. fervidus helixaF is stabilized via further hydrogen bonds invol-ving side-chains present in the close vicinity:His113ND1 . . . Ser119OG and Lys112NZ . . .Asp115OD2. In the S. solfataricus enzyme, similarhydrogen bonds do not exist because His113 andAsp115 are substituted with Ala and Val, respect-ively. Finally, the ®fth extra a-helix of archaealGAPDHs, named aC, occurs at the periphery ofthe tetrameric structure and does not appear toclearly contribute to stabilizing effects.
Figure 9 shows the sequence alignment deducedfrom the structural superimposition of Ca traces inthe ®ve GAPDHs examined. It ensues that 24 resi-dues are strictly conserved in both catalytic andcofactor binding domains. Yet, enlargement of thecomparison to GAPDH primary structures avail-able in the 99.1 release of the PRODOM database(Sonnhammer & Kahn, 1994) reveals that only nineof them actually remain invariant within this
whole enzyme family: Gly8, Gly10, Gly13, Ser139,Cys140, Thr142, Pro185, Pro214 and Glu301.
The Gly residues present at the N terminus ofthe cofactor-binding domain belong to the ®nger-print fragment characteristic of dinucleotide-bind-ing sites (Bork & Grunwald, 1990; Carugo &Argos, 1997; Wierenga et al., 1986), so, their sys-tematic requirement in all GAPDH sequences isnot surprising.
Three other strictly invariant residues criticallycontribute to the catalytic site: Ser139, Cys140 andThr142 in the M. fervidus enzyme. Among them,Cys140 corresponds to the residue essential for cat-alysis (see below), whereas Ser139 and Thr142appear to be similarly hydrogen-bonded in all ofthe ®ve compared GAPDH structures: Ser139-OG . . . Thr142OG1 and Ser139OG . . . Thr142N.Indeed, they certainly play a speci®c role withinthe catalytic site architecture during the wholeenzymatic reaction.
Finally, three strictly conserved residues in theGAPDH family do not appear to be crucial for theenzyme structure and function. Two of these corre-spond to Pro residues in positions 185 and 214, buttheir systematic presence does not arise from astructural requirement for a cis conformation of the
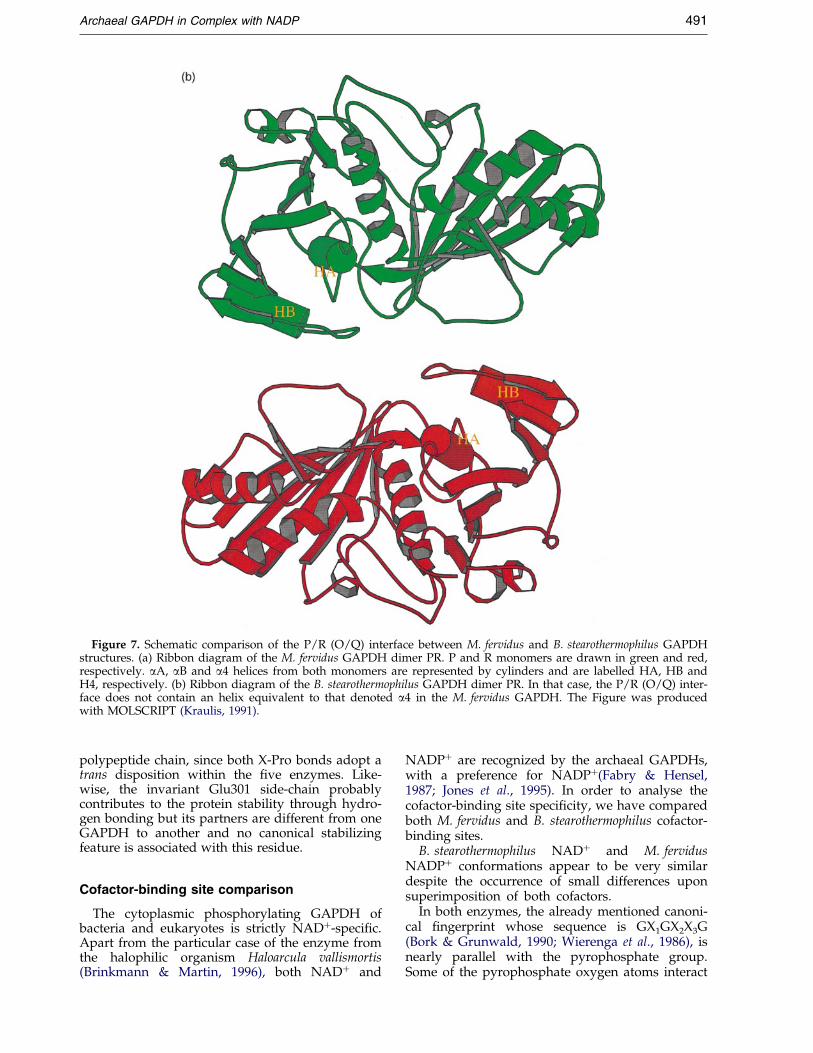
Figure 7. Schematic comparison of the P/R (O/Q) interface between M. fervidus and B. stearothermophilus GAPDHstructures. (a) Ribbon diagram of the M. fervidus GAPDH dimer PR. P and R monomers are drawn in green and red,respectively. aA, aB and a4 helices from both monomers are represented by cylinders and are labelled HA, HB andH4, respectively. (b) Ribbon diagram of the B. stearothermophilus GAPDH dimer PR. In that case, the P/R (O/Q) inter-face does not contain an helix equivalent to that denoted a4 in the M. fervidus GAPDH. The Figure was producedwith MOLSCRIPT (Kraulis, 1991).
Archaeal GAPDH in Complex with NADP 491
polypeptide chain, since both X-Pro bonds adopt atrans disposition within the ®ve enzymes. Like-wise, the invariant Glu301 side-chain probablycontributes to the protein stability through hydro-gen bonding but its partners are different from oneGAPDH to another and no canonical stabilizingfeature is associated with this residue.
Cofactor-binding site comparison
The cytoplasmic phosphorylating GAPDH ofbacteria and eukaryotes is strictly NAD�-speci®c.Apart from the particular case of the enzyme fromthe halophilic organism Haloarcula vallismortis(Brinkmann & Martin, 1996), both NAD� and
NADP� are recognized by the archaeal GAPDHs,with a preference for NADP�(Fabry & Hensel,1987; Jones et al., 1995). In order to analyse thecofactor-binding site speci®city, we have comparedboth M. fervidus and B. stearothermophilus cofactor-binding sites.
B. stearothermophilus NAD� and M. fervidusNADP� conformations appear to be very similardespite the occurrence of small differences uponsuperimposition of both cofactors.
In both enzymes, the already mentioned canoni-cal ®ngerprint whose sequence is GX1GX2X3G(Bork & Grunwald, 1990; Wierenga et al., 1986), isnearly parallel with the pyrophosphate group.Some of the pyrophosphate oxygen atoms interact
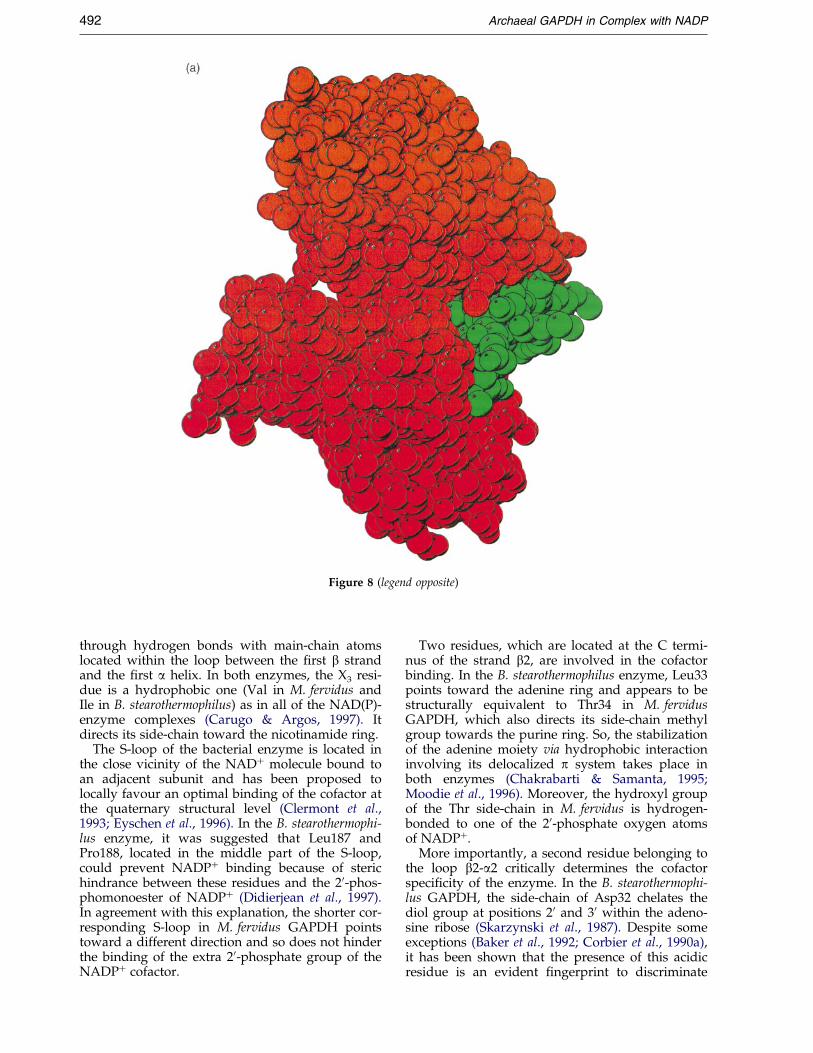
Figure 8 (legend opposite)
492 Archaeal GAPDH in Complex with NADP
through hydrogen bonds with main-chain atomslocated within the loop between the ®rst b strandand the ®rst a helix. In both enzymes, the X3 resi-due is a hydrophobic one (Val in M. fervidus andIle in B. stearothermophilus) as in all of the NAD(P)-enzyme complexes (Carugo & Argos, 1997). Itdirects its side-chain toward the nicotinamide ring.
The S-loop of the bacterial enzyme is located inthe close vicinity of the NAD� molecule bound toan adjacent subunit and has been proposed tolocally favour an optimal binding of the cofactor atthe quaternary structural level (Clermont et al.,1993; Eyschen et al., 1996). In the B. stearothermophi-lus enzyme, it was suggested that Leu187 andPro188, located in the middle part of the S-loop,could prevent NADP� binding because of sterichindrance between these residues and the 20-phos-phomonoester of NADP� (Didierjean et al., 1997).In agreement with this explanation, the shorter cor-responding S-loop in M. fervidus GAPDH pointstoward a different direction and so does not hinderthe binding of the extra 20-phosphate group of theNADP� cofactor.
Two residues, which are located at the C termi-nus of the strand b2, are involved in the cofactorbinding. In the B. stearothermophilus enzyme, Leu33points toward the adenine ring and appears to bestructurally equivalent to Thr34 in M. fervidusGAPDH, which also directs its side-chain methylgroup towards the purine ring. So, the stabilizationof the adenine moiety via hydrophobic interactioninvolving its delocalized p system takes place inboth enzymes (Chakrabarti & Samanta, 1995;Moodie et al., 1996). Moreover, the hydroxyl groupof the Thr side-chain in M. fervidus is hydrogen-bonded to one of the 20-phosphate oxygen atomsof NADP�.
More importantly, a second residue belonging tothe loop b2-a2 critically determines the cofactorspeci®city of the enzyme. In the B. stearothermophi-lus GAPDH, the side-chain of Asp32 chelates thediol group at positions 20 and 30 within the adeno-sine ribose (Skarzynski et al., 1987). Despite someexceptions (Baker et al., 1992; Corbier et al., 1990a),it has been shown that the presence of this acidicresidue is an evident ®ngerprint to discriminate
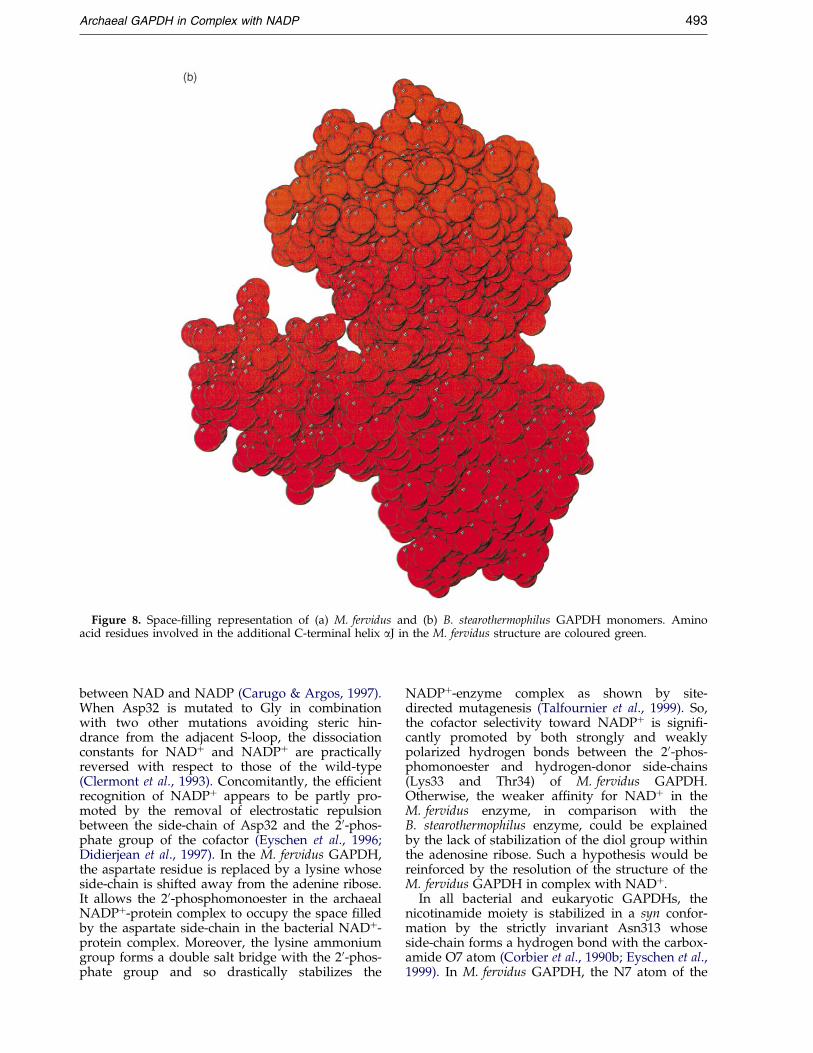
Figure 8. Space-®lling representation of (a) M. fervidus and (b) B. stearothermophilus GAPDH monomers. Aminoacid residues involved in the additional C-terminal helix aJ in the M. fervidus structure are coloured green.
Archaeal GAPDH in Complex with NADP 493
between NAD and NADP (Carugo & Argos, 1997).When Asp32 is mutated to Gly in combinationwith two other mutations avoiding steric hin-drance from the adjacent S-loop, the dissociationconstants for NAD� and NADP� are practicallyreversed with respect to those of the wild-type(Clermont et al., 1993). Concomitantly, the ef®cientrecognition of NADP� appears to be partly pro-moted by the removal of electrostatic repulsionbetween the side-chain of Asp32 and the 20-phos-phate group of the cofactor (Eyschen et al., 1996;Didierjean et al., 1997). In the M. fervidus GAPDH,the aspartate residue is replaced by a lysine whoseside-chain is shifted away from the adenine ribose.It allows the 20-phosphomonoester in the archaealNADP�-protein complex to occupy the space ®lledby the aspartate side-chain in the bacterial NAD�-protein complex. Moreover, the lysine ammoniumgroup forms a double salt bridge with the 20-phos-phate group and so drastically stabilizes the
NADP�-enzyme complex as shown by site-directed mutagenesis (Talfournier et al., 1999). So,the cofactor selectivity toward NADP� is signi®-cantly promoted by both strongly and weaklypolarized hydrogen bonds between the 20-phos-phomonoester and hydrogen-donor side-chains(Lys33 and Thr34) of M. fervidus GAPDH.Otherwise, the weaker af®nity for NAD� in theM. fervidus enzyme, in comparison with theB. stearothermophilus enzyme, could be explainedby the lack of stabilization of the diol group withinthe adenosine ribose. Such a hypothesis would bereinforced by the resolution of the structure of theM. fervidus GAPDH in complex with NAD�.
In all bacterial and eukaryotic GAPDHs, thenicotinamide moiety is stabilized in a syn confor-mation by the strictly invariant Asn313 whoseside-chain forms a hydrogen bond with the carbox-amide O7 atom (Corbier et al., 1990b; Eyschen et al.,1999). In M. fervidus GAPDH, the N7 atom of the

Figure 9. GAPDH multiple sequence alignment deduced from the three-dimensional structure superimposition ofenzymes from M. fervidus, S. solfataricus, T. maritima, B. stearothermophilus and E. coli. The 24 conserved residues areshown in bold. Strictly invariant residues within the whole GAPDH family are Gly8, Gly10, Gly13, Ser139, Cys140,Thr142, Pro185, Pro214 and Glu301. Numbering corresponds to the M. fervidus sequence.
494 Archaeal GAPDH in Complex with NADP

Archaeal GAPDH in Complex with NADP 495
nicotinamide moiety is hydrogen-bonded to theAsp171 side-chain. As Asp171 is strictly conservedin all known archaeal GAPDH sequences(Figure 1), it could be considered as the moleculardeterminant equivalent to bacterial Asn313 withrespect to the enzyme stereospeci®city of hydridetransfer. Asp171 was mutated to Asn in order toverify this hypothesis. The Asp171Asn mutationled to a 20-fold increase in the Km value forNADP� (39 mM for the Asp171Asn mutant versus2 mM for the wild type), while the Km values forother substrates, D-glyceraldehyde-3-phosphate(D-G3P) and inorganic phosphate, and the kcat
value were not signi®cantly modi®ed. Moreover,the 104-fold decrease of the kobs value of the stepsleading to thioacylenzyme formation with respectto the wild-type (2.2 sÿ1 versus 230 sÿ1) indicatedthat the ternary complex Asp171Asn mutant-NADP-D-G3P was less ef®cient when compared tothe wild-type. This most likely re¯ected a non-opti-mal positioning of the nicotinamide ring towardthe hemithioacetal intermediate, thus affecting theef®ciency of the hydride transfer. Moreover, theweaker intensity of the charge transfer transitionbetween the catalytic Cys140 and the pyridiniumring of NADP (Racker & Krimsky, 1952;Talfournier et al., 1999) determined for theAsp171Asn mutant when compared to the wild-type GAPDH, i.e. 200 Mÿ1 cmÿ1 versus 420 Mÿ1
cmÿ1, indicated that a signi®cant change occurredin the location and orientation of the nicotinamidering relative to the thiolate entity. Altogether theseresults strongly support an essential role of theside-chain of Asp171 in the syn positioning of thecofactor in archaeal GAPDHs. In the M. fervidusstructure, the Gln300 residue is structurally super-imposable upon the Asn313 residue from bacterialGAPDHs (Figure 9) and is therefore located in theclose vicinity of the nicotinamide moiety. Yet, itsside-chain does not point toward the carboxamidegroup of NADP�, since the terminal NE2 aminogroup is hydrogen-bonded to the side-chain ofAsp171. Finally, it is interesting to note that thesteric hindrance due to the side-chain of Ile304 pre-vents the nicotinamide moiety from adopting ananti disposition in the archaeal enzyme. Therefore,Ile304 plays a role similar to that observed for thestructurally equivalent Tyr or Phe residue(Figure 9) found in position 317 in eukaryotic andbacterial GAPDHs (Eyschen et al., 1999).
The present structure of M. fervidus GAPDH hasbeen obtained in the holo form in the presence ofthe natural cofactor NADP� whereas the structureof the S. solfataricus enzyme was solved in its apoform (Isupov et al., 1999). When compared to thermsd obtained from the superimposition of Ca
traces of single subunits (1.043 AÊ 2, see above), thermsd value arising from superimposition of thewhole tetramers is larger (1.461 AÊ 2) and likelyaccounts for a apo/holo conformational switch atthe quaternary level. On the contrary, rmsd valuesdeduced from the local superimposition of eithercatalytic domain (1.073 AÊ 2) or cofactor-binding
domain (0.899 AÊ 2) are of the same order as thatcorresponding to the whole subunit. Thus, in thatcase, the later results do not support a rigid-bodyrotation between domains within the monomers,as that described for the B. stearothermophilusenzyme (Skarzynski & Wonacott, 1988). A moredetailed analysis of the apo/holo transition withinthe tetramer may be tentatively deduced from thecompared superimposition of the three kinds ofdimers, i.e. PO (QR), PQ (OR) and PR (OQ). Thermsd value corresponding to the former dimer(1.088 AÊ 2) does not signi®cantly differ from thatassociated with single subunits, whereas the twoother dimers give rise to rmsd values (PQ,1.232 AÊ 2; PR, 1.317 AÊ 2) intermediate between thoseof the tetramer and the monomer superimposi-tions. Therefore, in agreement with ¯uorescencedata (Talfournier et al., 1999), it may be suggestedthat archaeal GAPDHs behave as a dimer ofdimers where the tightly associated PO and QRdimers could be considered as rigid bodies duringthe apo/holo transition. Yet, it must be noted thatthe present analysis does not relate to the sameenzyme in both apo and holo forms and thatM. fervidus and S. solfataricus GAPDHs share only49 % identity at the primary structure level.
Active site
GAPDHs (EC 1.2.1.12) catalyse the oxidativephosphorylation of D-G3P to form 1,3-diphospho-glycerate. Two essential amino acids of bacterialand eukaryotic GAPDHs are involved in thechemical mechanism: Cys149 directly participatesin the formation of the hemithioacetal and thio-acyl-enzyme intermediates (Mougin et al., 1988)and His176 acts as an acid-base catalyst during thewhole catalytic pathway (Talfournier et al., 1998).In M. fervidus GAPDH, no detectable activity wasobserved when Cys140 was substituted by Alawhich suggested an essential role for Cys140 in thecatalytic mechanism (Talfournier et al., 1998). Thepresent crystal structure de®nitively supports thelater proposal, since Cys140 of M. fervidus andS. solfataricus GAPDHs structurally superimpose toresidue Cys149 of bacterial GAPDHs.
In the crystal structure of the M. fervidusGAPDH, one His residue is found to be close tothe essential catalytic residue Cys140 (Figures 5and 10), like in the bacterial and eukaryoticGAPDHs. The distance between Cys140 SG andHis219 NE2 ranges from 3.49 AÊ to 3.55 AÊ accord-ing to the examined subunit. The His219 locationin the active site suggests a catalytic role of thisresidue in the same manner as His176 in the bac-terial and eukaryotic GAPDHs and site-directedmutagenesis was then used in order to test thishypothesis. The His219Asn mutation led to a 92-fold decrease of the kobs value of the steps leadingto thioacylenzyme formation when compared tothe wild-type (2.5 sÿ1 versus 230 sÿ1). The fact thatthe kobs was similar to the kcat value obtainedunder steady-state conditions (1.8 sÿ1) indicated

Figure 10. Stereo 2Fo ÿ Fc electron density map around the active site and NADP� (contoured at 1s). Bonds withinNADP� and the sulphate ion are drawn in green. Bonds within Cys140 and His219 side-chains are drawn in blue.The remaining protein bonds are drawn in red.
496 Archaeal GAPDH in Complex with NADP
that the rate-limiting step could now be associatedwith the acylenzyme formation, in contrast to whatwas described for the wild-type where the phos-phorolysis step is rate limiting (Talfournier et al.,1999). The isotopic effect of 2.2 observed with twoconcentrations of D-[1-2H]G3P at pH 8.5 for theHis219Asn mutant versus 1.1 for the wild-typeenzyme demonstrated that the limiting step wasassociated with the hydride transfer for theHis219Asn mutant. Therefore, the role of theHis219 residue in the catalytic mechanism is likelyto facilitate the hydride transfer, either as a basecatalyst or as a stabilizer of the deprotonated hemi-thioacetal intermediate. Another role of His219 inM. fervidus GAPDH is to decrease the pKapp ofCys140, since the mutation of His219 into Asnshifts it from 6.3 to 7.6 (Talfournier et al., 1999).Yet, in contrast to what is observed betweenCys149 and His176 in bacterial GAPDHs, no ionpair is formed between Cys140 and His219 inarchaeal GAPDHs (Talfournier et al., 1998). In thatcontext, it is noteworthy that the side-chain rota-meric states within essential residues Cys140 andHis219 probably do not re¯ect the actual active
conformation of these groups in the ternary com-plex. Indeed, in the crystal structure, hydrogenbonding of the Cys140 thiol group by the sulphateion is likely to be artefactual and would precludeHis219 from assisting the essential Cys140 duringthe catalytic mechanism.
Contrary to the two sulphate-binding siteswhich are observed in the catalytic site of the S. sol-fataricus GAPDH, only one sulphate ion is coordi-nated by the M. fervidus enzyme in a position quitesimilar to the site described as Pi in the formerstructure (Isupov et al., 1999). Moreover, with theexception of the thiol group of Cys140 (see above),chelation of the sulphate ion is carried out inexactly the same way within the two enzymeswith critical contributions from Ser139, Asn141,Arg167, His193 and His194 side-chains. Despitethe lack of a second sulphate ion in the sitedenoted Ps, both M. fervidus Arg167 and Arg168residues appear to be in convenient positions toputatively ®x the phosphate group of D-G3P andtopologically correspond to the equivalent arginylresidues assigned to this site in the S. solfataricusenzyme. The fact that mutating both Arg167 and

Table 4. Data collection statistics
Space group P21212Resolution ( AÊ ) 35-2.11Measured reflections 415,252Unique reflections 82,965Rsym (overall) 0.056Completeness (overall) 0.917Rsym (2.11-2.16 AÊ ) 0.151Completeness (2.11-2.16 AÊ ) 0.693I/s(I) (2.11-2.16 AÊ ) 4.0
Archaeal GAPDH in Complex with NADP 497
Arg168 into Asn was shown to strongly decreasethe ef®ciency of the acylation step (Talfournieret al., 1999) also supports these assignments.
In conjunction with conclusions deduced fromthe S. solfataricus GAPDH structure (Isupov et al.,1999), the overall view of the M. fervidus catalyticsite indicates that archaeal GAPDH bind theirligands in the same orientation as that observedwithin their bacterial and eukaryotic counterparts.Yet, most of the residues critical for enzymaticactivity are relocated along the polypeptide frag-ments that de®ne the site and only the triadSer139-Cys140-Thr142 lies in a similar position,whatever the GAPDH examined. In the particularcase of the essential archaeal His219, its locationclearly differs from the position occupied byHis176 in the bacterial and eukaryotic GAPDHsbut it surprisingly superimposes quite well withHis274 from the recently described structure ofaspartate-b-semialdehyde dehydrogenase fromE. coli (Had®eld et al., 1999), an enzyme that per-forms catalysis similar to that of GAPDH. Theevolutionary origin of GAPDH remains a subjectof controversy, since contradictory arguments havebeen proposed to support either the presence(Hensel et al., 1989) or the absence (Doolittle et al.,1990) of a common ancestor for the whole GAPDHfamily. Data from the M. fervidus enzyme con®rmthat the modular nature of GAPDHs makes themdif®cult to analyse from a phylogenetic point ofview (Fothergill-Gilmore & Michels, 1993). Never-theless, the present results rather argue in favourof the hypothesis that archaeal GAPDH could haveevolved from an ancestral enzyme possessing aGAPDH-like activity, but with a very broadspeci®city requiring only an essential cysteine forcatalysis.
Methods
Crystallographic investigations
The crystallographic methods used to solve the struc-ture of the M. fervidus GAPDH has been recently pub-lished as a short preliminary paper (Charron et al., 1999).Here, a detailed structural and functional analysis of theenzyme is presented.
Puri®cation and crystallization steps were carried outwith an enzyme genetically overexpressed in recombi-nant E. coli HB101 cells (Talfournier et al., 1998). Largecrystals suitable for X-ray analysis were obtained inexperimental conditions consisting of 18 % (v/v) 2-methyl-2,4-pentanediol, 12 mM magnesium acetate,2 mM EDTA, 1 mM DTT, 60 mM cacodylate buffer(pH 6.5) at 285 K. They belong to space group P21212with unit cell dimensions a � 136.7 AÊ , b � 153.3 AÊ , andc � 74.9 AÊ with one tetramer per asymmetric unit.
Data collection was carried out at 100 K on the beam-line D2AM at the European Synchrotron Radiation Facil-ity (ESRF) in Grenoble (France). Data were processedusing the MARXDS package (Klein, 1993; Kabsch, 1993)(Table 4; Charron et al., 1999).
The M. fervidus crystal structure was solved by mol-ecular replacement with the program AMoRe (Navaza,
1994) using the S. solfataricus GAPDH (Fleming et al.,1998; Isupov et al., 1999) as a template.
The re®nement was then carried out in the range 10-2.11 AÊ without NCS restraints using REFMAC(Murshudov et al., 1997). A total of 2 % of the data wereselected for Rfree calculations. Manual corrections of themodel were performed using O (Jones et al., 1991). Afterthe model was re®ned to an Rfactor of 26.5 %, electrondensity maps revealed density corresponding to theNADP� molecule in each subunit. The model wasfurther re®ned and water molecules were de®ned withARP (Lamzin & Wilson, 1993). In addition, one sulphateion was built into each of the subunits according to thedensity of the map. The model was re®ned to the ®nalRfactor of 19.4 % and Rfree of 25.7 % between 10 AÊ and2.11 AÊ resolution (Rfactor of 22.3 % and Rfree of 29.1 %between 2.22 AÊ and 2.11 AÊ resolution).
Coordinates
The structure factors and re®ned coordinates of theM. fervidus GAPDH have been deposited in the RCSBProtein Data Bank; the access code is 1cf2.
Enzymatic investigations
Steady-state kinetics
Initial rate measurements were carried out at 25 �C ona Kontron Uvikon 933 spectrophotometer by followingthe absorbance of NADPH at 340 nm. The temperatureof the solutions was maintained at 25 �C by thermostatedsample holders using a circulating water bath for all ofthe measurements. The forward reaction was carried outunder the following conditions: 50 mM Tris-HCl(pH 8.5), 100 mM (Asp171Asn mutant) or 400 mM(His219Asn mutant) K2HPO4, 2 mM EDTA, 2 mMNADP� and 2 mM or 4 mM D-G3P for Asp171Asnmutant or His219Asn mutant, respectively. The initialrate data were ®tted to the Michaelis-Menten relation-ship using least-squares regression analysis to determinekcat and Km. All the Km values were determined at satur-ating concentrations of the other substrates.
Deuterium isotopic effects
D-[1-2H]G3P was prepared as described (Corbier et al.,1990b). D-G3P was purchased from Sigma. The concen-trations of both were determined enzymatically. Isotopiceffects were measured in 50 mM Tris-HCl (pH 8.5),2 mM EDTA, 100 mM (wild-type) or 400 mM(His219Asn mutant) K2HPO4, by using the direct com-parison of the initial velocities observed for the oxidativephosphorylation of D-G3P and D-[1-2H]G3P (0.25 mMand 0.5 mM) in the presence of 2 mM NADP�.

498 Archaeal GAPDH in Complex with NADP
Analysis of the overall steps leading toacylenzyme formation
Fast kinetic experiments were carried out on a BiologicInstruments (SFM3) stopped-¯ow apparatus and datacollected from absorbance measurements were analysedwith the Biokine program using non-linear regressionanalysis. An average of at least eight runs was per-formed for each rate constant determination. Progresscurves of NADPH production for Asp171Asn andHis219Asn mutant enzymes were recorded at 25 �C and340 nm in 100 mM Tris-HCl (pH 8.5) and 2 mM EDTAin the absence of inorganic phosphate. One syringe was®lled with enzyme (15 mM in subunit) and NADP�
(1 mM) and the other contained D-G3P (2 mM or 4 mMfor Asp171Asn and His219Asn mutant enzymes, respect-ively). The given concentrations of reactants are those ofthe ®nal mixture.
Acknowledgements
This research was supported by the Centre Nationalde la Recherche Scienti®que, the University Henri Poin-care - Nancy I and the FR ProteÂines 42. We thank theEuropean Synchrotron Radiation Facility (ESRF) forbeamline access.
References
Arcari, P., Dello Russo, A., Ianniciello, G., Gallo, M. &Bocchini, V. (1993). Nucleotide sequence andmolecular evolution of the gene coding for glyce-raldehyde-3-phosphate dehydrogenase in the ther-moacidophilic archaebacterium Sulfolobussolfataricus. Biochem. Genet. 31, 241-251.
Baker, P. J., Britton, K. L., Rice, D. W., Rob, A. &Stillman, T. J. (1992). Structural consequences ofsequence patterns in the ®ngerprint region of thenucleotide binding fold. Implications for nucleotidespeci®city. J. Mol. Biol. 228, 662-671.
Barns, S. M., Fundyga, R. E., Jeffries, M. W. & Pace,N. R. (1994). Remarkable archaeal diversity detectedin a yellowstone park hot spring environment. Proc.Natl Acad. Sci. USA, 91, 1609-1613.
Biro, J., Fabry, S., Dietmaier, W., Bogedain, C. & Hensel,R. (1990). Engineering thermostability in archaebac-terial glyceraldehyde-3-phosphate dehydrogenase.Hints for the important role of interdomain contactsin stabilizing protein conformation. FEBS Letters,275, 130-134.
Bork, P. & Grunwald, C. (1990). Recognition of differentnucleotide-binding sites in primary structures usinga property-pattern approach. Eur. J. Biochem. 191,347-358.
Brinkmann, H. & Martin, W. (1996). Higher-plant chlor-oplast and cytosolic 3-phosphoglycerate kinases: acase of endosymbiotic gene replacement. Plant Mol.Biol. 30, 65-75.
Carugo, O. & Argos, P. (1997). NADP-dependentenzymes. I. Conserved stereochemistry of cofactorbinding. Proteins: Struct. Funct. Genet. 28, 10-28.
Chakrabarti, P. & Samanta, U. (1995). CH/p interactionin the packing of the adenine ring in protein struc-tures. J. Mol. Biol. 251, 9-14.
Charron, C., Talfournier, F., Isupov, M. N., Branlant, G.,Littlechild, J. A., Vitoux, B. & Aubry, A. (1999).Crystallization and preliminary X-ray diffractionstudies of D-glyceraldehyde-3-phosphate dehydro-genase from the hyperthermophilic archaeon Meth-anothermus fervidus. Acta Crystallog. sect. D, 55, 1353-1355.
Clermont, S., Corbier, C., Mely, Y., Gerard, D.,Wonacott, A. & Branlant, G. (1993). Determinants ofcoenzyme speci®city in glyceraldehyde-3-phosphatedehydrogenase: role of the acidic residue in the®ngerprint region of the nucleotide binding fold.Biochemistry, 32, 10178-10184.
Corbier, C., Clermont, S., Billard, P., Skarzynski, T.,Branlant, C., Wonacott, A. & Branlant, G. (1990a).Probing the coenzyme speci®city of glyceraldehyde-3-phosphate dehydrogenases by site-directed muta-genesis. Biochemistry, 29, 7101-7106.
Corbier, C., Mougin, A., Mely, Y., Adolph, H. W.,Zeppezauer, M., Gerard, D., Wonacott, A. &Branlant, G. (1990b). The nicotinamide subsite ofglyceraldehyde-3-phosphate dehydrogenase studiedby site-directed mutagenesis. Biochimie, 72, 545-554.
DeLong, E. F., Wu, K. Y., Pr'ezelin, B. B. & Jovine, R. V.(1994). High abundance of archaea in antartic mar-ine picoplankton. Nature, 371, 695-697.
Didierjean, C., Rahuel-Clermont, S., Vitoux, B.,Dideberg, O., Branlant, G. & Aubry, A. (1997).A crystallographic comparison between mutatedglyceraldehyde-3-phosphate dehydrogenases fromBacillus stearothermophilus complexed with eitherNAD� or NADP�. J. Mol. Biol. 268, 739-759.
Doolittle, R. F., Feng, D. F., Anderson, K. L. & Alberro,M. R. (1990). A naturally occurring horizontal genetransfer from an eukaryote to a prokaryote. J. Mol.Evol. 31, 383-388.
DueÂe, E., Olivier-Deyris, L., Fanchon, E., Corbier, C.,Branlant, G. & Dideberg, O. (1996). Comparison ofthe structures of wild-type and a N313T mutant ofEscherichia coli glyceraldehyde 3-phosphate dehy-drogenases: implication for NAD binding and coop-erativity. J. Mol. Biol. 257, 814-838.
Eyschen, J., Vitoux, B., Rahuel-Clermont, S., Marraud,M., Branlant, G. & Cung, M. T. (1996). Phosphorus-31 nuclear magnetic resonance studies on coenzymebinding and speci®city in glyceraldehyde-3-phos-phate dehydrogenase. Biochemistry, 35, 6064-6072.
Eyschen, J., Vitoux, B., Marraud, M., Cung, M. T. &Branlant, G. (1999). Engineered glycolytic glycer-aldehyde-3-phosphate dehydrogenase binds theanti conformation of NAD� nicotinamide but doesnot experience A-speci®c hydride transfer. Arch.Biochem. Biophys. 364, 219-227.
Fabry, S. & Hensel, R. (1987). Puri®cation and character-ization of D-glyceraldehyde-3-phosphate dehydro-genase from the thermophilic archaebacteriumMethanothermus fervidus. Eur. J. Biochem. 165, 147-155.
Fabry, S. & Hensel, R. (1988). Primary structure of gly-ceraldehyde-3-phosphate dehydrogenase deducedfrom the nucleotide sequence of the thermophilicarchaebacterium Methanothermus fervidus. Gene, 64,189-197.
Fabry, S., Lehmacher, A., Bode, W. & Hensel, R. (1988).Expression of the glyceraldehyde 3-phosphate dehy-drogenase gene from the extremely thermophilicarchaebacterium Methanothermus fervidus in E. coli.FEBS Letters, 237, 213-217.

Archaeal GAPDH in Complex with NADP 499
Fleming, T. M., Jones, C. E., Piper, P. W., Cowan,D. A., Isupov, M. N. & Littlechild, J. A. (1998).Characterisation, crystallization and preliminaryX-ray investigation of glyceraldehyde-3-phosphatedehydrogenase from the hyperthermophilicarchaeon Sulfolobus solfataricus. Acta Crystallog. sect.D, 54, 671-674.
Fothergill-Gilmore, L. A. & Michels, P. A. M. (1993).Evolution of glycolysis. Prog. Biophys. Mol. Biol. 59,105-235.
Gilboa, R., Bauer, A. J. & Shoham, G. (1998). Crystalliza-tion and preliminary crystallographic analysis ofglyceraldehyde 3-phosphate dehydrogenase fromSaccharomyces cerevisiae (baker's yeast). Acta Crystal-log. sect. D, 54, 1467-1470.
Grif®th, J. B., Lee, B., Murdock, A. L. & Amelunxen,R. E. (1983). Molecular symmetry of glyceralde-hyde-3-phosphate dehydrogenase from Bacillus coa-gulans. J. Mol. Biol. 169, 963-974.
Gunasekaran, K., Ramakrishnan, C. & Balaram, P.(1996). Disallowed Ramachandran conformations ofamino acid residues in protein structures. J. Mol.Biol. 264, 191-198.
Had®eld, A., Kryger, G., Ouyang, J., Petsko, G. A.,Ringe, D. & Viola, R. (1999). Structure of aspartate-b-semialdehyde dehydrogenase from Escherichia coli,a key enzyme in the aspartate family of amino acidbiosynthesis. J. Mol. Biol. 289, 991-1002.
Hensel, R., Zwickl, P., Fabry, S., Lang, J. & Palm, P.(1989). Sequence comparison of glyceraldehyde-3-phosphate dehydrogenases from the three urking-doms:evolutionary implications. Canad. J. Microbiol.35, 81-85.
Hutchinson, E. G. & Thornton, J. M. (1994). A revisedset of potentials for b-turn formation in proteins.Protein Sci. 3, 2207-2216.
Isupov, M. N., Fleming, T. M., Dalby, A. R., Crowhurst,G. S., Bourne, P. C. & Littlechild, J. A. (1999). Crys-tal structure of the glyceraldehyde 3-phosphatedehydrogenase from the hyperthermophilicarchaeon Sulfolobus solfataricus. J. Mol. Biol. 291, 651-660.
Jones, C. E., Fleming, T. M., Cowan, D. A., Littlechild,J. A. & Piper, P. W. (1995). The phosphoglyceratekinase and glyceraldehyde-3-phosphate dehydro-genase genes from the thermophilic archaeon Sulfo-lobus solfataricus overlap by 8-bp. Isolation,sequencing of the genes and expression in Escheri-chia coli. Eur. J. Biochem. 233, 800-808.
Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M.(1991). Improved methods for building models inelectron density maps and the location of errors inthese models. Acta Crystallog. Sect. A, 47, 110-119.
Kabsch, W. (1976). A solution for the best rotation torelate two sets of vectors. Acta Crystallog. sect. A, 32,922-923.
Kabsch, W. (1993). MARXDS, Version 1.0 Documentation.MAR Research GMBH, D-22547 Hamburg,Germany.
Kabsch, W. & Sander, C. (1983). Dictionary of proteinsecondary structure: pattern recognition of hydro-gen-bonded and geometrical features. Biopolymers,22, 2577-2637.
Karshikoff, A. & Ladenstein, R. (1998). Proteins fromthermophilic and mesophilic organisms essentiallydo not differ in packing. Protein Eng. 11, 867-872.
Kim, H., Feil, I. K., Verlinde, C. L. M. J., Petra, P. H. &Hol, W. G. J. (1995). Crystal structure of glycosomalglyceraldehyde-3-phosphate dehydrogenase from
Leishmania mexicana: implications for structure-baseddrug design and a new position for the inorganicphosphate binding site. Biochemistry, 34, 14975-14986.
Klein, C. (1993). MARXDS MARSCALE Manual. Version1.4. MAR Research GMBH, D-22547 Hamburg,Germany.
KorndoÈrfer, I., Steipe, B., Huber, R., Tomschy, A. &Jaenicke, R. (1995). The crystal structure of holo-glyceraldehyde-3-phosphate dehydrogenase fromthe hyperthermophilic bacterium Thermotoga mari-tima at 2.5 AÊ resolution. J. Mol. Biol. 246, 511-521.
Kraulis, P. J. (1991). MOLSCRIPT: a program to produceboth detailed and schematic plots of protein struc-tures. J. Appl. Crystallog. 24, 946-950.
Lamzin, V. S. & Wilson, K. S. (1993). Automated re®ne-ment of protein models. Acta Crystallog. sect. D, 49,129-147.
Laskowski, R. A., MacArthur, M. W., Moss, D. S. &Thornton, J. M. (1993). PROCHECK: a program tocheck the stereochemical quality of protein struc-tures. J. Appl. Crystallog. 26, 283-291.
Lesk, A. M. (1995). NAD-binding domains of dehydro-genases. Curr. Opin. Struct. Biol. 5, 775-783.
Mercer, W. D., Winn, S. I. & Watson, H. C. (1976).Twinning in crystals of human skeletal muscleD-glyceraldehyde-3-phosphate dehydrogenase.J. Mol. Biol. 104, 277-283.
Moodie, S. L., Mitchell, J. B. O. & Thornton, J. M. (1996).Protein recognition of adenylate: an example of afuzzy recognition template. J. Mol. Biol. 263, 486-500.
Moras, D., Olsen, K. W., Sabesan, M. N., Buehner, M.,Ford, G. C. & Rossmann, M. G. (1975). Studies ofasymmetry in the three-dimensional structure oflobster D-glyceraldehyde-3-phosphate dehydrogen-ase. J. Biol. Chem. 250, 9137-9162.
Mougin, A., Corbier, C., Soukri, A., Wonacott, A.,Branlant, C. & Branlant, G. (1988). Use of site-directed mutagenesis to probe the role of Cys149 inthe formation of charge-transfer transition in glycer-aldehyde-3-phosphate dehydrogenase. Protein Eng.2, 45-48.
Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997).Re®nement of macromolecular structures by themaximum likelihood method. Acta Crystallog. sect.D, 53, 240-255.
Navaza, J. (1994). AMoRe: an automated package formolecular replacement. Acta Crystallog. sect. A, 50,157-163.
Racker, E. & Krimsky, I. (1952). The mechanism of oxi-dation of aldehydes by glyceraldehyde-3-phosphatedehydrogenase. J. Biol. Chem. 198, 731-743.
Sabatino, P., Fermani, S., Ripamonti, A., Casseta, A.,Scagliarini, S. & Trost, P. (1999). Crystallization andpreliminary X-ray study of chloroplast glyceralde-hyde-3-phosphate dehydrogenase. Acta. Crystallog.sect. D, 55, 566-567.
Schulz, G. E. (1992). Binding of nucleotides by proteins.Curr. Opin. Struct. Biol. 2, 61-67.
Skarzynski, T. & Wonacott, A. J. (1988). Coenzyme-induced conformational changes in glyceraldehyde-3-phosphate dehydrogenase from Bacillus stearother-mophilus. J. Mol. Biol. 203, 1097-1118.
Skarzynski, T., Moody, P. C. E. & Wonacott, J. A.(1987). Structure of holo-glyceraldehyde-3-phos-phate dehydrogenase from Bacillus stearothermophi-lus at 1.8 AÊ resolution. J. Mol. Biol. 193, 171-187.

500 Archaeal GAPDH in Complex with NADP
Song, S., Li, J. & Lin, Z. (1998). Structure of holo-glycer-aldehyde-3-phosphate dehydrogenase from Pali-nurus versicolor re®ned at 2 AÊ resolution. ActaCrystallog. sect. D, 54, 558-569.
Sonnhammer, E. L. L. & Kahn, D. (1994). The modulararrangement of proteins as inferred from analysis ofhomology. Protein Sci. 3, 482-492.
Souza, D. H. F., Garratt, R. C., Araujo, A. P. U.,Guimaraes, B. G., Jesus, W. P. D., Michels, P. A. M.,Hannaert, V. & Oliva, G. (1998). Trypanosoma cruziglycosomal glyceraldehyde-3-phosphate dehydro-genase: structure, catalytic mechanism and targetedinhibitor design. FEBS Letters, 424, 131-135.
Stetter, K. O., Huber, R., BloÈchl, E., Kurr, M., Eden,R. D., Fielder, M., Cash, H. & Vance, I. (1993).Pyrobaculum Aerophilum sp. nov., a novel nitrate-reducing hyperthermophilic archaeum. Nature, 365,743-745.
Talfournier, F., Colloc'h, N., Mornon, J.-P. & Branlant,G. (1998). Comparative study of the catalyticdomain of phosphorylating glyceraldehyde-3-phos-phate dehydrogenases from bacteria and archaeavia essential cysteine probes and site-directed muta-genesis. Eur. J. Biochem. 252, 447-457.
Talfournier, F., Colloc'h, N., Mornon, J.-P. & Branlant,G. (1999). Functional characterization of the D-gly-ceraldehyde 3-phosphate dehydrogenase from the
archaeon Methanothermus fervidus by comparativemolecular modelling and site-directed mutagenesis.Eur. J. Biochem. 265, 93-104.
Tanner, J. J., Hecht, R. M. & Krause, K. L. (1996). Deter-minants of enzyme thermostability observed in themolecular structure of Thermus aquaticus D-glycer-aldehyde-3-phosphate dehydrogenase at 2.5 AÊ
resolution. Biochemistry, 35, 2597-2609.Vellieux, F. M. D., Hajdu, J., Verlinde, C. L. M. J.,
Groendijk, H., Read, R. J., Greenhough, T. J.,Campbell, J. W., Kalk, K. H., Littlechild, J. A.,Watson, H. C. & Hol, W. G. J. (1993). Structure ofglycosomal glyceraldehyde-3-phosphate dehydro-genase from Trypanosoma brucei determined fromLaue data. Proc. Natl Acad. Sci. USA, 90, 2355-2359.
Vieille, C. & Zeikus, J. G. (1996). Thermozymes: identify-ing molecular determinants of protein structuraland functional stability. Trends Biotechnol. 14, 183-190.
Wierenga, R. K., Terpstra, P. & Hol, W. G. J. (1986).Prediction of the occurrence of the ADP-bindingbab-fold in proteins, using an amino acid sequence®ngerprint. J. Mol. Biol. 187, 101-107.
Woese, C. R., Kandler, O. & Wheelis, M. L. (1990).Towards a natural system of organisms: proposalfor the domains archaea, bacteria, and eucarya.Proc. Natl Acad. Sci. USA, 87, 4576-4579.
Edited by R. Huber
(Received 27 September 1999; received in revised form 25 January 2000; accepted 25 January 2000)





























