Embed Size (px)
Citation preview

132
J Chir 2005,142, N°3 • © Masson, Paris, 2005
Prixdu Journalde Chirurgie
Benoît Malassagne2004
Revue de la littérature
Traitement chirurgical des tumeurs endocrines gastro-entéro-pancréatiques1. Traitement des tumeurs primitives
R. Kianmanesh, D. O’Toole, A. Sauvanet, P. Ruszniewski, J. BelghitiFédération d’hépato-gastroentérologie, Hôpital Beaujon – Clichy.
e-mail : [email protected]
Correspondance : R. Kianmanesh, Service de Chirurgie Générale et Digestive, Hôpital L. Mou-rier, F92700 Colombes.
Résumé/Abstract
Traitement chirurgical des tumeurs endocrines gastro-entéro-pancréatiquesR. Kianmanesh, D. O’Toole, A. Sauvanet, P. Ruszniewski, J. BelghitiLes tumeurs endocrines (TE), autrefois appelées tumeurs neuroendocrines, du tractus digestif sont rares. On distingue deux principaux types : les TE gastro-intestinales (autrefois appelées carcinoïdes) qui sont les plus fréquentes ; et les TE duodénopancréatiques. Lorsque la tumeur sécrète des polypeptides responsables de symptômes cliniques (syndrome hormonal), on utilise le terme de TE fonctionnelle. Le traitement des TE est multidisciplinaire. Les TE peu différenciées ont un très mauvais pronostic et sont traitées par la chimiothérapie systémique. La chirurgie d’exérèse est le seul traitement curatif des TE bien différenciées. Elle a comme objectifs de : 1) prolonger la survie en réséquant la tumeur primitive et ses éventuelles métastases ganglionnaires et/ou hépatiques ; 2) contrôler un éventuel syndrome hormonal et ; 3) prévenir ou traiter les complications locales. Parmi les TE gastro-intestinales, celles de l’appendice et du rectum sont les plus fréquentes. Elles sont souvent de petite taille (< 1 cm), bénignes, et découvertes de façon fortuite sur une pièce d’appendicectomie ou d’exérèse endoscopique. Les tumeurs iléales sont volontiers de petite taille, malignes, parfois multiples et se compliquent dans 30-50 % des cas (occlusion, mésentérite rétractile, hémorragie). Le syndrome carcinoïde, caractérisé par l’existence de douleurs abdominales, flushs, diarrhées, troubles tensionnels, bronchospasme et atteinte cardiaque droite est présent chez 10 % des malades et s’associe le plus souvent à des métastases hépatiques, de TE secrétant la sérotonine et ses dérivés (carcinoïde). Plus de la moitié des TE du pancréas sont non fonctionnelles. Elles sont généralement malignes et découvertes à un stade plus tardif, devant un syndrome de masse ou lors du bilan de métastases hépatiques. L’insulinome et le gastrinome (syndrome de Zollinger-Ellison) sont les TE duodénopancréatiques fonctionnelles les plus fréquentes. Pour les formes sporadiques, la taille tumorale, le potentiel malin et la localisation déterminent le type d’exérèse qui peut aller d’une énucléation à une pancréatectomie réglée, plus ou moins étendue. En cas de suspicion de gastrinome, une duodénotomie est systématiquement préconisée. Dans 10-20 % des cas, les TE duodénopancréatiques s’intègrent dans le cadre d’une néoplasie endocrinienne multiple type 1 (NEM-1) qui est une affection génétique, autosomique dominante, associant une atteinte endocrine plurifocale touchant de façon non constante les parathyroïdes, l’hypophyse, le pancréas et les surrénales. Pour les insulinomes- NEM-1, l’énucléation des lésions céphaliques associée à une pancréatectomie gauche semble être l’intervention la plus adaptée. Pour les gastrinomes-NEM-1, l’intérêt de la chirurgie d’exérèse reste controversé pour les tumeurs de moins de 2-3 cm, compte tenu essentiellement de la diffusion des lésions qui sont souvent de petite taille, de l’existence de taux élevé de récidive après exérèse et du bon pronostic spontané à long terme. Cependant pour prévenir la survenue de métastases hépatiques à distance, qui sont la principale cause de mortalité à long terme, on peut défendre une attitude chirurgicale plus agressive à un stade plus précoce.
Mots-clés : Pancréas. Estomac. Grêle. Traitement. Tumeur endocrine. Zollinger-Ellison. Carcinoïde.
Introduction
Les tumeurs endocrines (TE) autrefoisappelées tumeurs neuroendocrines, sontrares (1 % de l’ensemble des tumeurs)avec une incidence que l’on peut estimerde 1 à 10 pour 100 000 habitants par an[1-4]. Elles se manifestent généralementaux alentours de la cinquième ou lasixième décennie. Dans 3/4 des cas la tu-meur est située au niveau du tractus di-gestif et dans 1/4 des cas au niveau del’appareil pulmonaire [1, 3]. Elles sontréparties en 3 groupes selon leur sited’origine embryologique (tableau 1) :1) les TE dérivées de l’intestin anté-rieur, qui incluent les TE de thymus, dularynx, des bronches, et les TE duodé-nopancréatiques (TEP) ; 2) les tumeursde l’intestin moyen qui incluent les TEdu grêle, de l’appendice et du côlondroit et enfin ; 3) les TE de l’intestinpostérieur (rectum).Le foie représente le principal site demétastases (en dehors des ganglions pé-ritumoraux) [2, 5-10].
Tableau 1
Tumeurs endocrines gastro-entéro-pancréatiques selon leur origine embryologique.
Intestin Antérieur– Thymus, pharynx et bronches– Œsophage thoraco-abdominal– Estomac– Duodénum et pancréas
Intestin Moyen– Jéjunum– Iléon– Cæcum– Appendice– Colon
Intestin Postérieur– Rectum

133
J Chir 2005,142, N°3 • © Masson, Paris, 2005 Revue de la littérature
Surgical treatment of gastric, enteric, and pancreatic endocrine tumorsPart 1. Treatment of primary endocrine tumors
R. Kianmanesh, D. O’Toole, A. Sauvanet, P. Ruszniewski, J. Belghiti
Endocrine tumors (ET) of the digestive tract (formerly called neuroendocrine tumors) are rare. They are classified into two principal types : gastrointestinal ET’s (formerly called carcinoid tumors) which are the most common, and pancreaticoduodenal ET’s. Functioning ET’s secrete polypeptide hormones which cause characteristic hormonal syndromes. The management of ET is multidisciplinary. Poorly-differentiated ET’s have a poor prognosis and are treated by chemotherapy. Surgical excision is the only curative treatment of well-differentiated ET’s. The surgical goals are to : 1. prolong survival by resecting the primary tumor and any nodal or hepatic metastases. 2. control the symptoms related to hormonal secretion. 3. prevent or treat local complications.The most common sites of gastrointestinal ET’s ( carcinoids) are the appendix and the rectum ; these are often small (<1 cm.), benign, and discovered fortuitously at the time of appendectomy or colonoscopic removal. Ileal ET’s, even if small, are malignant, frequently multiple, and compicated in 30-50% of cases by bowel obstruction, mesenteric invasion, or bleeding. The carcinoid syndrome (consisting of abdominal pain, flushing, diarrhea, hypertension, bronchospasm, and right sided cardiac vegetations) is caused by the hypersecretion of serotonin into the systemic circulation; it occurs in 10% of cases and is usually associated with hepatic metastases.More than half of the cases of pancreatic ET are non-functional. They are usually malignant and of advanced stage at diagnosis presenting as a palpable or obstructing mass or as liver metastases. Insulinoma and gastrinoma (cause of the Zollinger-Ellison syndrome) are the most common functional ET’s. 80% are sporadic; in these cases, tumor size, location, and malignant potential determine the type of resection which may vary from a simple enucleation to a formal pancreatectomy. In 10-20% of cases, pancreaticoduodenal ET presents in the setting of multiple endocrine neoplasia (NEM type I), an autosomal-dominant genetic disease with multifocal endocrine involvement of the pituitary, parathyroid, pancreas, and adrenal glands. For insulinoma with NEM-I, enucleation of lesions in the pancreatic head plus a caudal pancreatectomy is the most appropriate procedure. For gastrinoma with NEM-I, the benefit of surgical resection for tumors less than 2-3 cm. in sizeis not clear. The lesions are frequently small, multiple, and widespread and recurrence is frequent after excision. The long-term prognosis is nevertheless fairly good. But the eventual development of liver metastases which are the most common cause of mortality still argues for an aggressive surgical approach in the early stages of the disease.
Key words: Pancreas. Stomach. Small intestine. Treatment. Endocrine carcinoma. Zollinger-Ellison. Carcinoid tumor.
Les TE gastro-entéro-pancréatiquesont souvent une évolution lente, avecdes taux de survie allant de quelquesmois à plusieurs dizaines d’années. Leurprise en charge est multidisciplinaire [3,4, 9, 11-17]. On distingue deux grandescatégories de TE gastro-entéro-pan-créatiques : les TEP et les TE digestivesnon duodénopancréatiques ou gastro-intestinales autrefois appelées tumeurs
carcinoïdes (estomac, jéjunum, iléon,appendice, côlon et rectum) [18]. Leterme « carcinoïde » est actuellementréservé aux TE secrétant de la séroto-nine et ses dérivés (sérotonine sériqueet urinaire, 5-HIA urinaires) [19]. Ondistingue selon leur capacité à secréterdes peptides responsables de symptô-mes cliniques, les TE fonctionnelles etTE non fonctionnelles. Les TE, qu’el-
Tableau 2
Marqueurs généraux des tumeurs endocrines.
• Chromogranine (A,B,C) : protéines monomériques d’acides solubles retrouvées dans les granu-les de sécrétion (la Chromogranine A est la plus utilisée).
• Enolase neuro-spécifique (NSE) : dimère de l’enzyme énolase, marqueur cytoplasmique de dif-férenciation neuroendocrine.
• Synaptophysine : glycoprotéine membranaire retrouvée dans les vésicules neuronales de cellules neuroendocrines.
les soient fonctionnelles ou non, secrè-tent d’autres peptides (polypéptide P)ou substances telles la chromogranine Aet la NSE (neuron specific enolase) quin’ont pas de traduction clinique maisqui sont des marqueurs utiles dans lediagnostic et le suivi (tableau 2) [3, 20].
Le diagnostic de malignité d’uneTE peut être difficile. Il repose essen-tiellement sur la présence (ou survenue)de métastases ganglionnaires ou hépati-ques (MH) et plus rarement sur l’enva-hissement locorégional de structures devoisinage telles que les structures vei-neuses [21]. En cas de doute, la biopsiepermet, dans la majorité des cas, d’affir-mer la nature endocrine de la lésion paranalyse histologique couplée à une ex-pression visible en immunomarquagede la chromogranine, de la synaptophy-sine et/ou du NSE [22, 23]. De plus ellepermet de juger de l’agressivité de la tu-meur par évaluation de la différencia-tion tumorale et de l’index de proliféra-tion cellulaire (nombre de mitoses,pourcentage de cellules exprimant l’an-tigène Ki 67 ou le pourcentage de MIB-1). Les TE peu différenciés représen-tent 10 % des TE, s’apparentent auxcarcinomes anaplasiques à petites cellu-les bronchiques, ont un très mauvaispronostic (taux de survie moyenne infé-rieure à 1 an) et sont traitées par la chi-miothérapie systémique (VP 16 et CD-DP) [11, 24, 25]. Pour les TE biendifférenciées (tableau 3), la chirurgied’exérèse est le seul traitement poten-tiellement curatif [4, 14, 17, 26-28]. Endehors du degré de différenciation cel-lulaire et l’activité proliférative (expres-sion de Ki 67, index mitotique, MIB-1),l’existence de métastases ganglionnaireset surtout hépatiques est le principalélément pronostique péjoratif [2, 8, 10,11, 26, 29-36].
Les TE digestives non-duodénopan-créatiques sont, sauf certaines TE gas-triques (ECLomes) toujours sporadi-ques. En revanche dans 10 à 25 % descas, les TEP s’intègrent dans le cadred’une néoplasie endocrinienne multiplede type 1 (NEM-1) ou syndrome deWermer [22, 33, 37, 38]. Il s’agit d’uneaffection génétique rare à transmissionautosomique dominante affectant le gè-ne de la ménine (chromosome 11q13)et qui favorise le développement de TEplurifocales [39, 40]. Le syndrome com-plet associe dans 85 à 95 % des cas unehyperparathyroïdie par hyperplasie del’ensemble des glandes parathyroïdes ;

Traitement chirurgical des tumeurs endocrines gastro-entéro-pancréatiques R. Kianmanesh et al.
134 un adénome hypophysaire dans 16 à40 % des cas (dont 2/3 de prolactino-mes), une atteinte surrénalienne dans10 à 40 % des cas et des TEP dans 30à 75 % des cas [22, 41, 42]. Les TEPsurvenant dans le cadre de NEM-1 sontsouvent multiples et de petite taille etpeuvent secréter plusieurs polypeptides.Parmi les malades ayant une NEM-1,les gastrinomes (50 % des cas) puis lesinsulinomes (20 % des cas) sont les plusfréquents ; les autres TEP représentemoins de 10 % des cas (tableau 4) [43].La maladie de von Hippel Lindau quiest une affection rare à transmissionautosomique dominante, prédisposeégalement à la survenue de TEP (dans2/3 des cas) et d’autres tumeurs kysti-ques du pancréas, des hémangioblas-tomes (cervelet, rétine), au phéo-chromocytome (souvent bilatéral), àl’adénocarcinome du rein et à des tu-meurs épithéliales de l’oreille interne[22, 44, 45].
Tumeurs endocrines duodénopancréatiques primitives (TEP)
Les TEP sont rares, représentent moinsde 5 % des tumeurs pancréatiques, etont une incidence de 0.4 à 1 pour100 000 habitants par an [46]. Elles sont
pour la plupart des tumeurs solides. Lesformes kystiques à contenu hétérogèneet nécrotique représentent moins de25 % des cas [47-49]. Les TEP « carci-noïdes » (secrétant la sérotonine et sesdérivés) sont très rares (moins de 5 %des cas) [1, 50, 51].
La plupart des TEP sont de bas gra-de de malignité. La classification histo-logique de l’Organisation Mondiale dela Santé (WHO) 2000 qui est celle ac-tuellement utilisée [19, 23, 52], distin-gue 3 types de TEP ; 1) les TE bien dif-férenciées ayant un comportementbénin ou incertain ; 2) les TE malignesde bas grade ou carcinome endocrinebien différenciées et ; 3) les TE mali-gnes de haut grade ou carcinomes en-docrines peu différenciés (tableau 3).
Plus de la moitié des TEP sont nonfonctionnelles et découvertes à un stadetardif d’évolution tumorale devant lessymptômes liés au syndrome de masse(douleurs, ictère, hypertension portalesegmentaire, plus rarement hémorragiedigestive), ou lors du bilan de lésions se-condaires hépatiques [47, 48, 53-55].Cependant, avec les progrès de l’image-rie médicale de plus en plus de TEPnon fonctionnelles de petite taille sontdiagnostiquées de façon fortuite ou de-vant des symptômes non spécifiques.
Tableau 3
Classification histopronostique des tumeurs endocrines.
Tumeurs endocrines bien différenciées– Comportement bénin : tumeur confinée au pancréas, pas d’angioinvasion, taille < 2 cm, ≤ 2 mito-
ses/10 CFG* et ≤ 2 % Ki 67**+/10 CFG• Fonctionnelles*** : insulinomes seulement****• Non fonctionnelles
– Comportement incertain : tumeur confinée au pancréas, angioinvasion ou taille ≥ 2 cm, > 2 mito-ses/10 CFG, > 2 % KI 67+/10 CFG.• Fonctionnelles : insulinomes, gastrinomes, vipome, glucagonome, somatostatinome ou syn-drome hormonal inapproprié• Non fonctionnelle
Carcinome endocrine bien différencié : tumeur maligne de bas grade– Seuls critères formels de malignité : invasion d’organes contigus et/ou métastases.– Critères très souvent présents, non formels : angioinvasion, engainements périnerveux, taille
≥ 3 cm, 2 à 10 mitoses CFG, > 5 % Ki 67+/10 CFG– Non fonctionnelle
Carcinome endocrine peu différencié : tumeur maligne de haut grade– Invasion d’organe contigus et métastases à distance : très fréquente– Angioinvasion et engainement périnerveux nombreux, nécrose, > 10 mitoses/10 CFG, > 15 % Ki
67+, p53+ : critères très souvent présents
* CFG : champs au fort grossissement. ** Ki 67 : indice de prolifération. *** Fonctionnelle : associée à un syndrome clinique. **** À l’exception des insulinomes, les tumeurs fonctionnelles ne sont jamais classées parmi les tumeurs endocrines à comportement bénin.
Parmi les TEP fonctionnelles, les insu-linomes et gastrinomes (syndrome deZollinger-Ellison) sont les plus fré-quentes [17, 21, 53, 56]. Les tumeurs se-crétant du glucagon (glucagonome), deVIP (vipome ou syndrome de Verner-Morrison), de la somatostatine (soma-tostatinome) ou d’autres substancesACTH, GRF, ou substance hypercalcé-miantes (PTH-related peptide ou calci-tonine) sont beaucoup plus rares(tableau 4) [53]. Que la TEP soit fonc-tionnelle ou non, l’interrogatoire etl’étude des antécédents familiaux (pré-sence d’antécédents personnel ou fami-liaux de polyendocrinopathie) la prati-que d’un screening biologique etmorphologique et la recherche généti-que de la mutation du gène de méninesont systématiques devant tout patientprésentant une TEP [22].
La possibilité d’une exérèse chirur-gicale doit toujours être discutée lors deconcertations multidiscipilinaires. Onpeut schématiquement définir trois ty-pes de chirurgie pour TE : 1) l’exérèsesà visée « curative ». Elle s’applique aussibien à la tumeur primitive qu’à ses éven-tuelles métastases ganglionnaires et hé-patiques ; 2) l’exérèse à visée cytoréduc-trice ou « debulking » qui est plusrarement indiquée pour traiter unsyndrome sécrétoire hormonal noncontrôlé par le traitement médical. Ce-pendant, pour être efficace sur les symp-tômes, plus de 80 à 90 % de la massetumorale, y compris les métastases gan-glionnaires et hépatiques, doivent êtreréséqués [12, 35, 57] ; 3) l’exérèse pourprévenir ou traiter d’éventuelles compli-cations locales. Cette dernière est sou-vent indiquée pour les TE iléales qui ontun taux de complications locales élevé(30 à 50 %). Il s’agit le plus souvent decomplications hémorragiques ou occlu-sives. L’occlusion peut être en rapportavec une obstruction endoluminale, in-vagination, ou secondaire à des adhéren-ces dues à une mésentérite rétractile.Cette indication est plus rare pour lesTEP qui se compliquent dans moins de10 à 20 % des cas (ictère, hémorragieduodénale, envahissement veineux avechypertension portale segmentaire) [14,26, 58, 59].
Le type d’exérèse dépend principa-lement de la taille, de la localisation(TEP ou TE gastro-intestinale), dunombre et des rapports de la lésion avecles structures avoisinantes, de l’existen-
J Chir 2005,142, N°3 • © Masson, Paris, 2005
135
Revue de la littérature
Tableau 4
Principales caractéristiques des tumeurs endocrines digestives.
Tumeur(incidence)
Clinique Biologie Taille des lésions (fréquence)
Localisation(fréquence)
AssociationNEM1(%)
Métastases(ganglionset/ou foie) (%)
Guérisonpar la chirurgie(%)
Insulinome(1-10/an/million)
Hypoglycémieà jeun, à l’effortmanifestation neuro-psychiquesrégression symptômes après charge en glucose
HypoglycémieHyperinsulinémieAugmentationpeptide CÉpreuve de jeune
Petite(85-90 %)
Pancréas(> 98 %)
5-8 8-17 90
Gastrinome(0.5-2/an/million)
Syndrome de Zollinger-EllisonMaladie ulcéreuse diarrhées
Débit acide basalHypergastrinémieTest à la secrétine
Petite(40-50 %)
Duodénum 40-70 %Pancréas 30-60 %Autres (< 5 %)
20-25 25-70 30-50
Vipome(0.05-0.2/an/million)
Syndrome de Verner-MorissonDiarrhéesHypokaliémieAchlorydrie
Augmentationde la vipémie
Grosses(60-100 %)
Pancréas 90 %– corps-queue (75 %)Rétropérotineou duodénum < 10 %
5-15 25-80 20
Glucagonome(0.02-0.1/an/million)
DiabèteÉrythème nécrolytique migransCachéxie
Augmentationdu glucagonémie
Grosse(80-100 %)
Pancréas 1-20 40-90 30
Somatostatinome(< 0.05/an/million)
DiarrhéesStéatorrhéesLithiase vésiculaire
Augmentationde la somatosta-tinémie
Grosses(80-100 %)
Pancréas 55 %Duodénum 40 %
5-40 50-90
Tumeurs carcinoïde(10-20/an/million)
Complications (20-40 %) :– hémorragie, occlusion,
mésentérite rétractile
Augmentationde la sérotoninesérique et les 5HIA urinaires,
Petite taille, adénopathie,Lésions multiples (20-30 %)
Iléon, appendice, rectum > 50 % des cas Duodéno-pancréas < 5 %
Non (sauf TE gastriques)
30-100
Syndrome carcinoïde (10 %) :– diarrhées (30-80 %),
flushs (60-75 %), bronchospasme(5-15 %), atteinte cardiaque droite (10-40 %)
Tachykinine et prostaglandines
Tumeurvolumineuseou le plus souvent présence de métas-tases hépatiques
Tumeur non-fonction-nelles pancréatiques(1-2 an/million)
Fortuite, amaigrissement (20-40 %)Douleurs (30-60 %)Syndrome tumoral (40-50 %)Complication locale (< 10 %)
Chromogranine ANSE
Variable Pancréas tête 50 %Corps-queue 50 %
10-30 50-80 < 40-50
Tumeurs rares(< 0.001 an/million)PpomeGRFomeACTHomePTHrpCalcitoninome
Fonction de l’hormone secrétéeAmaigrissementAcromégalieSyndrome de CushingHypercalcémie
Ppome (pancréas)GRFome(pancreas 33 %, poumon 53 %, grêle 10 %),ACTHome(pancréas 4-16 %)
Ppome 18-44GRFome 16
Ppome > 60GRFome 30ACTHOme> 95

Traitement chirurgical des tumeurs endocrines gastro-entéro-pancréatiques R. Kianmanesh et al.
136
ce de métastases ganglionnaires et sur-tout hépatiques synchrones (voir à ce su-jet la partie 2. de cette mise au point dansle prochain numéro), du terrain (comor-bidité) et de l’existence d’une NEM as-sociée [17, 26, 60, 61]. Ceci soulignel’importance du bilan morphologiquepré-opératoire qui doit être exhaustif.
Bilan morphologiqueLa détection pré-opératoire de la TEprimitive et de ses éventuelles métasta-ses repose essentiellement sur le scannerhélicoïdal thoraco-abdominal, l’écho-endoscopie et la scintigraphie des ré-cepteurs de la somatostatine (octréos-can) [62, 63].
La sensibilité du scanner hélicoïdal,avec des coupes fines, dans le diagnosticdes TEP de moins 15 mm est de l’ordrede 80 à 90 % [48, 64, 65]. Les appareilde la dernière génération (scanner mul-tibarrettes) pourront probablement dé-tecter des lésions de plus petite taille[48, 66, 67].
L’utilisation de l’écho-endoscopieest particulièrement importante pour ladétection de petites tumeurs duodéna-les (sensibilité supérieure de 50 à 60 %pour les gastrinomes) qui ne sont habi-tuellement pas visibles sur le scanner.La combinaison de l’écho-endoscopieet de l’octréoscan a une sensibilité d’en-viron 90 % pour localiser les gastrino-mes pancréatiques [68-71].
La sensibilité de l’octréoscan estnettement moindre (inférieure à 50 %)pour détecter les insulinomes qui expri-ment peu les récepteurs de la somatos-tatine [72, 73]. Dans ce cas, l’écho-en-doscopie et le scanner sont les examensles plus utiles [69, 74].
Pour les tumeurs non fonctionnel-les, et en cas de doute diagnostique, uneponction sous écho-endoscopie doitêtre réalisée [69, 75].
L’imagerie par résonance magnéti-que est parfois nécessaire pour mieuxcaractériser la nature et les rapports dela tumeur avec les structures de voisina-ge surtout le canal de Wirsung. Cepen-dant du fait de sa faible résolution spa-tiale, sa sensibilité pour la détection deTEP de petite taille (notamment desganglions et des MH) reste encore in-férieure à celle des scanners de la der-nière génération [65, 67, 76].
Les TEP se présentent à l’échogra-phie comme une masse hypoéchogène,parfois hétérogène avec une zone liqui-dienne en son sein (dégénérescence kys-
tique entouré d’un liseré tissulaire). Lesformes purement kystiques représen-tent moins de 5 % de l’ensemble des lé-sions kystiques du pancréas [65, 77].
L’échographie per opératoire est unexamen sensible pour la détection depetites tumeurs pancréatiques, notam-ment des insulinomes.
De façon exceptionnelle, les exa-mens plus invasifs telle que l’artériogra-phie, le cathétérisme veineux étagé,couplé à un test de simulation intra-ar-térielle (calcium) sont indiquées pourlocaliser la tumeur [78, 79].
Malgré les progrès du bilan mor-phologiques, près de 10 % des TEPprimitives ne sont pas localisées par lesexplorations pré-opératoires [80]. Ils’agit le plus souvent de formes infra-centimétriques, souvent extra-pancréa-tiques (gastrinomes duodénaux ou gan-glionnaires) voire dans certains cas deformes multiples de microadénomespancréatiques qui s’intègrent dans le ca-dre de NEM-1 [48, 80].
Explorations per opératoiresCes explorations sont souvent guidéespar le bilan morphologique pré-opéra-toire et dépendent de la nature de laTEP.
Devant tout syndrome de Zollin-ger-Ellison, surtout sans lésion cible àl’octréoscan et écho-endoscopie, l’en-semble de la cavité abdominale est ex-plorée afin de ne pas méconnaître uneexceptionnelle localisation ectopiquequi représente moins de 2 % des cas(pancréas aberrant, grêle, ovaires, esto-mac ou la rate) [2]. La fréquence desMH, même pour les gastrinomes de pe-tite taille, implique la palpation et lapratique d’une échographie hépatiqueper opératoire.
Pour toute TE pancréatique cépha-lique, le bloc duodénopancréatique esttotalement mobilisé pour faciliter l’ex-ploration manuelle et surtout échogra-phique (sonde de 7.5 ou 10 MHz) [79,81, 82]. Lorsque la tumeur est isthmi-que ou corporéale, la mobilisation dubord inférieur et supérieur du pancréaspermet une meilleure exposition de lapartie médiane et gauche du pancréas.
Pour les tumeurs multiples (surtoutdans le cadre d’une NEM-1) certainspréconisent d’explorer tout le paren-chyme pancréatique [79-84]. La sensi-bilité de l’échographie peropératoire estplus élevée pour détecter des lésions
pancréatiques qu’extrapancréatiques(90 % versus 58 %) [85-87].
Pour les gastrinomes de la paroiduodénale, qui sont souvent de petitetaille et parfois non détectés sur le bilanmorphologique préopératoire, la palpa-tion est suivie d’une endoscopie axialeper opératoire avec transillumination etsurtout d’une duodénotomie systémati-que, ce qui permet de détecter plus de95 % des nodules [2, 9, 26, 69, 80, 85].
Type d’exérèseLes interventions les plus pratiquéessont les énucléations et pancréatecto-mies gauches. Le choix dépend essen-tiellement de la nature maligne de la lé-sion, de la taille, de la localisation, et desrapports de la tumeur avec le canal pan-créatique principal, et enfin du terrainet du caractère sporadique ou non de lamaladie (multiplicité des lésions).
• Énucléation
Elle est proposée pour les tumeurs spo-radiques, présumées bénignes et situéesà distance du canal pancréatique princi-pal [88, 89]. L’information concernantla distance séparant la lésion du canalpancréatique est fondamentale et doitêtre précisée sur le bilan morphologi-que préopératoire (écho-endoscopie etIRM). L’échographie per opératoire estsystématiquement effectuée au mieuxavec une sonde de 7.5 ou de 10-MHz.L’avantage principal de l’énucléationest la préservation du parenchyme pan-créatique permettant d’éviter le risquede diabète et d’insuffisance pancréati-que à long terme. Il est à noter quel’énucléation est praticable dans certai-nes conditions sous vidéo-laparoscopie[90-93]. Après énucléation, la loge del’exérèse est en général drainée. Laprincipale complication est la fistule (oucollection profonde riche en amylase)qui s’observe dans 20 % à 40 % des cas.
• Exérèse pancréatique médiane ou gauche
Elle est réservée aux tumeurs localiséesen dehors de la tête du pancréas, non ac-cessibles à une énucléation, essentielle-ment en raison de leur taille (supérieureà 2-3 cm) et surtout de leur rapportétroit avec le canal pancréatique princi-pal. Lorsque la tumeur est située surl’isthme ou sur la partie droite du corps,une pancréatectomie médiane peut êtreproposée [61, 94, 95]. C’est l’interven-

J Chir 2005,142, N°3 • © Masson, Paris, 2005
137
Revue de la littérature
tion qui s’approche le plus d’une énu-cléation, en terme de préservation duparenchyme pancréatique, et qui per-met de préserver les structures avoisi-nantes (voie biliaire, duodénum, esto-mac, rate). La tranche pancréatiquecéphalique est suturée sur elle-même.Le moignon gauche est en règle géné-rale anastomosé à l’estomac ou au jéju-num (sur une anse en Y). Lorsque l’exé-rèse est plus étendue vers la gauche ousituée d’emblée dans la partie gauche ducorps, le moignon restant est laissé enplace et drainé sans anastomose pan-créatico-digestive. Lorsque l’exérèse dupancréas gauche est complète ou cauda-le et que les vaisseaux spléniques sontconservés, il s’agit une pancréatectomiegauche sans splénectomie [96]. Ces exé-rèse gauches avec ou sans splénectomiepeuvent, dans certaines conditions, s’ef-fectuer sous vidéo-laparoscopie [90, 92,93, 97-102].
Dans la série multicentrique françai-se rapportant les résultats des pancréa-tectomies médianes, sur 53 malades, 17avaient une TEP [61]. La taille moyen-ne de pancréas réséquée était de5 ± 2,2 cm (2 à 15 cm). Le taux de fis-tule pancréatique était de 30 % et lamortalité de 2 %. En l’absence de pan-créatopathie sous jacente, le taux de dia-bète de novo a été de 2 %. La morbiditéglobale des exérèses gauches était del’ordre de 10 % à 3 % et la mortalitéétait inférieure à 1 % [53, 94]. Les com-plications les plus fréquentes sont lesfistules, collections postopératoires,l’hémorragie et le diabète. La morbiditéde la pancréatectomie médiane est plusélevée que celle des pancréatectomiesgauches, probablement du fait de l’ac-cumulation du risque de la fuite pan-créatique du moignon droit et de celuid’une anastomose pancréatico-digestive(sur un parenchyme pancréatique fria-ble). Les pancréatectomies gauches ex-posent, en fonction de l’importance dusacrifice parenchymateux, à un risquede diabète à long terme estimé entre 2et 10 % et qui augmente avec l’étenduede la pancréatectomie [103, 104].
L’influence de la préservation splé-nique sur la morbidité des pancréatec-tomies gauches reste controversée chezl’adulte non-immunodéprimé [105-107]. Cependant, lorsque la tumeur estbénigne et de petite taille (inférieure à2 cm), une conservation splénique estsouhaitable compte tenu d’une plusgrande fréquence et d’une plus grande
gravité des complications infectieusesbactériennes et thromboemboliques àdistance, d’autant plus que le malade estjeune ou immunodéprimé [106-108].Les malades doivent être préférentielle-ment vaccinés contre les souches deStreptococcus pneumoniae idéalement plu-sieurs semaines avant l’intervention ouun mois après l’intervention. Certainsproposent une vaccination contre lesautres bactéries encapsulées commel’Hemophilus influenzae B et le Neisseiriameningococcus (surtout chez les enfants etles malades potentiellement immuno-déprimés) [107, 108]. En tous les cas, etmalgré l’absence de consensus, les adul-tes non immunodéprimés reçoivent uneantibioprophylaxie par une pénicilli-ne A pendant les 2 ans suivant l’inter-vention ou au minimum couvrant la pé-riode postopératoire (le temps que lesvaccins produisent leur effet protec-teur). Le rappel du vaccin est en généralnécessaire tous les 4 à 5 ans. Le maladedoit être averti de consulter un médecinen urgence pour recevoir une antibio-thérapie adaptée en cas d’épisode infec-tieux d’origine bactérienne suspectée(surtout ORL ou méningé) ou prouvée[106-108].
• Duodéno-pancréatectomie céphalique (DPC)
Cette intervention, est réservée aux tu-meurs céphaliques généralement mali-gnes, ou à potentiel malin, ou bénignesmais situées en profondeur à proximitédu canal pancréatique principal. Dansles centres chirurgicaux à gros débit, letaux de mortalité est inférieur à 5 % [53,109-112]. La mortalité augmente signi-ficativement en fonction de l’âge et del’existence de comorbidité (diabète,bronchopathie chronique obstructive etcoronaropathie) [113]. La perméabilitédu tronc coeliaque et la persistance d’unflux en sens normal dans l’artère gastro-duodénale (du territoire coeliaque versla mésentérique supérieure) sont néces-saires pour pratiquer la DPC [114]. Letaux de morbidité de la DPC est de l’or-dre de 20 à 70 % [113]. Les complica-tions postopératoires sont principale-ment représentées par les fistulespancréatico-digestives (10 à 30 %), lestroubles de vidange gastrique (10 à30 %), les collections infectieuses (5 à15 %) et l’hémorragie (1 à 10 %) [53,109, 113, 115, 116]. Le risque de fistuleest plus élevé lorsque le parenchymepancréatique est sain (friable) et quand
le canal de Wirsung est non dilaté [117-120], ce qui est très souvent le cas desTEP qui ne s’accompagnent pas de di-latation canalaire ni de phénomènes depancréatite d’amont. L’intérêt de l’uti-lisation des dérivés de la somatostatinedans la prévention de fistule pancréa-tico-digestive reste discuté, mais la der-nière étude multicentrique randomiséfrançaise est en faveur de l’utilité del’octréotide lorsque le pancréas est fria-ble et chez les malades ayant un canalde Wirsung non dilaté [118].
À long terme, la période post-DPC(évaluée essentiellement pour l’adéno-carcinome du pancréas) est associéedans 65 à 80 % des cas à un amaigrisse-ment transitoire de 5 à 10 kg [103]. Cetamaigrissement se corrige au delà d’unan, et se stabilise à environ 5 % sous lepoids de forme, sauf en cas de récidivetumorale [103]. En l’absence de diabètepré-opératoire, celui-ci ne compliqueque 0 à 7 % des DPC. Après DPC avecanastomose pancréatico-digestive, 30 à60 % des malades doivent être suppléésen enzymes pancréatiques pour corrigerune stéatorrhée clinique [103, 121,122]. Le risque d’ulcère anastomotiqueà distance (5 à 20 %) nécessite la pres-cription systématique d’antisecrétoiresdans les mois suivants la DPC [103,121-123]. L’intérêt fonctionnel de laconservation pylorique reste controver-sé [103, 111, 116, 124]. Afin d’évitertout rebond et le risque d’ulcère anas-tomotique, les malades ayant eu uneDPC pour gastrinome sont traités defaçon transitoire dans la période posto-pératoire par des inhibiteurs de la pom-pe à protons.
Traitement chirurgical en fonction de la nature de la tumeur primitive duodénopancréatiquePour les TE fonctionnelles, la chirurgiepermet la guérison des insulinomesdans 70 à 90 % des cas. En revanche,les gastrinomes ne sont guéris par lachirurgie que dans une proportion al-lant de 20 % à 70 % des cas [2, 125].Les tumeurs plus volumineuses ont sou-vent une extension métastatique (gan-glionnaire et/ou hépatique) et posent leproblème d’une résécabilité au prixd’attitude chirurgicale plus agressive[10, 126]. La recherche de NEM-1 estsystématique devant une TEP [22, 39,127]. Si la place de chirurgie dans le ca-dre d’insulinome-NEM-1 est peu con-

Traitement chirurgical des tumeurs endocrines gastro-entéro-pancréatiques R. Kianmanesh et al.
138
troversée, en revanche elle reste encorecontroversée pour les gastrinomes sur-venant dans le cadre d’une NEM-1 [16,128-133].
• Tumeurs fonctionnelles
InsulinomeSon incidence annuelle est de 0.1 à2 cas pour 100 000 habitants. L’âgemoyen est entre 40 et 60 ans (excep-tionnellement diagnostiquées avantl’âge de 15 ans) avec une incidenceégale dans les 2 sexes [4, 30, 134-136].L’insulinome provoque des manifesta-tions liées à l’hypoglycémie organiquequi est parfois de diagnostic difficile.Elle associe la neuroglycopénie (étatconfusionnel, stupeur, troubles visuels,amnésie, crises convulsives, tremble-ments, céphalées, signe de Babinski,paresthésies, irritabilité, hémiplégie,voire coma) à des signes liés à la répon-se adrénergique (asthénie, sueurs froi-des, pâleur, tachycardie, palpitations).L’augmentation de la prise d’alimentssucrés et l’insulinémie élevée provo-que en règle générale une prise depoids. Lors des crises, le dosage d’uneglycémie basse et une insulinémie etpeptide C paradoxalement élevés et larégression des symptômes après admi-nistration de glucose affirment le dia-gnostic.
Insulinome sporadiqueLa tumeur siège dans 98 % des cas surle pancréas et dans 2 % ailleurs (pan-créas aberrant, duodénum, antre, hilede la rate) [135, 136]. Dans plus de90 % des cas, il s’agit d’une forme spo-radique avec une tumeur unique de pe-tite taille (0,5 et 3 cm) [4, 30, 134-136].
Pour ces formes, l’énucléation, (si elleest techniquement possible) est le trai-tement de choix. Dans 10 % des cas,l’insulinome est malin et se présentesous forme d’une tumeur volumineuseavec métastases (ganglionnaires et/ouhépatiques) ou plus rarement l’évolu-tion est marquée par la survenue demétastases. Dans ces cas, le pronosticdépend surtout de la possibilité d’exé-rèse complète de l’ensemble des lésionstumorales. La localisation des insulino-mes est pour 1/3 des cas dans la tête et/ou le crochet, 1/3 dans l’isthme ou lecorps, et 1/3 dans la queue du pancréas[135, 136]. La tumeur est infra-centi-métrique dans 40 % des cas, elle mesu-re entre 1 et 3 cm et dans près dans50 % des cas, et elle est supérieure à3 cm dans 10 % des cas. Lorsque l’in-sulinome mesure plus de 2 cm et sur-tout quand il est proche du canal deWirsung, une pancréatectomie est in-diquée. La persistance d’une hypogly-cémie postopératoire qui concernemoins de 10 % des cas, peut être due àla persistance de lésions multiples et/ouméconnues, la présence de métastasesnon détectées ou l’existence d’une hy-perplasie diffuse de la glande (nesidio-blastose) [134, 137].
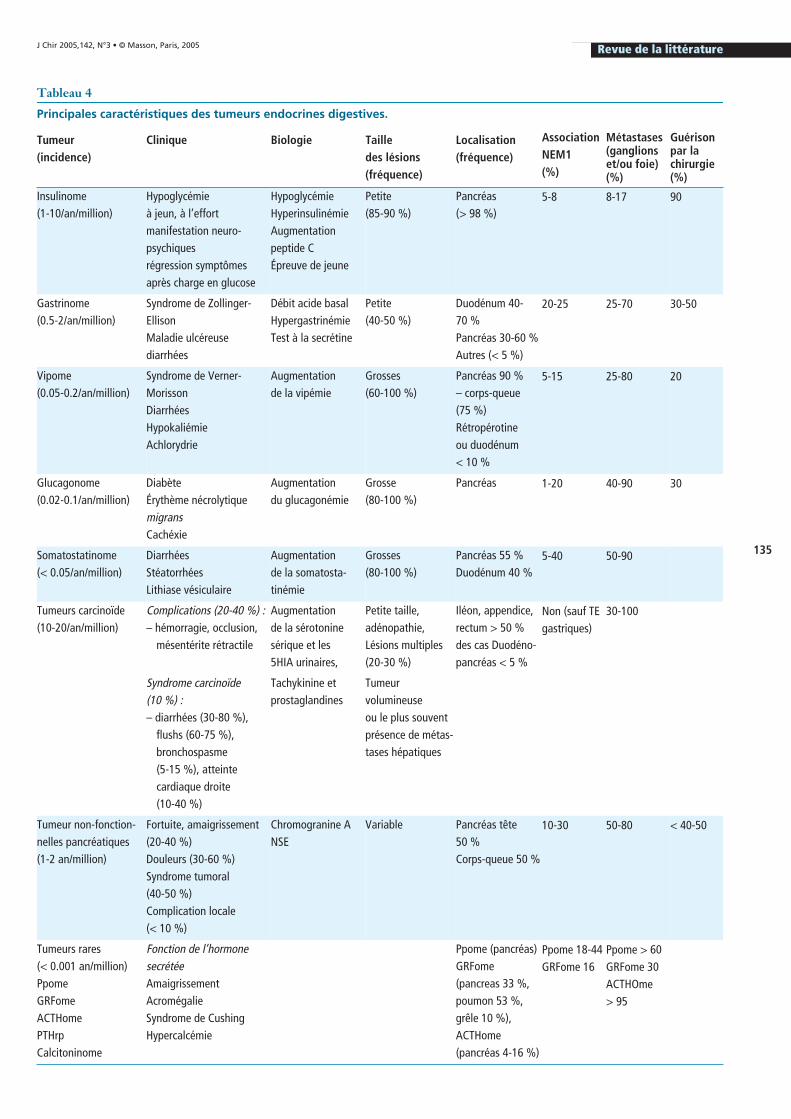
Les explorations pré-opératoires ac-tuelles (écho-endoscopie, scanner mul-tiphasique hélicoïdal et IRM) permet-tent de localiser la lésion dans plus de90 % des cas [74, 135]. En cas de lésionunique, la probabilité de guérison, par-fois au prix d’une réintervention, est su-périeure de 70 à 80 % et les taux de sur-vie sans récidive à 5 ans sont supérieursà 90 % (figure 1) [78, 135, 138, 139]. Lasurveillance per opératoire de la glycé-
Figure 1 : Insulinome de la tête du pancréas visible sur le scanner en phase artérielle venant au contact de la voie biliaire intrapancréatique.
mie (toutes les 10 min) permet d’éviterla survenue d’hypoglycémie per opéra-toire, qui n’a pas de traduction clinique,mais qui peut entraîner des lésions neu-rologiques irréversibles [56].
Insulinome s’intégrant dans le cadre d’une NEM-1Dans moins de 10 % des cas, l’insuli-nome s’intègre dans le cadre d’uneNEM-1 et nécessite une prise en char-ge spécifique [134, 135, 138]. La stra-tégie chirurgicale devant un insulinomedans le cadre d’une NEM-1 est pluscomplexe car il s’agit dans 70 % des casde lésions multiples et de petite taille etpouvant correspondre à des microadé-nomes échappant à la palpation et àl’échographie per opératoire [138].L’indication opératoire est justifiéedans certains cas devant le risque vitald’une hypoglycémie non contrôlée parle traitement médical. L’attitude chi-rurgicale est variable selon le nombreet la localisation des lésions, mais d’unefaçon générale, l’énucléation des lé-sions visibles est associée à une inciden-ce élevée de récidive (40 %) alors quela pancréatectomie subtotale gauche(isthmo-corporéo-caudale) associée àl’énucléation des lésions céphaliques,donne un taux de récidive inférieur à10 % à 5 ans [140, 141]. Il faut souli-gner l’importance de la prise en charge,en première intention, de l’hyperpara-thyroïdie qui est présente dans 90 %des cas [142].
GastrinomeSon incidence annuelle est d’environ0,5 à 3 cas par million d’habitants. Lesexe ratio est de 2 hommes pour unefemme. L’âge moyen de découverte estentre 45 et 50 ans (extrêmes 9 à 90 ans).L’hypergastrinémie provoque une hy-persécrétion acide gastrique qui est res-ponsable des symptômes du syndromede Zollinger-Ellison. Les symptômessont ceux de la maladie ulcéreuse quiest souvent récidivante, compliquée ouchronique, et la diarrhée chronique (2/3des cas), cédant à la prise d’inhibiteursde la pompe à protons. Les ulcères peu-vent siéger sur l’ensemble du tractus di-gestif haut, il peut s’agir d’une oesopha-gite sévère, des ulcères gastriques ouduodénaux bulbaires ou post-bulbaires,siégeant souvent au-delà du genus supe-rius voire sur le jéjunum. L’absenced’infection à l’Helicobacter Pylori (en de-hors d’un traitement antisécrétoire parinhibiteurs de la pompe à protons) est
139
J Chir 2005,142, N°3 • © Masson, Paris, 2005 Revue de la littérature
très évocatrice. Près de 9 malades sur10 ont des ulcérations du tractus diges-tif haut, la moitié d’entre eux ont uneoesophagite peptique [131, 133, 143,144].
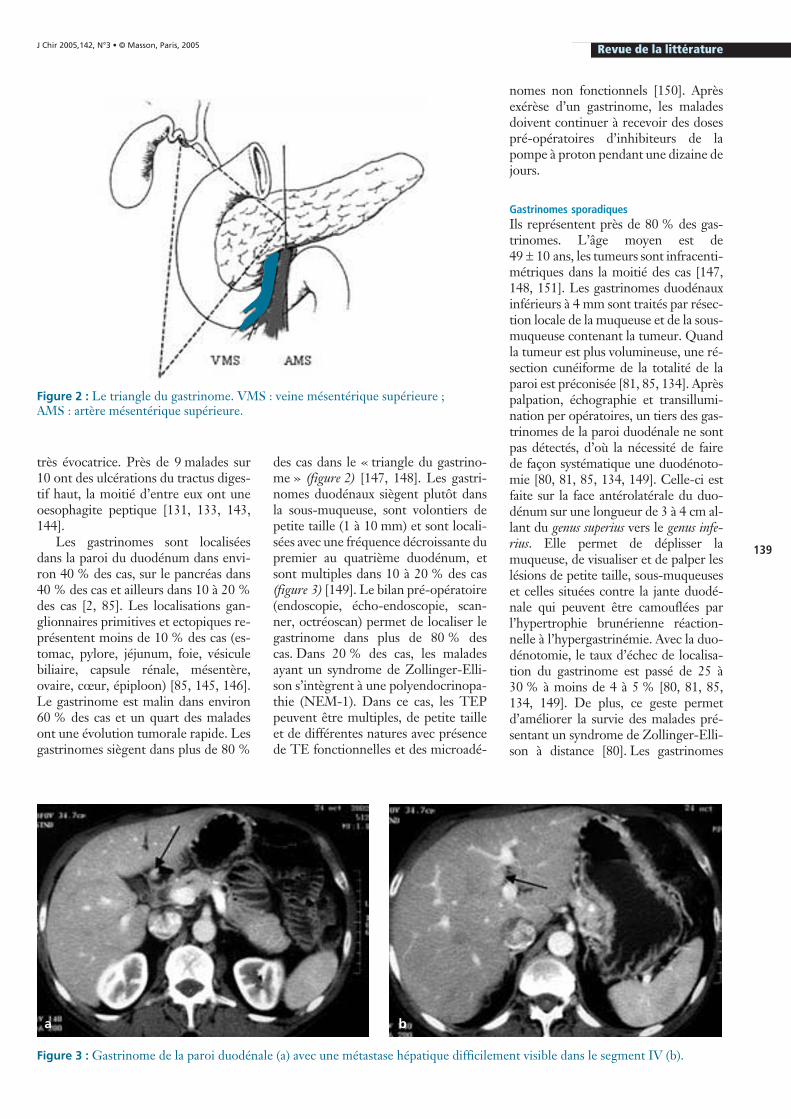
Les gastrinomes sont localiséesdans la paroi du duodénum dans envi-ron 40 % des cas, sur le pancréas dans40 % des cas et ailleurs dans 10 à 20 %des cas [2, 85]. Les localisations gan-glionnaires primitives et ectopiques re-présentent moins de 10 % des cas (es-tomac, pylore, jéjunum, foie, vésiculebiliaire, capsule rénale, mésentère,ovaire, cœur, épiploon) [85, 145, 146].Le gastrinome est malin dans environ60 % des cas et un quart des maladesont une évolution tumorale rapide. Lesgastrinomes siègent dans plus de 80 %
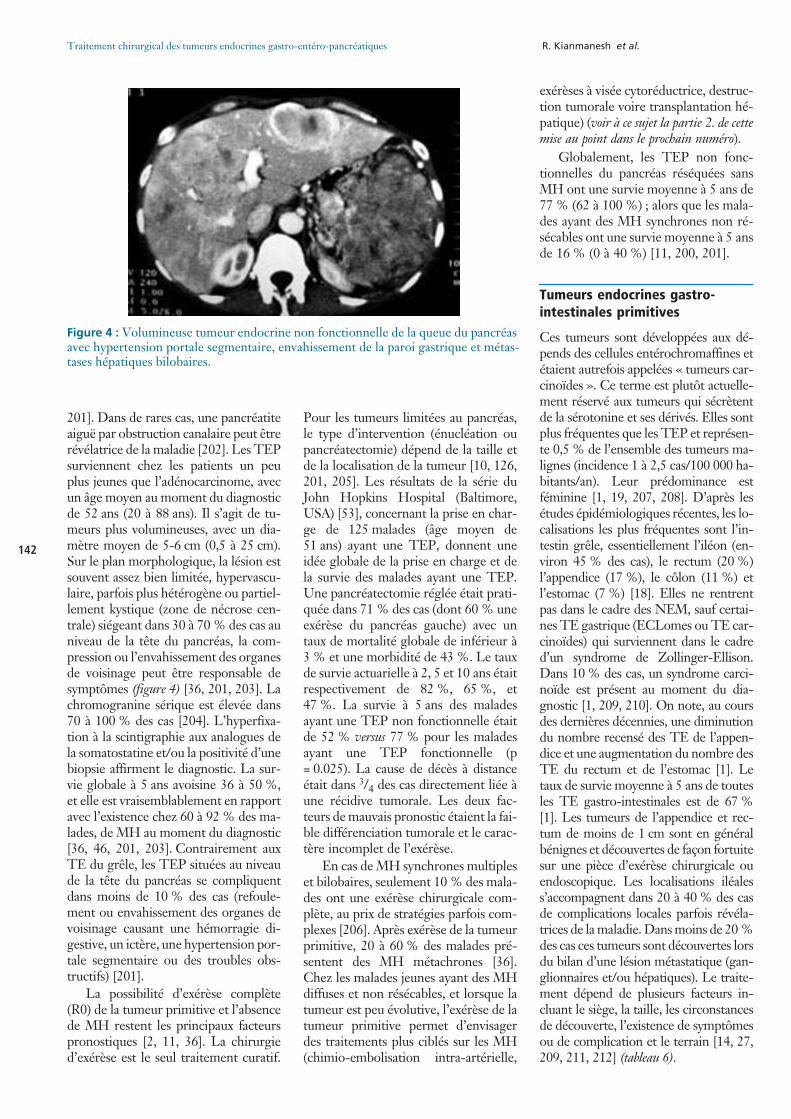
des cas dans le « triangle du gastrino-me » (figure 2) [147, 148]. Les gastri-nomes duodénaux siègent plutôt dansla sous-muqueuse, sont volontiers depetite taille (1 à 10 mm) et sont locali-sées avec une fréquence décroissante dupremier au quatrième duodénum, etsont multiples dans 10 à 20 % des cas(figure 3) [149]. Le bilan pré-opératoire(endoscopie, écho-endoscopie, scan-ner, octréoscan) permet de localiser legastrinome dans plus de 80 % descas. Dans 20 % des cas, les maladesayant un syndrome de Zollinger-Elli-son s’intègrent à une polyendocrinopa-thie (NEM-1). Dans ce cas, les TEPpeuvent être multiples, de petite tailleet de différentes natures avec présencede TE fonctionnelles et des microadé-
Figure 2 : Le triangle du gastrinome. VMS : veine mésentérique supérieure ; AMS : artère mésentérique supérieure.
Figure 3 : Gastrinome de la paroi duodénale (a) avec une métastase hépatique difficilement visible dans le segment IV (b).
a b
nomes non fonctionnels [150]. Aprèsexérèse d’un gastrinome, les maladesdoivent continuer à recevoir des dosespré-opératoires d’inhibiteurs de lapompe à proton pendant une dizaine dejours.
Gastrinomes sporadiquesIls représentent près de 80 % des gas-trinomes. L’âge moyen est de49 ± 10 ans, les tumeurs sont infracenti-métriques dans la moitié des cas [147,148, 151]. Les gastrinomes duodénauxinférieurs à 4 mm sont traités par résec-tion locale de la muqueuse et de la sous-muqueuse contenant la tumeur. Quandla tumeur est plus volumineuse, une ré-section cunéiforme de la totalité de laparoi est préconisée [81, 85, 134]. Aprèspalpation, échographie et transillumi-nation per opératoires, un tiers des gas-trinomes de la paroi duodénale ne sontpas détectés, d’où la nécessité de fairede façon systématique une duodénoto-mie [80, 81, 85, 134, 149]. Celle-ci estfaite sur la face antérolatérale du duo-dénum sur une longueur de 3 à 4 cm al-lant du genus superius vers le genus infe-rius. Elle permet de déplisser lamuqueuse, de visualiser et de palper leslésions de petite taille, sous-muqueuseset celles situées contre la jante duodé-nale qui peuvent être camouflées parl’hypertrophie brunérienne réaction-nelle à l’hypergastrinémie. Avec la duo-dénotomie, le taux d’échec de localisa-tion du gastrinome est passé de 25 à30 % à moins de 4 à 5 % [80, 81, 85,134, 149]. De plus, ce geste permetd’améliorer la survie des malades pré-sentant un syndrome de Zollinger-Elli-son à distance [80]. Les gastrinomes

Traitement chirurgical des tumeurs endocrines gastro-entéro-pancréatiques R. Kianmanesh et al.
140
pancréatiques sont localisés de façonéquivalente dans la queue et la régionisthmo-corporéo-céphalique du pan-créas [2, 152]. Les localisations pure-ment ganglionnaires représentent danscertaines séries jusqu’à 10 % des cas [2,148]. Les gastrinomes pancréatiques(40 % des cas) de moins de 2 cm et si-tués à distance du canal pancréatiquepeuvent être énucléés, les autres cas né-cessitent une exérèse pancréatique ré-glée. Quelle que soit la taille et la loca-lisation de la tumeur primitive, lafréquence élevée des métastases gan-glionnaires justifie la réalisation systé-matique d’un curage ganglionnaire desaires de drainage [81, 85, 134]. La survieglobale sans récidive après exérèse estde 40 % à 5 ans et de 34 % à 10 ans [2,85, 134]. Dans l’étude de Norton et al.[85], le gastrinome était sporadiquechez 123/151 malades (81 %). Uneréintervention était nécessaire chez15 % des malades (180 opérations pour151 malades avec 23 malades ayant eudeux ou trois opérations). Le gastrino-me était localisé lors de la première ex-ploration chirurgicale dans 88 % descas, et dans 100 % des cas lors des ex-plorations ultérieures (pas de différenceentre les malades ayant un gastrinomesporadique versus NEM-1).
Gastrinomes s’intégrant dans le cadre d’une NEM-1Ils représentent environ 20 % des gas-trinomes et plus de la moitié des tu-meurs endocrines duodénopancréati-ques découvertes chez les malades ayantune NEM-1 (tableau 5) [129, 153-155].L’âge moyen est en général d’une dizai-ne d’années inférieur à celui des formessporadiques. La tumeur peut être isolée,multiple ou associé à d’autres types deTEP (notamment des insulinomes et
des tumeurs non fonctionnelles). La lo-calisation pancréatique semble être plusfréquente que dans les cas sporadiques[85]. La place de la chirurgie pour cesformes reste très controversée essentiel-lement pour trois raisons : 1) l’efficacitéimportante des antisécrétoires et labonne tolérance à long terme du traite-ment médical et ; 2) la bonne surviespontanée à distance des malades et en-fin ; 3) l’absence de proportion de ma-lades guéris par la chirurgie dans cetteindication (taux de récidive à 5 et10 ans de 96 et 100 % respectivement)[2, 85, 128, 129, 134, 153-156].Dans la série de Sheppard et al. [138],parmi 65 malades ayant une TEP surNEM-1, 7 (11 %) avaient ou ont déve-loppés des MH et 3 sont décédés direc-tement de l’évolution de leurs MH.Dans la série de Weber et al. (suivimoyen de 7,1 ans), 6 % des maladesavaient des MH synchrones et 9 % desmalades ont développé des MH méta-chrones [33]. Dans la série de Cadiot etal. [132] parmi 77 gastrinomes NEM-1(suivi moyen de 8,5 ans), 10 % des ma-lades (n = 8) ont développé des MH mé-tachrones et 2 décès (2.6 %) étaient di-rectement liés à l’évolution des MH.Ces éléments sont des arguments pourne proposer un geste d’exérèse pancréa-tique que lorsque la taille du gastrinomeest de plus de 2 à 3 cm [2, 33, 128, 132,154, 157]. Cependant pour éviter la sur-venue des MH, on peut avoir une atti-tude plus agressive [150]. En effet, dansla série de Doherty et al. [150], parmiles 103 malades ayant une TEP surNEM-1, 46 % des décès étaient dus àl’évolution maligne des TEP. Dans cel-le de Lowney et al. [158], parmi 43NEM-1 ayant 48 TEP, une évolutionmétastatique à distance a été observée
Tableau 5
Incidence relative des tumeurs endocrines duodénopancréatiques parmi les malades ayant une néoplasie endocrinienne multiple de type 1*.
Fréquence (%) Survie à 10 ans
• Gastrinome 58 82
• Insulinome 3 91
• Gastrinomes et Insulinomes 5
• Glucagonome 1,6
• Vipome 1 54
• Somatostatinome 0,6
• Tumeurs non-fonctionnelles 19 62
* D’après Lévy-Bohbot N et al. Registre du groupe des Tumeurs Endocrines (GTE).
chez un tiers des cas ayant une tumeurde moins d’ un cm. Cette évolution mé-tastatique potentiellement fatale restedonc le principal argument en faveurd’une attitude plus agressive de certai-nes équipes vis-à-vis des gastrinomessurvenant dans le cadre d’une NEM-1[150, 156, 158]. Un sous-groupe de ma-lades ayant un syndrome de Zollinger-Ellison a une évolution métastatiquehépatique rapide et d’autres non.L’existence d’une mutation dans lesexons 3 à 8 du gène de la ménine, sem-ble être associée à une plus faible mali-gnité [159]. Dans tous les cas, comptetenu de la fréquence élevée d’atteintepancréatique multiple, le geste d’exérè-se pancréatique n’est pas encore stan-dardisé et dépend de la localisation dela majorité des lésions. Il peut s’agird’une exploration complète du blocduodénopancréatique avec duodé-notomie et une pancréatectomie distale,l’énucléation des lésions de la tête oud’une DPC avec énucléation des lésionscorporéo-caudales [156]. Lorsqued’autres TEP fonctionnelles sont pré-sentes, parfois avec présence simultanéede gastrinome et d’insulinome, l’indica-tion chirurgicale est plus facilement po-sée [160-162]. Dans tous les cas, malgrél’existence de cas d’hyperparathyroïdienormocalcémiques (jusqu’à 40 % desgastrinomes-NEM-1), la recherche etle traitement chirurgical d’une atteintedes parathyroïdes (hyperplasie) avant lachirurgie pancréatique est systématique[157, 163].
Autres tumeurs endocrines duodénopancréatiques fonctionnellesLes autres TE fonctionnelles (glucago-nome, VIPome, somatostatinome, GR-Fome, ACTHome, tumeurs serétant laPTH-rP, calcitoninome) sont beau-coup plus rares [53, 125, 164, 165]. Lestumeurs sont en général volumineuses,malignes, avec une extension loco-régionale et/ou une atteinte hépatique(tableau 4). Leur exérèse, lorsqu’elle estpossible, nécessite parfois une gesteélargi aux organes adjacents.
GlucagonomeIl représente moins de 5 % desTEP. L’âge de survenue moyen est de65 ans. Les manifestations associent deslésions cutanées caractéristiques : unérythème nécrotique migrateur (60 à90 %), un amaigrissement important(80 %), un diabète (87 %), des manifes-tations thrombo-emboliques (40 %),

J Chir 2005,142, N°3 • © Masson, Paris, 2005
141
Revue de la littérature
une diarrhée (20 %), des signes neurop-sychiques (dépression, insomnie), uneanémie (50 à 90 %) et une hypoaminoa-cidémie (30 à 90 %) [56]. Le glucagonsérique est élevé (parfois de façon mo-dérée, x 2N). La tumeur est toujourspancréatique, dans 2/3 des cas corpo-réo-caudale et près de 50 % dans laqueue [166]. Elle mesure moins de20 mm dans 20 % des cas, entre 3 à10 cm dans 60 % des cas, et elle est ma-ligne dans 60 à 80 % des cas [53, 56,166-171]. Le traitement est chirurgicalpour les tumeurs résécables. Une cyto-réduction tumorale est parfois indiquéepour contrôler le syndrome hormonal.Les dérivés de la somatostatine amélio-rent les symptômes dans 90 % des cas[172-175]. La survie globale à 10 ans estde 64 % pour les malades sans dissémi-nation métastatiques et de 51 % pourceux ayant des métastases [166].
VipomeLe Vipome ou syndrome de Verner-Morrison ou « WDHA syndrom »(watery diarrhea hypokaliemia achlo-ridria) représente moins de 5 % desTEP et s’observe à tout âge avec unpic à la quatrième décennie [176, 177].Il est 3 fois plus fréquent chez la fem-me. Il se caractérise par l’existence dediarrhées profuses (90 %) parfois su-périeures à 1 litre/jour, continue ouintermittentes, ainsi qu’une déshydra-tation et un amaigrissement (44 à72 %). On observe des troubles hy-droélectrolytiques dans 70 % des cas(hypokaliémie, acidose métabolique,hypochlorhydrie) [178]. Une hyper-calcémie de cause multifactorielle estfréquente (27 à 75 %). Le VIP sériqueélevé fait le diagnostic [179]. La lésionest en général de grande taille (supé-rieure à 3 cm) souvent pancréatique(supérieure à 90 % des cas), et dans lamoitié des cas située dans la queue[56]. Elle peut être exceptionnelle-ment de localisation intrahépatique[180]. Trente à 80 % des malades ontdes MH au moment du diagnostic[179]. La chirurgie est indiquée pourles tumeurs localisées après une réé-quilibration hydroélectrolytique souscouverture des analogues de la soma-tostatine qui permettent d’améliorerles symptômes dans 50 à 60 % des cas[179]. Environ 1/3 des malades ontune exérèse curative. La surviemoyenne est de 3,6 ans mais des sur-vies au-delà de 15 ans ont été obser-vées après exérèse.
SomatostatinomeIl est une tumeur exceptionnelle [56,181-184]. L’âge moyen de diagnosticest de 50 ans. Il est plus fréquent chezl’homme. Les symptômes associent dia-bète, lithiase biliaire, amaigrissement,diarrhée et parfois stéatorrhée. La tu-meur est de localisation duodénale(35 %), pancréatique (55 %) ou ampul-laire (inférieure à 10 %). Les localisa-tions pancréatiques fréquentes sont latête et la queue. Il s’agit souvent d’unetumeur large (5 cm en moyenne) de larégion céphalique du pancréas avec mé-tastases locorégionales (ganglions, veineporte, foie) [56, 185]. Dans 50 % desformes duodénales, il existe une maladiede von Recklinghausen associée [186,187]. Le diagnostic est fait devant unesomatostatinémie souvent très élevée(supérieure à 40N). Le traitement estchirurgical, le plus souvent il s’agitd’une DPC, mais environ 40 % des ma-lades ne sont pas réséqués (évolution lo-cale et/ou présence de métastases). Encas de dissémination métastatique, lesanalogues de la somatostatine peuventêtre utiles. La survie globale à 5 ans estentre 40 % et 60 % ; en l’absence demétastases, elle est proche de 100 %[56, 184, 185].
GRFomeLe GRFome responsable de survenued’une acromégalie est très rare [17, 188,189]. L’âge de survenue est de 40 ans(15 à 60 ans). Il existe une prédominan-ce féminine. Il peut être associée à uneNEM-1 [190]. Le GH sérique est élevé.L’augmentation de la prolactinémie estinconstante. Il n’existe pas de tumeurhypophysaire. Le dosage de GRF séri-que confirme le diagnostic. Le but dubilan est de localiser la lésion pancréa-tique et d’éventuelles lésions associées(recherche de TE grêle, thymus, para-thyroïdes, TE bronchique). L’exérèsechirurgicale est couverte par un traite-ment aux analogues de la somatostatine.
Tumeurs hypercalcémiantesElles sont très rares [191]. Comme pourcertaines tumeurs (exemple du cancerdu sein), l’hypercalcémie peut être laconséquence d’une lyse osseuse tumo-rale ou paranéoplasique secondaire à lasécrétion de substance PTH-like (Para-thyroid hormone-related protein, ouPTH-rP) [191-193]. Les manifestationscliniques sont celles de l’hypercalcémieet peuvent inclure des signes neuropsy-chiques. Le diagnostic repose sur la mi-
se en évidence d’une hypercalcémie etl’absence de cause évidente d’hypercal-cémie (pas d’adénome parathyroïdien,PTH normale, pas de lésions secondai-res osseuses à la scintigraphie). Le do-sage de PTH-rP élevé affirme le dia-gnostic. Les lésions sont souventmalignes, volumineuses, avec souventprésence de MH multiples. Le traite-ment consiste à contrôler l’hypercalcé-mie qui peut menacer le pronostic vital(réhydratation et cures par diphospho-nates) et de proposer, si possible, uneexérèse chirurgicale à visée curative ouplus rarement à visée cytoréductricepour aider à un meilleur contrôle dusyndrome hormonal par le traitementmédical.
ACTHomeC’est une tumeur pancréatique très raresecrétant de l’ACTH qui peut être res-ponsable d’un syndrome de Cushing[53]. Elle représente moins de 10 % dessyndromes de Cushing ectopiques ets’accompagne très souvent de métasta-ses, le plus souvent hépatiques (plus de95 % des cas) [194, 195].
StérotoninomeLe Sérotoninome du pancréas est ex-ceptionnel et peut être responsable desyndrome carcinoïde (moins de 1 %,une soixantaine de cas publiés) [51, 196,197].
CalcitoninomeC’est une TEP très rare qui peut s’ac-compagner de diarrhées [198].
NeurotensinomeEnfin 35 cas de Neurotensinome (tu-meur secrétant de la neurotensine) ontété apportés [199].
• Tumeurs non fonctionnelles
Ces tumeurs représentent 30 à 60 % desTEP et sont malignes dans 80 % des cas[4, 26, 46, 134, 200]. Elles peuvent sur-venir dans le cadre de NEM-1 (environ10 % des cas) parfois en association avecdes TE fonctionnelles. Compte tenu del’absence de symptômes hormonaux, el-les sont découvertes à un stade plus tar-dif que les TEP fonctionnelles, devantdes manifestations liées au syndrome demasse ou lors du bilan de lésions métas-tatiques souvent hépatiques. Les princi-paux symptômes révélateurs du dia-gnostic sont les douleurs abdominales(40 à 70 %), une altération de l’état gé-néral et l’ictère (20 à 50 %) [53,
Traitement chirurgical des tumeurs endocrines gastro-entéro-pancréatiques R. Kianmanesh et al.
142
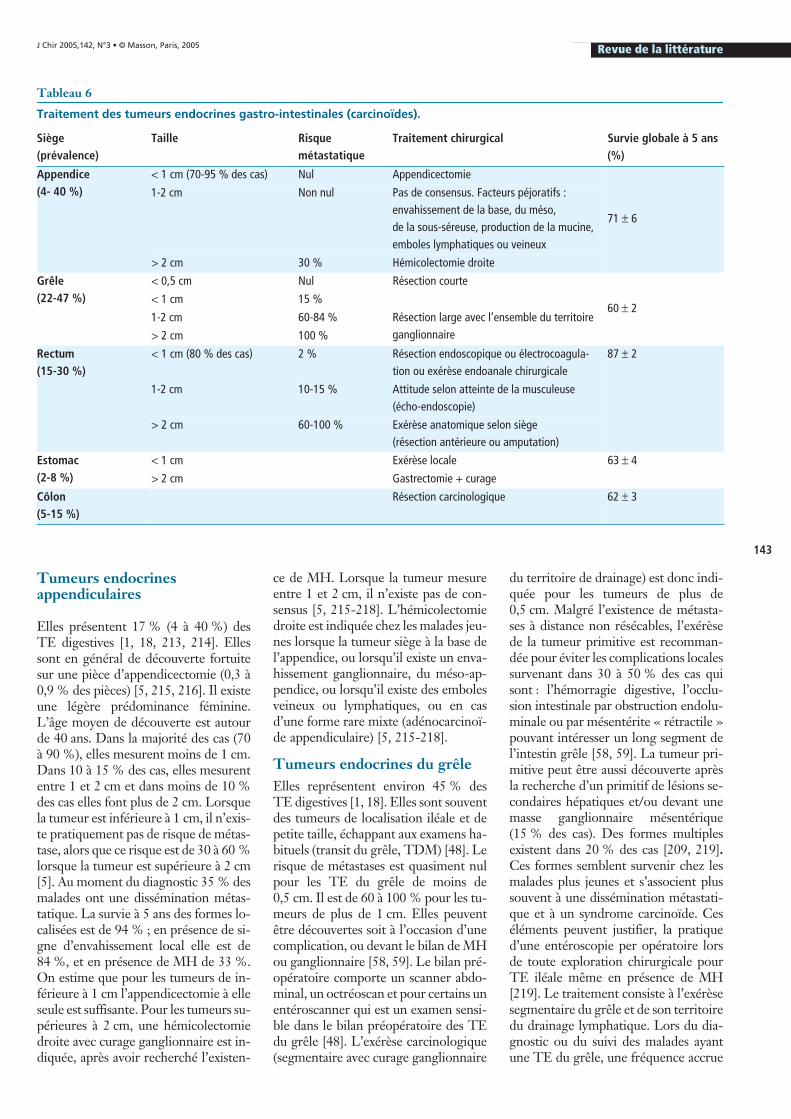
201]. Dans de rares cas, une pancréatiteaiguë par obstruction canalaire peut êtrerévélatrice de la maladie [202]. Les TEPsurviennent chez les patients un peuplus jeunes que l’adénocarcinome, avecun âge moyen au moment du diagnosticde 52 ans (20 à 88 ans). Il s’agit de tu-meurs plus volumineuses, avec un dia-mètre moyen de 5-6 cm (0,5 à 25 cm).Sur le plan morphologique, la lésion estsouvent assez bien limitée, hypervascu-laire, parfois plus hétérogène ou partiel-lement kystique (zone de nécrose cen-trale) siégeant dans 30 à 70 % des cas auniveau de la tête du pancréas, la com-pression ou l’envahissement des organesde voisinage peut être responsable desymptômes (figure 4) [36, 201, 203]. Lachromogranine sérique est élevée dans70 à 100 % des cas [204]. L’hyperfixa-tion à la scintigraphie aux analogues dela somatostatine et/ou la positivité d’unebiopsie affirment le diagnostic. La sur-vie globale à 5 ans avoisine 36 à 50 %,et elle est vraisemblablement en rapportavec l’existence chez 60 à 92 % des ma-lades, de MH au moment du diagnostic[36, 46, 201, 203]. Contrairement auxTE du grêle, les TEP situées au niveaude la tête du pancréas se compliquentdans moins de 10 % des cas (refoule-ment ou envahissement des organes devoisinage causant une hémorragie di-gestive, un ictère, une hypertension por-tale segmentaire ou des troubles obs-tructifs) [201].
La possibilité d’exérèse complète(R0) de la tumeur primitive et l’absencede MH restent les principaux facteurspronostiques [2, 11, 36]. La chirurgied’exérèse est le seul traitement curatif.
Pour les tumeurs limitées au pancréas,le type d’intervention (énucléation oupancréatectomie) dépend de la taille etde la localisation de la tumeur [10, 126,201, 205]. Les résultats de la série duJohn Hopkins Hospital (Baltimore,USA) [53], concernant la prise en char-ge de 125 malades (âge moyen de51 ans) ayant une TEP, donnent uneidée globale de la prise en charge et dela survie des malades ayant une TEP.Une pancréatectomie réglée était prati-quée dans 71 % des cas (dont 60 % uneexérèse du pancréas gauche) avec untaux de mortalité globale de inférieur à3 % et une morbidité de 43 %. Le tauxde survie actuarielle à 2, 5 et 10 ans étaitrespectivement de 82 %, 65 %, et47 %. La survie à 5 ans des maladesayant une TEP non fonctionnelle étaitde 52 % versus 77 % pour les maladesayant une TEP fonctionnelle (p= 0.025). La cause de décès à distanceétait dans 3/4 des cas directement liée àune récidive tumorale. Les deux fac-teurs de mauvais pronostic étaient la fai-ble différenciation tumorale et le carac-tère incomplet de l’exérèse.
En cas de MH synchrones multipleset bilobaires, seulement 10 % des mala-des ont une exérèse chirurgicale com-plète, au prix de stratégies parfois com-plexes [206]. Après exérèse de la tumeurprimitive, 20 à 60 % des malades pré-sentent des MH métachrones [36].Chez les malades jeunes ayant des MHdiffuses et non résécables, et lorsque latumeur est peu évolutive, l’exérèse de latumeur primitive permet d’envisagerdes traitements plus ciblés sur les MH(chimio-embolisation intra-artérielle,
Figure 4 : Volumineuse tumeur endocrine non fonctionnelle de la queue du pancréas avec hypertension portale segmentaire, envahissement de la paroi gastrique et métas-tases hépatiques bilobaires.
exérèses à visée cytoréductrice, destruc-tion tumorale voire transplantation hé-patique) (voir à ce sujet la partie 2. de cettemise au point dans le prochain numéro).
Globalement, les TEP non fonc-tionnelles du pancréas réséquées sansMH ont une survie moyenne à 5 ans de77 % (62 à 100 %) ; alors que les mala-des ayant des MH synchrones non ré-sécables ont une survie moyenne à 5 ansde 16 % (0 à 40 %) [11, 200, 201].
Tumeurs endocrines gastro-intestinales primitives
Ces tumeurs sont développées aux dé-pends des cellules entérochromaffines etétaient autrefois appelées « tumeurs car-cinoïdes ». Ce terme est plutôt actuelle-ment réservé aux tumeurs qui sécrètentde la sérotonine et ses dérivés. Elles sontplus fréquentes que les TEP et représen-te 0,5 % de l’ensemble des tumeurs ma-lignes (incidence 1 à 2,5 cas/100 000 ha-bitants/an). Leur prédominance estféminine [1, 19, 207, 208]. D’après lesétudes épidémiologiques récentes, les lo-calisations les plus fréquentes sont l’in-testin grêle, essentiellement l’iléon (en-viron 45 % des cas), le rectum (20 %)l’appendice (17 %), le côlon (11 %) etl’estomac (7 %) [18]. Elles ne rentrentpas dans le cadre des NEM, sauf certai-nes TE gastrique (ECLomes ou TE car-cinoïdes) qui surviennent dans le cadred’un syndrome de Zollinger-Ellison.Dans 10 % des cas, un syndrome carci-noïde est présent au moment du dia-gnostic [1, 209, 210]. On note, au coursdes dernières décennies, une diminutiondu nombre recensé des TE de l’appen-dice et une augmentation du nombre desTE du rectum et de l’estomac [1]. Letaux de survie moyenne à 5 ans de toutesles TE gastro-intestinales est de 67 %[1]. Les tumeurs de l’appendice et rec-tum de moins de 1 cm sont en généralbénignes et découvertes de façon fortuitesur une pièce d’exérèse chirurgicale ouendoscopique. Les localisations iléaless’accompagnent dans 20 à 40 % des casde complications locales parfois révéla-trices de la maladie. Dans moins de 20 %des cas ces tumeurs sont découvertes lorsdu bilan d’une lésion métastatique (gan-glionnaires et/ou hépatiques). Le traite-ment dépend de plusieurs facteurs in-cluant le siège, la taille, les circonstancesde découverte, l’existence de symptômesou de complication et le terrain [14, 27,209, 211, 212] (tableau 6).
J Chir 2005,142, N°3 • © Masson, Paris, 2005
143
Revue de la littérature
Tumeurs endocrines appendiculaires
Elles présentent 17 % (4 à 40 %) desTE digestives [1, 18, 213, 214]. Ellessont en général de découverte fortuitesur une pièce d’appendicectomie (0,3 à0,9 % des pièces) [5, 215, 216]. Il existeune légère prédominance féminine.L’âge moyen de découverte est autourde 40 ans. Dans la majorité des cas (70à 90 %), elles mesurent moins de 1 cm.Dans 10 à 15 % des cas, elles mesurententre 1 et 2 cm et dans moins de 10 %des cas elles font plus de 2 cm. Lorsquela tumeur est inférieure à 1 cm, il n’exis-te pratiquement pas de risque de métas-tase, alors que ce risque est de 30 à 60 %lorsque la tumeur est supérieure à 2 cm[5]. Au moment du diagnostic 35 % desmalades ont une dissémination métas-tatique. La survie à 5 ans des formes lo-calisées est de 94 % ; en présence de si-gne d’envahissement local elle est de84 %, et en présence de MH de 33 %.On estime que pour les tumeurs de in-férieure à 1 cm l’appendicectomie à elleseule est suffisante. Pour les tumeurs su-périeures à 2 cm, une hémicolectomiedroite avec curage ganglionnaire est in-diquée, après avoir recherché l’existen-
ce de MH. Lorsque la tumeur mesureentre 1 et 2 cm, il n’existe pas de con-sensus [5, 215-218]. L’hémicolectomiedroite est indiquée chez les malades jeu-nes lorsque la tumeur siège à la base del’appendice, ou lorsqu’il existe un enva-hissement ganglionnaire, du méso-ap-pendice, ou lorsqu’il existe des embolesveineux ou lymphatiques, ou en casd’une forme rare mixte (adénocarcinoï-de appendiculaire) [5, 215-218].
Tumeurs endocrines du grêleElles représentent environ 45 % desTE digestives [1, 18]. Elles sont souventdes tumeurs de localisation iléale et depetite taille, échappant aux examens ha-bituels (transit du grêle, TDM) [48]. Lerisque de métastases est quasiment nulpour les TE du grêle de moins de0,5 cm. Il est de 60 à 100 % pour les tu-meurs de plus de 1 cm. Elles peuventêtre découvertes soit à l’occasion d’unecomplication, ou devant le bilan de MHou ganglionnaire [58, 59]. Le bilan pré-opératoire comporte un scanner abdo-minal, un octréoscan et pour certains unentéroscanner qui est un examen sensi-ble dans le bilan préopératoire des TEdu grêle [48]. L’exérèse carcinologique(segmentaire avec curage ganglionnaire
du territoire de drainage) est donc indi-quée pour les tumeurs de plus de0,5 cm. Malgré l’existence de métasta-ses à distance non résécables, l’exérèsede la tumeur primitive est recomman-dée pour éviter les complications localessurvenant dans 30 à 50 % des cas quisont : l’hémorragie digestive, l’occlu-sion intestinale par obstruction endolu-minale ou par mésentérite « rétractile »pouvant intéresser un long segment del’intestin grêle [58, 59]. La tumeur pri-mitive peut être aussi découverte aprèsla recherche d’un primitif de lésions se-condaires hépatiques et/ou devant unemasse ganglionnaire mésentérique(15 % des cas). Des formes multiplesexistent dans 20 % des cas [209, 219].Ces formes semblent survenir chez lesmalades plus jeunes et s’associent plussouvent à une dissémination métastati-que et à un syndrome carcinoïde. Ceséléments peuvent justifier, la pratiqued’une entéroscopie per opératoire lorsde toute exploration chirurgicale pourTE iléale même en présence de MH[219]. Le traitement consiste à l’exérèsesegmentaire du grêle et de son territoiredu drainage lymphatique. Lors du dia-gnostic ou du suivi des malades ayantune TE du grêle, une fréquence accrue
Tableau 6
Traitement des tumeurs endocrines gastro-intestinales (carcinoïdes).
Siège(prévalence)
Taille Risque métastatique
Traitement chirurgical Survie globale à 5 ans (%)
Appendice(4- 40 %)
< 1 cm (70-95 % des cas) Nul Appendicectomie
71 ± 6
1-2 cm Non nul Pas de consensus. Facteurs péjoratifs : envahissement de la base, du méso, de la sous-séreuse, production de la mucine, emboles lymphatiques ou veineux
> 2 cm 30 % Hémicolectomie droite
Grêle(22-47 %)
< 0,5 cm Nul Résection courte
60 ± 2< 1 cm 15 %
1-2 cm 60-84 % Résection large avec l’ensemble du territoire ganglionnaire> 2 cm 100 %
Rectum(15-30 %)
< 1 cm (80 % des cas) 2 % Résection endoscopique ou électrocoagula-tion ou exérèse endoanale chirurgicale
87 ± 2
1-2 cm 10-15 % Attitude selon atteinte de la musculeuse (écho-endoscopie)
> 2 cm 60-100 % Exérèse anatomique selon siège (résection antérieure ou amputation)
Estomac(2-8 %)
< 1 cm Exérèse locale 63 ± 4> 2 cm Gastrectomie + curage
Côlon(5-15 %)
Résection carcinologique 62 ± 3

Traitement chirurgical des tumeurs endocrines gastro-entéro-pancréatiques R. Kianmanesh et al.
144
d’autres tumeurs associées (adénome ouadénocarcinome colique ou de la vessie)de l’ordre de 30 % a été rapportée [1,209]. La survie globale à 5 ans est situéeentre 55 et 76 %. La survie des formeslocalisées est comprise entre 70 et 95 %et celle des formes métastatiques entre32 et 51 % [18].
Tumeurs endocrines du côlonElles représentent environ 10 % (5 à15 %) des TE digestives [1, 18]. Dans2/3 cas la tumeur est située sur le côlondroit [220]. La taille est de plus de 2 cmdans 85 % des cas. Près de 2/3 des ma-lades ont des métastases ganglionnaireset/ou hépatiques au moment du dia-gnostic. Le pronostic est plus mauvaisque les autres TE digestives. Le traite-ment chirurgical consiste à réaliser uneexérèse segmentaire avec curage gan-glionnaire du territoire du drainagelymphatique [14, 221, 222]. La survieglobale à 5 ans est comprise entre 42 et70 %, les formes métastatiques ont unesurvie à 5 ans comprise entre 5 et 30 %[18].
Tumeurs endocrines du rectumElles représentent environ 20 % (15 à30 %) des TE digestives [1, 18]. Le dia-gnostic est fait dans la moitié des cas surune pièce d’exérèse endoscopique. Ellessont dans 75 % des cas infracentrimé-triques [18, 209, 222]. L’attitude théra-peutique est assez comparable à celledes TE de l’appendice. La taille et l’en-vahissement en profondeur qui sont aumieux appréciée par l’écho-endoscopieainsi que le degré de la différentiationtumorale déterminent le choix théra-peutique et le pronostic. Lorsque la tu-meur est bien différenciée, avec unetaille inférieure à 2 cm et limitée à lasous-muqueuse (ce qui est apprécié aumieux par l’écho-endoscopie rectale),l’exérèse locale par voie endoscopiquepeut être suffisante (risque global demétastase inférieur à 15 %). Dans le cascontraire une exérèse chirurgicale aveccurage ganglionnaire est nécessaire. Lestumeurs de plus de 2 cm ont une diffu-sion métastatique locale ou à distancedans 80 à 100 % des cas [31]. La survieà 5 ans est proche de 95 % pour les tu-meurs bien différenciées de moins de1 cm limitées à la sous-muqueuse, et de47 % en présence de métastases gan-glionnaires et inférieures à 20 % en pré-sence de MH [18]. La diffusion métas-
tatique et le mauvais pronostic des TEde plus de 2 cm du rectum incitent àtenter de préserver l’appareil sphincté-rien [222].
Tumeurs endocrines gastriquesElles représentent moins de 10 % desTE digestives et sont appelées ECLo-mes (tumeurs à cellules entérochromaf-fin-like) [223]. Elles sont le plus souventasymptomatiques et très rarement dé-couvertes à l’occasion d’un saignement[224]. Il en existe 4 types. Le type I (73à 75 % des cas) est représenté par lesECLomes survenant dans le cadred’une gastrite chronique atrophique. Letype II (13 à 20 %) est représenté pardes ECLomes survenant au cours d’unsyndrome de Zollinger-Ellison associéà une NEM-1. Ces 2 types surviennentdans un contexte d’hypergastrinémie,sont souvent multiples, de petite taille(< 1 cm) et bénignes. L’hypergastriné-mie est liée soit à une atrophie gastrique(gastrite atrophiante chronique de ty-pe A, gastropathie de la maladie deBiermer), plus rarement à un syndromede Zollinger-Ellison [225]. Les types IIIet IV sont rares (inférieurs à 10 % desECLomes), sporadiques et de mauvaispronostic. La tumeur est en généralunique, de grande taille et a le plus sou-vent un potentiel invasif et métastatique(ganglions, foie) [226]. Pour simplifier,on peut retenir qu’après un bilan com-plet (endoscopie, écho-endoscopie etTDM), les tumeurs de plus de 1 cm ouinfiltrant la musculeuse sont générale-ment traitées par une exérèse gastriquecarcinologique avec curage ganglion-naire. Les tumeurs infracentimétriquessont soit surveillées soit détruites ou ré-séquées endoscopiquement [224]. Lestumeurs de type IV sont des carcinomesendocrines peu différenciés uniques lar-ges (diamètre moyen de 4 cm), de trèsmauvais pronostic et se comportentcomme des adénocarcinomes gastri-ques. La survie globale à 5 ans est supé-rieure à 70 %, celle des formes métas-tatiques est de moins de 20 % [18].
Prise en charge du syndrome carcinoïdeLe syndrome carcinoïde (ou carcinoï-dien) est le plus souvent secondaire à lasécrétion de grande quantité de subs-tances sérotoninergiques et d’autresdérivées (5 hydroxytryptamine, tachy-kinines, 5 hydroxytryptophane, brady-
kinines ou prostaglandines) par une tu-meur carcinoïde volumineuse ou par lesMH, dépassant la capacité de métabo-lisation hépatique de la sérotonine [4,210, 227]. L’origine de la TE primitiveest iléale dans 10 à 15 % (appendice ex-ceptionnellement), pancréatique (20 %)ou bronchique (13 %). Les flushs sontprésents chez 2/3 des malades, unediarrhée d’allure motrice est associéeaux flushs dans 60 % et isolée dans15 % des cas [4, 12]. On observe éga-lement des douleurs abdominales par-fois liées à un syndrome occlusif et/ouune mésentérite rétractile (par évolu-tion fibrosante du mésentère) liée àl’évolution de la TE primitive, une ins-tabilité hémodynamique, un bronchos-pasme, voire des signes de cardiopathiecarcinoïde (atteinte des valves tricuspi-de et pulmonaire) qui est observée chez30 à 50 % des malades et peut être res-ponsable de décès [228-230]. L’atteintecardiaque est irréversible témoignantsouvent de la gravité du syndrome hor-monal. Elle est due à l’épaississementfibreux de l’endocarde (aspect lisse enéchographie cardiaque) responsabled’une insuffisance tricuspide, parfoisassociée à un rétrécissement pulmonai-re avec signes d’insuffisance cardiaquedroite [229]. Le bilan de la TE primi-tive et celle des MH doit comporter unbilan cardiaque qui peut nécessiter uneprise en charge spécifique (par exempleremplacement valvulaire avant une chi-rurgie hépatique qui est dangereuse encas de fuite tricuspide ou insuffisancecardiaque).
La base du traitement chirurgical estl’exérèse des lésions tumorales (primiti-ve et MH). Certaines manifestationspeuvent survenir ou s’aggraver lors del’intervention chirurgicale d’où la né-cessité de préparer les malades par lesanalogues de la somatostatine [231]. Lachirurgie cytoréductrice (debulking)permet de contrôler le syndrome hor-monal et peut aider la prise en chargemédicale [57]. La base du traitementmédical est l’inhibition de la sécrétionhormonale par les analogues de la so-matostatine (formes retard) [4, 14, 232-235]. Ce traitement au long cours est li-thogène avec un risque global de cho-lécystite aiguë lithiasique d’environ7 %, cependant il ne justifie pas la pra-tique de cholécystectomie préventive,sauf lors de la chirurgie sur la tumeurprimitive et/ou lors d’une chirurgie hé-patique cytoréductive [236].

J Chir 2005,142, N°3 • © Masson, Paris, 2005
145
Revue de la littérature
ConclusionLes tumeurs endocrines (TE) du tractusdigestif sont rares. Le traitement desTE est multidisciplinaire. La chirurgied’exérèse représente le seul traitementcuratif. Les objectifs de la chirurgie sontde prolonger la survie, de contrôler unéventuel syndrome hormonal, et deprévenir ou traiter les complications lo-cales. Parmi les TE gastro-intestinales,celles de l’appendice et du rectum sontsouvent infracentimétriques, bénigneset découvertes sur une pièce d’appendi-cectomie ou d’exérèse endoscopique.Les tumeurs iléales sont de petite tailleet malignes et parfois multiples. Elles secaractérisent par un taux élevé de com-plications (occlusion, mésentérite ré-tractile, hémorragie). Le syndrome car-cinoïde est présent chez 10 % desmalades, il est quasiment toujours asso-ciée à la présence de métastases hépati-ques et nécessite une prise en charge leplus souvent multimodale (analogues dela somatostatine, chimiothérapie systé-mique, chimio-embolisation ou embo-lisation intra-artérielles, chirurgie). LesTEP fonctionnelles les plus fréquentessont l’insulinome et le gastrinome (syn-drome de Zollinger-Ellison). Les TEnon fonctionnelles du pancréas repré-sentent plus de la moitié des TEP etsont le plus souvent découvertes à unstade plus tardif devant un syndrome demasse ou lors du bilan de métastases hé-patiques. Les TEP et quelques rares casde TE gastriques peuvent s’intégrerdans le cadre d’une polyendocrinopa-thie (NEM de type 1) dont la recherchedoit être systématique.
Références
1. Modlin IM, Lye KD, Kidd M. A 5-deca-de analysis of 13,715 carcinoid tumors.Cancer 2003;97:934-959.
2. Jensen RT. Natural history of digestiveendocrine tumors. In Mignon M, Co-lombel JF (eds). “Recent Advances in thepathology and management of inflam-matory bowel disease and digestive endo-crine tumors.” Paris: 1999;192-219.
3. Baudin E, Ducreux M, Sabourin JC et al.Les tumeurs neuroendocrines gastro-en-téropancréatiques. Médecine Thérapeu-tique Endocrinologie 1999;1:165-174.
4. Ruszniewski P, O’Toole D. Clinicalspectrum of digestive neuroendocrinetumors. Rev Prat 2002;52:262-267.
5. Moertel CG, Weiland LH, NagorneyDM, Dockerty MB. Carcinoid tumor ofthe appendix: treatment and prognosis.N Engl J Med 1987;317:1699-1701.
6. Moertel CG, Johnson CM, McKusickMA et al. The management of patientswith advanced carcinoid tumors and isletcell carcinomas. Ann Intern Med1994;120:302-309.
7. Kulke MH, Mayer RJ. Carcinoid tumors.N Engl J Med 1999;340:858-868.
8. Sarmiento JM, Heywood G, Rubin J etal. Surgical treatment of neuroendocrinemetastases to the liver: a plea for resec-tion to increase survival. J Am Coll Surg2003;197:29-37.
9. Jensen RT. Current diagnosis and mana-gement of gastrointestinal neuroendo-crine tumours. Dig Liver Dis2001;33:212-214.
10. Norton JA, Warren RS, Kelly MG et al.Aggressive surgery for metastatic liverneuroendocrine tumors. Surgery2003;134:1057-1063.
11. Madeira I, Terris B, Voss M et al. Pro-gnostic factors in patients with endocrinetumours of the duodenopancreatic area.Gut 1998;43:422-427.
12. Madeira I, Ruszniewski P. Digestive sys-tem carcinoid tumors: treatment. RevMed Interne 1999;20: 421-426.
13. Schlumberger M, Baudin E. Neuroen-docrine tumors. Ann Endocrinol (Paris)1997;58:95-99.
14. Ducreux M, Baudin E, Schlumberger M.Treatment strategy of neuroendocrinetumors. Rev Prat 2002;52:290-296.
15. Elias D, Lasser P, Ducreux M et al. Liverresection (and associated extrahepatic re-sections) for metastatic well-differentia-ted endocrine tumors: a 15-year singlecenter prospective study. Surgery2003;133: 375-382.
16. Mignon M. Natural history of neuroen-docrine enteropancreatic tumors. Diges-tion 2000;62 Suppl 1:51-58.
17. Partensky C. Prise en charge des tumeursendocrines duodéno-pancréatiques. JChir (Paris) 2000;137:142-150.
18. Maggard MA, O’Connell JB, Ko CY.Updated population-based review of car-cinoid tumors. Ann Surg 2004;240:117-122.
19. Oberg K. Diagnosis and treatment ofcarcinoid tumors. Expert Rev AnticancerTher 2003;3:863-877.
20. Baudin E, Gigliotti A, Ducreux M et al.Neuron-specific enolase and chromogra-nin A as markers of neuroendocrine tu-mours. Br J Cancer 1998;78:1102-1107.
21. Jensen RT. Pancreatic endocrine tu-mors: recent advances. Ann Oncol1999;10 Suppl 4:170-176.
22. Calender A. Genetics of neuroendocrinetumors. Rev Prat 2002;52: 256-261.
23. Saint-Andre JP, Dupre F, Chenue F,Guyetant S. Histopathology of neuroen-docrine tumors. Ann Endocrinol (Paris)1997;58:101-111.
24. Mitry E, Baudin E, Ducreux M et al.Treatment of poorly differentiated neu-roendocrine tumours with etoposide andcisplatin. Br J Cancer 1999;81:1351-1355.
25. Mitry E, Rougier P. Treatment strategyfor pancreatic endocrine tumours. Gas-troenterol Clin Biol 2003;27:S31-S36.
26. O’Toole D, Kianmanesh R, Farges O,Ruszniewski P. Treatment of pancreatic-duodenal endocrine tumors. Rev Prat2002;52:1546-1553.
27. Ahlman H. Surgical treatment of carci-noid tumours of the stomach and smallintestine. Ital J Gastroenterol Hepatol1999;31 Suppl 2: S198-S201.
28. Plockinger U, Wiedenmann B. Neu-roendocrine tumors of the gastro-en-tero-pancreatic system: the role of earlydiagnosis, genetic testing and preventivesurgery. Dig Dis 2002;20:49-60.
29. O’Toole D, Maire F, Ruszniewski P.Ablative therapies for liver metastases ofdigestive endocrine tumours. EndocrRelat Cancer 2003;10:463-468.
30. Ruszniewski P, Terris B. Natural historyand prognosis of pancreatic endocrinetumors. Gastroenterol Clin Biol2003;27:S20-S25.
31. Roche A, Lasser P. Locoregional therapyof neuroendocrine tumors. Rev Prat2002;52:274-278.
32. Sutliff VE, Doppman JL, Gibril F et al.Growth of newly diagnosed, untreatedmetastatic gastrinomas and predictors ofgrowth patterns. J Clin Oncol1997;15:2420-2431.
33. Weber HC, Venzon DJ, Lin JT et al. De-terminants of metastatic rate and survivalin patients with Zollinger-Ellison syn-drome: a prospective long-term study.Gastroenterology 1995;108:1637-1649.
34. Jaeck D, Oussoultzoglou E, Bachellier Pet al. Hepatic metastases of gastroente-ropancreatic neuroendocrine tumors: sa-fe hepatic surgery. World J Surg2001;25:689-692.
35. Que FG, Sarmiento JM, Nagorney DM.Hepatic surgery for metastatic gastroin-testinal neuroendocrine tumors. CancerControl 2002;9:67-79.
36. Chu QD, Hill HC, Douglass HO, Jr. etal. Predictive factors associated withlong-term survival in patients with neu-roendocrine tumors of the pancreas. AnnSurg Oncol 2002;9: 855-862.
37. Calender A, Giraud S, Porchet N et al.[Clinicogenetic study of MEN1: recentphysiopathological data and clinical ap-plications. Study Group of Multiple En-docrine Neoplasia (GENEM)]. Ann En-docrinol (Paris) 1998;59:444-451.
38. Aubert-Petit G, Baudin E, Cailleux AF etal. Neuro-endocrine tumors and vonHippel-Lindau disease. 3 cases. PresseMed 1999;28:1231-1234.
39. Chandrasekharappa SC, Guru SC, Ma-nickam P et al. Positional cloning of thegene for multiple endocrine neoplasia-type 1. Science 1997; 276:404-407.
40. Agarwal SK, Lee BA, Sukhodolets KE etal. Molecular pathology of the MEN1gene. Ann N Y Acad Sci 2004;1014:189-198.
41. Calender A, Cadiot G, Mignon M. Mul-tiple endocrine neoplasia type 1: geneticand clinical aspects. Gastroenterol ClinBiol 2001;25: B38-B48.
42. Brandi ML, Gagel RF, Angeli A et al.Guidelines for diagnosis and therapy ofMEN type 1 and type 2. J Clin Endocri-nol Metab 2001;86: 5658-5671.
43. Goudet P, Peschaud F, Mignon M et al.[Gastrinomas in multiple endocrine neo-plasia type-1. A 127-case cohort studyfrom the endocrine tumor group(GTE)]. Ann Chir 2004;129: 149-155.
44. Hammel P, Beigelman C, Chauveau D etal. [Variety of pancreatic lesions obser-ved in von Hippel-Lindau disease. Apro-

Traitement chirurgical des tumeurs endocrines gastro-entéro-pancréatiques R. Kianmanesh et al.
146
pos of 8 cases]. Gastroenterol Clin Biol1995;19: 1011-1017.
45. Hammel PR, Vilgrain V, Terris B et al.Pancreatic involvement in von Hippel-Lindau disease. The Groupe Francopho-ne d’Étude de la Maladie de von Hippel-Lindau. Gastroenterology2000;119:1087-1095.
46. Eriksson B, Oberg K. Neuroendocrinetumours of the pancreas. Br J Surg2000;87:129-131.
47. Guth A. Tumeurs endocrines du pan-créas. In Vilgrain V, Menu Y (eds):« Imagerie du foie, des voies biliaires, dupancréas et de la rate. » Paris: Flamma-rion, 2002;349-359.
48. Baudin E, Dromain C. Biology and ima-ging of neuroendocrine tumors. Rev Prat2002;52:268-273.
49. Pereira PL, Wiskirchen J. Morphologi-cal and functional investigations of neu-roendocrine tumors of the pancreas. EurRadiol 2003;13:2133-2146.
50. Kirshbom PM, Kherani AR, OnaitisMW et al. Carcinoids of unknown origin:comparative analysis with foregut, mid-gut, and hindgut carcinoids. Surgery1998;124:1063-1070.
51. Akerstrom G. Management of carcinoidtumors of the stomach, duodenum, andpancreas. World J Surg 1996;20:173-182.
52. Couvelard A, Felce-Dachez M, DegottC. Histological classification of endocri-ne tumors of the pancreas. GastroenterolClin Biol 2003;27: S15-S19.
53. Phan GQ, Yeo CJ, Hruban RH et al. Sur-gical experience with pancreatic and pe-ripancreatic neuroendocrine tumors: re-view of 125 patients. J Gastrointest Surg1998;2: 472-482.
54. Lam KY, Lo CY. Pancreatic endocrinetumour: a 22-year clinico-pathologicalexperience with morphological, immu-nohistochemical observation and a re-view of the literature. Eur J Surg Oncol1997;23: 36-42.
55. Dromain C, de Baere T, Baudin E et al.MR imaging of hepatic metastases cau-sed by neuroendocrine tumors: compa-ring four techniques. AJR Am J Roentge-nol 2003;180: 121-128.
56. Azimuddin K, Chamberlain RS. Thesurgical management of pancreatic neu-roendocrine tumors. Surg Clin NorthAm 2001;81:511-525.
57. Sarmiento JM, Que FG. Hepatic surgeryfor metastases from neuroendocrine tu-mors. Surg Oncol Clin N Am2003;12:231-242.
58. Akerstrom G, Makridis C, Johansson H.Abdominal surgery in patients with mid-gut carcinoid tumors. Acta Oncol1991;30:547-553.
59. Akerstrom G. Aspects on surgical strate-gy in midgut carcinoids and endocrinepancreatic tumors. In Mignon M, Co-lombel JF (eds): «Recent advances in thepathophysiology and management of in-flammatory bowel disease and digestiveendocrine tumors. » Paris: 1999;330-334.
60. Partensky C, Berger F, Owono P et al.[Cephalic duodenopancreatectomy forendocrine tumor of the ampulla of Vaterand of the minor papilla]. GastroenterolClin Biol 1999;23:832-836.
61. Sauvanet A, Partensky C, Sastre B et al.Medial pancreatectomy: a multi-institu-tional retrospective study of 53 patientsby the French Pancreas Club. Surgery2002;132:836-843.
62. Ricke J, Klose KJ. Recommendations forstandardised imaging in neuroendocrinetumours. In Mignon M, Colombel JF(eds): «Recent advances in the pathophy-siology and management of inflammato-ry bowel disease and digestive endocrinetumors. » Paris: 1999;298-301.
63. Ricke J, Klose KJ, Mignon M et al. Stan-dardisation of imaging in neuroendocri-ne tumours: results of a European delphiprocess. Eur J Radiol 2001;37:8-17.
64. Rodallec M, Vilgrain V, Zins M et al. He-lical CT of pancreatic endocrine tumors.J Comput Assist Tomogr 2002: 26:728-733.
65. Zins M, Sauvanet A. Diagnosis of pan-creatic tumors. Rev Prat 2002; 52:1525-1531.
66. Ichikawa T, Peterson MS, Federle MP etal. Islet cell tumor of the pancreas: bi-phasic CT versus MR imaging in tumordetection. Radiology 2000;216:163-171.
67. Kalra MK, Maher MM, Mueller PR, Sai-ni S. State-of-the-art imaging of pan-creatic neoplasms. Br J Radiol2003;76:857-865.
68. Palazzo L, Roseau G, Salmeron M. En-doscopic ultrasonography in the preope-rative localization of pancreatic endocri-ne tumors. Endoscopy 1992;24 Suppl1:350-353.
69. O’Toole D, Palazzo L, Ruszniewski P.Role of endoscopic ultrasonography inendocrine tumours of the duodenopan-creatic region. In Mignon M, ColombelJF (eds): “Recent Advances in the patho-logy and management of inflammatorybowel disease and digestive endocrine tu-mors.” Paris: 1999;229-240.
70. Gauger PG, Scheiman JM, WamstekerEJ et al. Role of endoscopic ultrasonogra-phy in screening and treatment of pan-creatic endocrine tumours in asympto-matic patients with multiple endocrineneoplasia type 1. Br J Surg 2003;90:748-754.
71. Chiti A, Fanti S, Savelli G et al. Compa-rison of somatostatin receptor imaging,computed tomography and ultrasound inthe clinical management of neuroendo-crine gastro-entero-pancreatic tumours.Eur J Nucl Med 1998;25:1396-1403.
72. de Herder WW, Hofland LJ, van derLely AJ, Lamberts SW. Somatostatin re-ceptors in gastroentero-pancreatic neu-roendocrine tumours. Endocr RelatCancer 2003;10:451-458.
73. van der Lely AJ, de Herder WW, Kren-ning EP, Kwekkeboom DJ. Octreoscanradioreceptor imaging. Endocrine2003;20:307-311.
74. Proye C, Malvaux P, Pattou F et al. No-ninvasive imaging of insulinomas andgastrinomas with endoscopic ultrasono-graphy and somatostatin receptor scinti-graphy. Surgery 1998; 124:1134-1143.
75. Voss M, Hammel P, Molas G et al. Valueof endoscopic ultrasound guided fineneedle aspiration biopsy in the diagnosisof solid pancreatic masses. Gut2000;46:244-249.
76. Fidler JL, Johnson CD. Imaging of neu-roendocrine tumors of the pancreas. IntJ Gastrointest Cancer 2001;30:73-85.
77. Régent D. Tumeur endocrine du pan-créas. In Régent D, Schmutz G, GéninG (eds). «Imagerie du foie, des voies bi-liaires et du pancréas. » Paris: 1994;174-176.
78. Simon D, Starke A, Goretzki PE, RoeherHD. Reoperative surgery for organic hy-perinsulinism: indications and operativestrategy. World J Surg 1998;22:666-671.
79. Hiramoto JS, Feldstein VA, LaBerge JM,Norton JA. Intraoperative ultrasoundand preoperative localization detects alloccult insulinomas ; discussion 1025-6.Arch Surg 2001;136:1020-1025.
80. Norton JA, Alexander HR, Fraker DL etal. Does the use of routine duodenotomy(DUODX) affect rate of cure, develop-ment of liver metastases, or survival inpatients with Zollinger-Ellison syndro-me? Ann Surg 2004;239:617-625.
81. Norton JA. Gastrinoma: advances in lo-calization and treatment. Surg OncolClin N Am 1998;7:845-861.
82. Norton JA. Intra-operative proceduresto localize endocrine tumours of the pan-creas and duodenum. Ital J GastroenterolHepatol 1999;31 Suppl 2:S195-S197.
83. Thompson GB. Diagnosis and manage-ment if insulinomas. Endocr Pract2002;8:385-386.
84. Norton JA, Shawker TH, Doppman JLet al. Localization and surgical treatmentof occult insulinomas. Ann Surg1990;212:615-620.
85. Norton JA, Fraker DL, Alexander HR etal. Surgery to cure the Zollinger-Ellisonsyndrome. N Engl J Med 1999;341:635-644.
86. Farley DR, van Heerden JA, Grant CS,Thompson GB. Extrapancreatic gastri-nomas. Surgical experience. Arch Surg1994;129:506-511.
87. Grant CS. Gastrointestinal endocrinetumours. Insulinoma. Baillieres ClinGastroenterol 1996;10:645-671.
88. Correnti S, Liverani A, Antonini G et al.Intraoperative ultrasonography for pan-creatic insulinomas. Hepatogastroente-rology 1996;43:207-211.
89. Jordan PH, Jr. A personal experiencewith pancreatic and duodenal neuroen-docrine tumors. J Am Coll Surg1999;189:470-482.
90. Chapuis Y, Bigourdan JM, Massault PPet al. [Videolaparoscopic excision of insu-linoma. A study of 5 cases]. Chirurgie1998;123:461-467.
91. Mabrut JY, Boulez J, Peix JL et al. Lapa-roscopic pancreatic resections. Ann Chir2003;128:425-432.
92. Jaroszewski DE, Schlinkert RT, Thomp-son GB, Schlinkert DK. Laparoscopiclocalization and resection of insulinomas.Arch Surg 2004;139:270-274.
93. Masson B, Sa-Cunha A, Laurent C et al.[Laparoscopic pancreatectomy: report of22 cases]. Ann Chir 2003;128:452-456.
94. Ikeda S, Matsumoto S, Maeshiro K et al.Segmental pancreatectomy for the dia-gnosis and treatment of small lesions inthe neck or body of the pancreas. Hepa-togastroenterology 1995;42:730-733.

J Chir 2005,142, N°3 • © Masson, Paris, 2005
147
Revue de la littérature
95. Rotman N, Fagniez PL. Medial pancrea-tectomy. J Hepatobiliary Pancreat Surg2000;7:453-455.
96. Pradère B, Carrère N, Gonzi JL. Lespancréatectomies gauches. JChir 1999;136:324-331.
97. Iihara M, Kanbe M, Okamoto T et al.Laparoscopic ultrasonography for resec-tion of insulinomas. Surgery2001;130:1086-1091.
98. Fernandez-Cruz L, Saenz A, Astudillo Eet al. Outcome of laparoscopic pancreaticsurgery: endocrine and nonendocrine tu-mors. World J Surg 2002;26:1057-1065.
99. Gramatica L, Jr., Herrera MF, Mercado-Luna A et al. Videolaparoscopic resectionof insulinomas: experience in two insti-tutions. World J Surg 2002;26:1297-1300.
100. Barlehner E, Anders S, Schwetling R.Laparoscopic resection of the left pan-creas: technique and indication. DigSurg 2002;19:507-510.
101. Park AE, Heniford BT. Therapeutic la-paroscopy of the pancreas. Ann Surg2002;236:149-158.
102. Finlayson E, Clark OH. Surgical treat-ment of insulinomas. Surg Clin NorthAm 2004;84:775-785.
103. Sauvanet A. Functional results of pan-creatic surgery. Rev Prat 2002; 52:1572-1575.
104. Fabre JM, Houry S, Manderscheid JC etal. Surgery for left-sided pancreatic can-cer. Br J Surg 1996;83:1065-1070.
105. Benoist S, Dugue L, Sauvanet A et al. Isthere a role of preservation of the spleenin distal pancreatectomy? J Am Coll Surg1999;188:255-260.
106. Brigden ML, Pattullo A, Brown G.Pneumococcal vaccine administrationassociated with splenectomy: the needfor improved education, documentation,and the use of a practical checklist. Am JHematol 2000;65:25-29.
107. Sumaraju V, Smith LG, Smith SM. In-fectious complications in asplenic hosts.Infect Dis Clin North Am 2001;15:551-65.
108. Lynch AM, Kapila R. Overwhelmingpostsplenectomy infection. Infect DisClin North Am 1996; 10:693-707.
109. Yeo CJ, Cameron JL, Sohn TA et al. Sixhundred fifty consecutive pancreatico-duodenectomies in the 1990s: pathology,complications, and outcomes. Ann Surg1997;226:248-257.
110. Sohn TA, Yeo CJ, Cameron JL et al. Re-sected adenocarcinoma of the pancreas-616 patients: results, outcomes, and pro-gnostic indicators. J Gastrointest Surg2000;4:567-579.
111. Schmidt CM, Powell ES, YiannoutsosCT et al. Pancreaticoduodenectomy: a20-year experience in 516 patients. ArchSurg 2004;139:718-725.
112. Fabre JM, Arnaud JP, Navarro F et al.Results of pancreatogastrostomy afterpancreatoduodenectomy in 160 consecu-tive patients. Br J Surg 1998;85:751-754.
113. Schafer M, Mullhaupt B, Clavien PA.Evidence-based pancreatic head resec-tion for pancreatic cancer and chronicpancreatitis. Ann Surg 2002;236:137-148.
114. Machado MC, Penteado S, MontagniniAL, Machado MA. An alternative tech-
nique in the treatment of celiac axis ste-nosis diagnosed during pancreaticoduo-denectomy. HPB Surg 1998;10:371-373.
115. Yeo CJ, Barry MK, Sauter PK et al.Erythromycin accelerates gastric emp-tying after pancreaticoduodenectomy. Aprospective, randomized, placebo-con-trolled trial. Ann Surg 1993;218:229-237.
116. Yeo CJ, Cameron JL, Maher MM et al.A prospective randomized trial of pan-creaticogastrostomy versus pancreatico-jejunostomy after pancreaticoduodenec-tomy. Ann Surg 1995; 222:580-588.
117. Suc B, Msika S, Fingerhut A et al. Tem-porary fibrin glue occlusion of the mainpancreatic duct in the prevention of in-tra-abdominal complications after pan-creatic resection: prospective randomi-zed trial. Ann Surg 2003;237:57-65.
118. Suc B, Msika S, Piccinini M et al. Octreo-tide in the prevention of intra-abdominalcomplications following elective pan-creatic resection: a prospective, multi-center randomized controlled trial. ArchSurg 2004;139:288-294.
119. Buchler MW, Friess H, Wagner M et al.Pancreatic fistula after pancreatic headresection. Br J Surg 2000;87:883-889.
120. Cullen JJ, Sarr MG, Ilstrup DM: Pan-creatic anastomotic leak after pancreati-coduodenectomy: incidence, significan-ce, and management. Am J Surg1994;168:295-298.
121. Lemaire E, O’Toole D, Sauvanet A et al.Functional and morphological changesin the pancreatic remnant following pan-creaticoduodenectomy with pancreatico-gastric anastomosis. Br J Surg2000;87:434-438.
122. Yamaguchi K, Tanaka M, Chijiiwa K etal. Early and late complications of pylo-rus-preserving pancreatoduodenectomyin Japan 1998. J Hepatobiliary PancreatSurg 1999; 6:303-311.
123. Sakaguchi T, Nakamura S, Suzuki S et al.Marginal ulceration after pylorus-pre-serving pancreaticoduodenectomy. J He-patobiliary Pancreat Surg 2000;7:193-197.
124. Horstmann O, Markus PM, GhadimiMB, Becker H. Pylorus preservation hasno impact on delayed gastric emptyingafter pancreatic head resection. Pancreas2004;28:69-74.
125. Matthews BD, Smith TI, Kercher KW etal. Surgical experience with functioningpancreatic neuroendocrine tumors. AmSurg 2002;68:660-665.
126. Norton JA, Kivlen M, Li M et al. Mor-bidity and mortality of aggressive resec-tion in patients with advanced neuroen-docrine tumors. Arch Surg2003;138:859-866.
127. Agarwal SK, Kester MB, Debelenko LVet al. Germline mutations of the MEN1gene in familial multiple endocrine neo-plasia type 1 and related states. Hum MolGenet 1997;6:1169-1175.
128. Veldhuis JD, Norton JA, Wells SA, Jr. etal. Surgical versus medical managementof multiple endocrine neoplasia (MEN)type I. J Clin Endocrinol Metab1997;82:357-364.
129. Ruszniewski P, Podevin P, Cadiot G etal. Clinical, anatomical, and evolutivefeatures of patients with the Zollinger-Ellison syndrome combined with type I
multiple endocrine neoplasia. Pancreas1993;8:295-304.
130. Mignon M, Ruszniewski P, Podevin P etal. Current approach to the managementof gastrinoma and insulinoma in adultswith multiple endocrine neoplasia ty-pe I. World J Surg 1993;17:489-497.
131. Mignon M, Cadiot G. Diagnostic andtherapeutic criteria in patients with Zol-linger-Ellison syndrome and multipleendocrine neoplasia type 1. J Intern Med1998;243:489-494.
132. Cadiot G, Vuagnat A, Doukhan I et al.Prognostic factors in patients with Zol-linger-Ellison syndrome and multipleendocrine neoplasia type 1. Grouped’Étude des Neoplasies EndocriniennesMultiples (GENEM and groupe de Re-cherche et d’Étude du Syndrome de Zol-linger-Ellison (GRESZE). Gastroente-rology 1999;116:286-293.
133. Mignon M, Cadiot G. Natural history ofgastrinoma: lessons from the past. Ital JGastroenterol Hepatol 1999;31 Suppl2:S98-103.
134. Norton JA. Surgical treatment of gas-trointestinal endocrine tumors. In Mi-gnon M, Colombel JF (eds): “Recent Ad-vances in the pathology and managementof inflammatory bowel disease and diges-tive endocrine tumors.” Paris: 1999;248-259.
135. Doherty GM, Doppman JL, ShawkerTH et al. Results of a prospective strategyto diagnose, localize, and resect insulino-mas. Surgery 1991;110:989-996.
136. Rothmund M, Angelini L, Brunt LM etal. Surgery for benign insulinoma: an in-ternational review. World J Surg1990;14:393-398.
137. Proye C, Stalnikiewicz G, Wemeau JL etal. Genetically-driven or supposed gene-tic-related insulinomas in adults: valida-tion of the surgical strategy proposed bythe A.F.C.E./G.E.N.E.M.. Ann Endo-crinol (Paris) 2004;65:149-161.
138. Sheppard BC, Norton JA, Doppman JLet al. Management of islet cell tumors inpatients with multiple endocrine neopla-sia: a prospective study. Surgery1989;106:1108-1117.
139. Proye C, Pattou F, Carnaille B et al. In-traoperative insulin measurement duringsurgical management of insulinomas.World J Surg 1998;22:1218-1224.
140. O’Riordain DS, O’Brien T, van HeerdenJA et al. Surgical management of insuli-noma associated with multiple endocrineneoplasia type I. World J Surg1994;18:488-493.
141. Lo CY, Lam KY, Fan ST. Surgical stra-tegy for insulinomas in multiple endocri-ne neoplasia type I. Am J Surg1998;175:305-307.
142. O’Riordain DS, O’Brien T, Grant CS etal. Surgical management of primary hy-perparathyroidism in multiple endocrineneoplasia types 1 and 2. Surgery1993;114:1031-1037.
143. Mignon M, Lamorthe B, Cadiot G. Zol-linger-Ellison syndrome. Rev Prat1994;44:1620-1628.
144. Bonnaud G, Cadiot G, Mignon M. [Dia-gnostic and therapeutic strategy of Zol-linger-Ellison syndrome. Groupe de Re-cherches et d’Études du Syndrome deZollinger-Ellison (GRESZE)]. Ann Chir1994;48:535-546.

Traitement chirurgical des tumeurs endocrines gastro-entéro-pancréatiques R. Kianmanesh et al.
148
145. Herrmann ME, Ciesla MC, Chejfec G etal. Primary nodal gastrinomas. Arch Pa-thol Lab Med 2000;124:832-835.
146. Norton JA, Alexander HR, Fraker DL etal. Possible primary lymph node gastri-noma: occurrence, natural history, andpredictive factors: a prospective study.Ann Surg 2003;237:650-657.
147. Stabile BE, Morrow DJ, Passaro E Jr.The gastrinoma triangle: operative im-plications. Am J Surg 1984;147:25-31.
148. Passaro E Jr, Howard TJ, Sawicki MP etal. The origin of sporadic gastrinomaswithin the gastrinoma triangle: a theory.Arch Surg 1998;133:13-16.
149. Sugg SL, Norton JA, Fraker DL et al. Aprospective study of intraoperativemethods to diagnose and resect duodenalgastrinomas. Ann Surg 1993;218:138-144.
150. Doherty GM, Olson JA, Frisella MM etal. Lethality of multiple endocrine neo-plasia type I. World J Surg 1998;22:581-586.
151. Howard TJ, Zinner MJ, Stabile BE, Pas-saro E Jr. Gastrinoma excision for cure.A prospective analysis. Ann Surg1990;211:9-14.
152. Soga J, Yakuwa Y. The gastrinoma/Zol-linger-Ellison syndrome: statistical eva-luation of a Japanese series of 359 cases.J Hepatobiliary Pancreat Surg1998;5:77-85.
153. Lehy T, Cadiot G, Mignon M et al. In-fluence of multiple endocrine neoplasiatype 1 on gastric endocrine cells in pa-tients with the Zollinger-Ellison syndro-me. Gut 1992;33:1275-1279.
154. Cadiot G, Mignon M. Duodenopancrea-tic endocrine tumors in MEN I patients:influence on natural history and manage-ment. In Mignon M, Colombel JF (eds):“Recent advances in the pathophysiologyand management of inflammatory boweldisease and digestive endocrine tumors.”Paris: 1999:312-314.
155. Mignon M, Cadiot G. Importance ofMEN I association in patients with di-gestive neuroendocrine tumors. Whichscreening to recommend? In Mignon M,Colombel JF (eds): “Recent advances inthe pathophysiology and management ofinflammatory bowel disease and digesti-ve endocrine tumors.” Paris: 1999;315-317.
156. Lairmore TC, Chen VY, DeBenedettiMK et al. Duodenopancreatic resectionsin patients with multiple endocrine neo-plasia type 1. Ann Surg 2000;231:909-918.
157. Fraker DL, Norton JA. The role of sur-gery in the management of islet cell tu-mors. Gastroenterol Clin North Am1989;18:805-830.
158. Lowney JK, Frisella MM, Lairmore TC,Doherty GM. Pancreatic islet cell tumormetastasis in multiple endocrine neopla-sia type 1: correlation with primary tu-mor size. Surgery 1998;124:1043-8, dis-cussion.
159. Bartsch DK, Langer P, Wild A et al.Pancreaticoduodenal endocrine tumorsin multiple endocrine neoplasia type 1:surgery or surveillance? Surgery2000;128:958-966.
160. Skogseid B, Oberg K, Akerstrom G et al.Limited tumor involvement found atmultiple endocrine neoplasia type I pan-
creatic exploration: can it be predicted bypreoperative tumor localization? WorldJ Surg 1998;22:673-677.
161. Thompson NW. Management of pan-creatic endocrine tumors in patients withmultiple endocrine neoplasia type 1.Surg Oncol Clin N Am 1998;7:881-891.
162. Thompson NW. Current concepts inthe surgical management of multiple en-docrine neoplasia type 1 pancreatic-duo-denal disease. Results in the treatment of40 patients with Zollinger-Ellison syn-drome, hypoglycaemia or both. J InternMed 1998;243:495-500.
163. Cadiot G, Houillier P, Allouch A et al.Oral calcium tolerance test in the earlydiagnosis of primary hyperparathyroi-dism and multiple endocrine neoplasiatype 1 in patients with the Zollinger-El-lison syndrome. Groupe de Recherche etd’Étude du Syndrome de Zollinger-Elli-son. Gut 1996;39:273-278.
164. Brentjens R, Saltz L. Islet cell tumors ofthe pancreas: the medical oncologist’sperspective. Surg Clin North Am2001;81:527-542.
165. Tomassetti P, Migliori M, Lalli S et al.Epidemiology, clinical features and dia-gnosis of gastroenteropancreatic endo-crine tumours. Ann Oncol 2001;12 Suppl2:S95-S99.
166. Soga J, Yakuwa Y. Glucagonomas/diabe-tico-dermatogenic syndrome (DDS): astatistical evaluation of 407 reported ca-ses. J Hepatobiliary Pancreat Surg1998;5:312-319.
167. Guillausseau PJ, Guillausseau C, Villet Ret al. Glucagonomas. Clinical, biological,anatomopathological and therapeutic as-pects (general review of 130 cases. Gas-troenterol Clin Biol 1982;6:1029-1041.
168. Boden G. Glucagonomas and insulino-mas. Gastroenterol Clin North Am1989;18:831-845.
169. Rothmund M, Stinner B, Arnold R. En-docrine pancreatic carcinoma. Eur J SurgOncol 1991;17:191-199.
170. Chastain MA. The glucagonoma syndro-me: a review of its features and discussionof new perspectives. Am J Med Sci2001;321:306-320.
171. Mansour JC, Chen H. Pancreatic endo-crine tumors. J Surg Res 2004; 120:139-161.
172. Bouin M, Aoust LD. Clinical response ofan atypical glucagonoma treated with along-acting somatostatin analog. Gas-troenterol Clin Biol 2002;26:926-929.
173. Arnold R, Frank M, Kajdan U. Manage-ment of gastroenteropancreatic endocri-ne tumors: the place of somatostatin ana-logues. Digestion 1994;55 Suppl 3:107-113.
174. Eriksson B, Janson ET, Bax ND et al.The use of new somatostatin analogues,lanreotide and octastatin, in neuroendo-crine gastro-intestinal tumours. Diges-tion 1996;57 Suppl 1:77-80.
175. Wermers RA, Fatourechi V, Kvols LK.Clinical spectrum of hyperglucagonemiaassociated with malignant neuroendocri-ne tumors. Mayo Clin Proc1996;71:1030-1038.
176. Bloom SR, Polak JM, Welbourn RB.Pancreatic apudomas. World J Surg1979;3:587-595.
177. Krejs GJ. VIPoma syndrome. Am J Med1987;82:37-48.
178. Grier JF. WDHA (watery diarrhea, hy-pokalemia, achlorhydria) syndrome: cli-nical features, diagnosis, and treatment.South Med J 1995;88:22-24.
179. Peng SY, Li JT, Liu YB et al. Diagnosisand treatment of VIPoma in China: (casereport and 31 cases review) diagnosis andtreatment of VIPoma. Pancreas2004;28:93-97.
180. Lundstedt C, Linjawi T, Amin T. LiverVIPoma: report of two cases and litera-ture review. Abdom Imaging1994;19:433-437.
181. Harris GJ, Tio F, Cruz AB, Jr. Somatos-tatinoma: a case report and review of theliterature. J Surg Oncol 1987;36:8-16.
182. Masson B, Desfourneaux V, RaymondJM et al. Somatostatinome. À proposd’un cas. J Chir (Paris) 1997;134:22-26.
183. Hamy A, Heymann MF, Bodic J et al.[Duodenal somatostatinoma. Anatomic/clinical study of 12 operated cases]. AnnChir 2001;126:221-226.
184. Green BT, Rockey DC. Duodenal soma-tostatinoma presenting with completesomatostatinoma syndrome. J Clin Gas-troenterol 2001;33:415-417.
185. House MG, Yeo CJ, Schulick RD. Pe-riampullary pancreatic somatostatinoma.Ann Surg Oncol 2002;9:869-874.
186. Patel VG, Henderson VJ, FairweatherDA et al. Malignant duodenal somatos-tatinoma presenting in association withvon Recklinghausen disease. Am Surg2003;69:1077-1082.
187. Mao C, Shah A, Hanson DJ, HowardJM. Von Recklinghausen’s disease asso-ciated with duodenal somatostatinoma:contrast of duodenal versus pancreaticsomatostatinomas. J Surg Oncol1995;59:67-73.
188. Berger G, Trouillas J, Bloch B et al. Mul-tihormonal carcinoid tumor of the pan-creas. Secreting growth hormone-re-leasing factor as a cause of acromegaly.Cancer 1984;54:2097-2108.
189. Losa M, von Werder K. Pathophysiolo-gy and clinical aspects of the ectopic GH-releasing hormone syndrome. Clin En-docrinol (Oxf) 1997;47:123-135.
190. Metz DC. Diagnosis of non-Zollinger-Ellison syndrome, non-carcinoid syn-drome, enteropancreatic neuroendocri-ne tumours. Ital J Gastroenterol Hepatol1999;31 Suppl 2:S153-S159.
191. Kaassis M, Duquenne M, Croue A et al.[Calcitonin-secreting neuroendocrinecarcinoma of the pancreas with splenicinvasion and paraneoplastic hypercalce-mia]. Presse Med 2001;30:24.
192. Rankin W, Grill V, Martin TJ. Parathy-roid hormone-related protein and hyper-calcemia. Cancer 1997;80:1564-1571.
193. Abraham P, Ralston SH, Hewison M etal. Presentation of a PTHrP-secretingpancreatic neuroendocrine tumour, withhypercalcaemic crisis, pre-eclampsia, andrenal failure. Postgrad Med J2002;78:752-753.
194. Kitchens CS, Alexander RW. Cushing’ssyndrome secondary to a neuroendocri-ne tumor: relapse after bilateral adrena-lectomy. Cancer 1981;48:1873-1876.
195. Ruszniewski P, Girard F, Benamouzig Ret al. Long acting somatostatin treatmentof paraneoplastic Cushing’s syndrome ina case of Zollinger-Ellison syndrome.Gut 1988; 29:838-842.

J Chir 2005,142, N°3 • © Masson, Paris, 2005
149
Revue de la littérature
196. Deixonne B, Duchene D, Eledjam JJ etal. Cancer du pancréas endocrine à EC-cell responsable d’une diarrhée chroni-que. J Chir 1991;128:415-418.
197. Mao C, el Attar A, Domenico DR et al.Carcinoid tumors of the pancreas. Statusreport based on two cases and review ofthe world’s literature. Int J Pancreatol1998;23:153-164.
198. Fleury A, Flejou JF, Sauvanet A et al. Cal-citonin-secreting tumors of the pancreas:about six cases. Pancreas 1998;16:545-550.
199. Vinik AI, Strodel WE, Eckhauser FE etal. Somatostatinomas, PPomas, neuro-tensinomas. Semin Oncol 1987;14:263-281.
200. Bartsch DK, Schilling T, Ramaswamy Aet al. Management of nonfunctioning is-let cell carcinomas. World J Surg2000;24:1418-1424.
201. Solorzano CC, Lee JE, Pisters PW et al.Nonfunctioning islet cell carcinoma ofthe pancreas: survival results in a con-temporary series of 163 patients. Surgery2001;130:1078-1085.
202. Simpson WF, Adams DB, Metcalf JS,Anderson MC. Nonfunctioning pan-creatic neuroendocrine tumors presen-ting as pancreatitis: report of four cases.Pancreas 1988;3:223-231.
203. Madura JA, Cummings OW, Wiebke EAet al. Nonfunctioning islet cell tumors ofthe pancreas: a difficult diagnosis but oneworth the effort. Am Surg 1997;63:573-577.
204. Nobels FR, Kwekkeboom DJ, Coop-mans W et al. Chromogranin A as serummarker for neuroendocrine neoplasia:comparison with neuron-specific enolaseand the alpha-subunit of glycoproteinhormones. J Clin Endocrinol Metab1997;82:2622-2628.
205. Park BJ, Alexander HR, Libutti SK et al.Operative management of islet-cell tu-mors arising in the head of the pancreas.Surgery 1998;124:1 056-1061.
206. Kianmanesh R, Farges O, Abdalla EK etal. Right portal vein ligation: a new plan-ned two-step all-surgical approach forcomplete resection of primary gastroin-testinal tumors with multiple bilateral li-ver metastases. J Am Coll Surg2003;197:164-170.
207. Schnirer II, Yao JC, Ajani JA. Carcinoid--a comprehensive review. Acta Oncol2003;42:672-692.
208. Oberg K, Eriksson B. Digestive endocri-ne tumor management ; medical advan-ced disease. In Mignon M, Colombel JF(eds): “Recent Advances in the pathologyand management of inflammatory boweldisease and digestive endocrine tumors.”Paris: 1999;260-270.
209. Shebani KO, Souba WW, FinkelsteinDM et al. Prognosis and survival in pa-tients with gastrointestinal tract carci-noid tumors. Ann Surg 1999;229:815-821.
210. O’Toole D, Ducreux M, Bommelaer Get al. Treatment of carcinoid syndrome:a prospective crossover evaluation of lan-reotide versus octreotide in terms of ef-ficacy, patient acceptability, and toleran-ce. Cancer 2000;88:770-776.
211. Jensen RT. Carcinoid and pancreatic en-docrine tumors: recent advances in mo-lecular pathogenesis, localization, andtreatment. Curr Opin Oncol2000;12:368-377.
212. Goede AC, Winslet MC. Surgery forcarcinoid tumours of the lower gastroin-testinal tract. Colorectal Dis 2003;5:123-128.
213. Modlin IM, Sandor A. An analysis of8305 cases of carcinoid tumors. Cancer1997;79:813-829.
214. Sandor A, Modlin IM. A retrospectiveanalysis of 1570 appendiceal carcinoids.Am J Gastroenterol 1998; 93:422-428.
215. Goede AC, Caplin ME, Winslet MC.Carcinoid tumour of the appendix. Br JSurg 2003;90:1317-1322.
216. Rindi G, Capella C, Solcia E. Endocrinetumors of the stomach, intestine and ap-pendix. In Mignon M, Colombel JF(eds): “Recent advances in the pathophy-siology and management of inflammato-ry bowel disease and digestive endocrinetumors.” Paris: 1999;274-277.
217. Rossi G, Valli R, Bertolini F et al. Doesmesoappendix infiltration predict a wor-se prognosis in incidental neuroendocri-ne tumors of the appendix? A clinicopa-thologic and immunohistochemicalstudy of 15 cases. Am J Clin Pathol2003;120:706-711.
218. Roggo A, Wood WC, Ottinger LW.Carcinoid tumors of the appendix. AnnSurg 1993;217:385-390.
219. Yantiss RK, Odze RD, Farraye FA, Ro-senberg AE. Solitary versus multiple car-cinoid tumors of the ileum: a clinical andpathologic review of 68 cases. Am J SurgPathol 2003;27:811-817.
220. Ballantyne GH, Savoca PE, Flannery JTet al. Incidence and mortality of carci-noids of the côlon. Data from the Con-necticut Tumor Registry. Cancer1992;69:2400-2405.
221. Simon SR, Fox K. Neuroendocrine car-cinoma of the côlon. Correct diagnosis isimportant. J Clin Gastroenterol1993;17:304-307.
222. Bernick PE, Klimstra DS, Shia J et al.Neuroendocrine carcinomas of the côlonand rectum. Dis Côlon Rectum2004;47:163-169.
223. Modlin IM, Lye KD, Kidd M. Carcinoidtumors of the stomach. Surg Oncol2003;12:153-172.
224. Modlin IM, Kidd M, Tang LH. Surgicalmanagement of gastric carcinoid tumors.In Mignon M, Colombel JF (eds): “Re-cent advances in the pathophysiologyand management of inflammatory boweldisease and digestive endocrine tumors.”Paris: 1999;341-343.
225. Lehy T, Roucayrol AM, Mignon M,Lewin MJ. Gastric carcinoid tumors inZollinger-Ellison syndrome and perni-cieuous anaemia: prevalence and biology.In Mignon M, Colombel JF (eds): “Re-cent advances in the pathophysiologyand management of inflammatory boweldisease and digestive endocrine tumors.”Paris: 1999;286-287.
226. Gilligan CJ, Lawton GP, Tang LH et al.Gastric carcinoid tumors: the biologyand therapy of an enigmatic and contro-versial lesion. Am J Gastroenterol1995;90:338-352.
227. Lips CJ, Lentjes EG, Hoppener JW.The spectrum of carcinoid tumours andcarcinoid syndromes. Ann Clin Biochem2003;40:612-627.
228. Quaedvlieg PF, Lamers CB, Taal BG.Carcinoid heart disease: an update.Scand J Gastroenterol Suppl 2002:66-71.
229. Botero M, Fuchs R, Paulus DA, LindDS. Carcinoid heart disease: a case re-port and literature review. J Clin Anesth2002;14:57-63.
230. Anderson AS, Krauss D, Lang R. Cardio-vascular complications of malignant car-cinoid disease. Am Heart J1997;134:693-702.
231. Vaughan DJ, Brunner MD. Anesthesiafor patients with carcinoid syndrome. IntAnesthesiol Clin 1997;35:129-142.
232. Ruszniewski P, Ducreux M, ChayvialleJA et al. Treatment of the carcinoid syn-drome with the longacting somatostatinanalogue lanreotide: a prospective studyin 39 patients. Gut 1996;39:279-283.
233. Arnold R. Medical treatment of metasta-sizing carcinoid tumors. World J Surg1996;20:203-207.
234. Bax ND, Woods HF, Batchelor A, Jen-nings M. Octreotide therapy in carcinoiddisease. Anticancer Drugs 1996;7 Suppl1:17-22.
235. Ducreux M, Ruszniewski P, ChayvialleJA et al. The antitumoral effect of thelong-acting somatostatin analog lanreo-tide in neuroendocrine tumors. Am JGastroenterol 2000;95:3276-3281.
236. Trendle MC, Moertel CG, Kvols LK.Incidence and morbidity of cholelithiasisin patients receiving chronic octreotidefor metastatic carcinoid and malignantislet cell tumors. Cancer 1997;79:830-834.





























