Embed Size (px)
Citation preview

Chapitre
3Tumeurs rares et prédisposition génétique
C. Lasset, S. Giraud, V. Bonadona
Un certain nombre de prédispositions hérédi-taires au cancer pour lesquelles un diagnostic
moléculaire existe, c’est-à-dire que les gènes res-ponsables sont identifiés, sont décrites avec en corollaire la possibilité d’identifier des personnes asymptomatiques à haut risque de cancer auxquel-les doit être proposée une prise en charge préven-tive adaptée. Le tableau I présente les principales prédispositions connues dans ce cadre.Les prédispositions héréditaires pour lesquelles le cancer est au premier plan sont détaillées.
Prédispositions héréditaires aux cancers fréquents et tumeurs rares
Deux cancers fréquents — le cancer du sein et le cancer colorectal — sont concernés, dans une fai-ble proportion (10 % au plus) par une prédisposi-tion héréditaire en relation avec la transmission de type autosomique dominant d’une anomalie constitutionnelle d’un gène majeur connu, de type suppresseur de tumeur. Le spectre tumoral de ces prédispositions comporte des cancers peu fré-quents : ovaire, sein chez l’homme, intestin grêle, des voies urinaires, etc. Rarement observés, ces cancers quand ils sont associés au cancer fréquent (sein ou côlon-rectum) sont spécifiques du risque héréditaire, indiquant la présence probable d’un gène de prédisposition connu muté.
Prédispositions héréditaires au cancer du sein
Environ 7 % des cancers du sein sont d’origine hé-réditaire et deux gènes sont identifiés : BRCA1 et BRCA2 (OMIM # 114480, 113705, 600185). Une prédisposition héréditaire est suspectée si dans la même branche parentale au moins deux personnes apparentées au 1er ou 2e degré sont atteintes de can-cer du sein dont un diagnostiqué avant 40 ans. Plus
rarement, un cancer de l’ovaire ou un cancer du sein chez l’homme est observé, suggérant fortement la présence d’un gène BRCA muté et/ou orientant vers l’un des deux : 90 % des syndromes « sein-ovaire » sont liés au gène BRCA1 (contre seulement 30 % des syndromes « sein seul ») ; la présence d’un cancer du sein chez l’homme oriente vers BRCA2 (1).Chez la femme, le risque de développer un can-cer du sein avant 70 ans est d’environ 60 % pour BRCA1 et 45 % pour BRCA2 (contre 14 % dans la population générale). Les risques annuels de se-cond cancer du sein sont de 5 % et 3 % respecti-vement (soit une femme sur deux sur dix ans). Le risque de cancer de l’ovaire avant 70 ans est estimé à 40 % pour BRCA1 et 11 % pour BRCA2. Le risque de cancer de la prostate est modérément augmenté après 50 ans. Pour BRCA2, le risque de cancer du sein chez l’homme avant 70 ans est de 5 % et une augmentation de risque de cancer du pancréas est observée (2).Les cancers du sein « héréditaires » surviennent à un âge au diagnostic plus jeune (43 ans contre 60 ans). Liés à BRCA1, ils sont plus souvent de grade SBR élevé, non hormonodépendants et n’ex-primant pas HER2 (type basal). Liés à BRCA2, ils n’ont pas de caractéristique particulière (3). Les cancers de l’ovaire « héréditaires » sont plus préco-ces (55 ans contre 65 ans en moyenne) et de type séreux. Les cancers de l’ovaire de type mucineux ou « borderline » ne font pas partie du spectre tumo-ral des gènes BRCA.La prise en charge préventive des femmes porteuses d’un gène BRCA muté fait l’objet de recommen-dations (1). Au niveau mammaire, deux options existent. Associé à un examen clinique 2 à 3 fois par an, le dépistage annuel associe mammogra-phie standard, échographie mammaire et IRM du sein, réalisées par des radiologues expérimentés. L’alternative est la chirurgie à type de mastectomie bilatérale avec reconstruction immédiate. Cette in-tervention, capable de réduire le risque de cancer du sein d’au moins 90 %, est recommandée sous
Chapitre
16 Tumeurs malignes rares
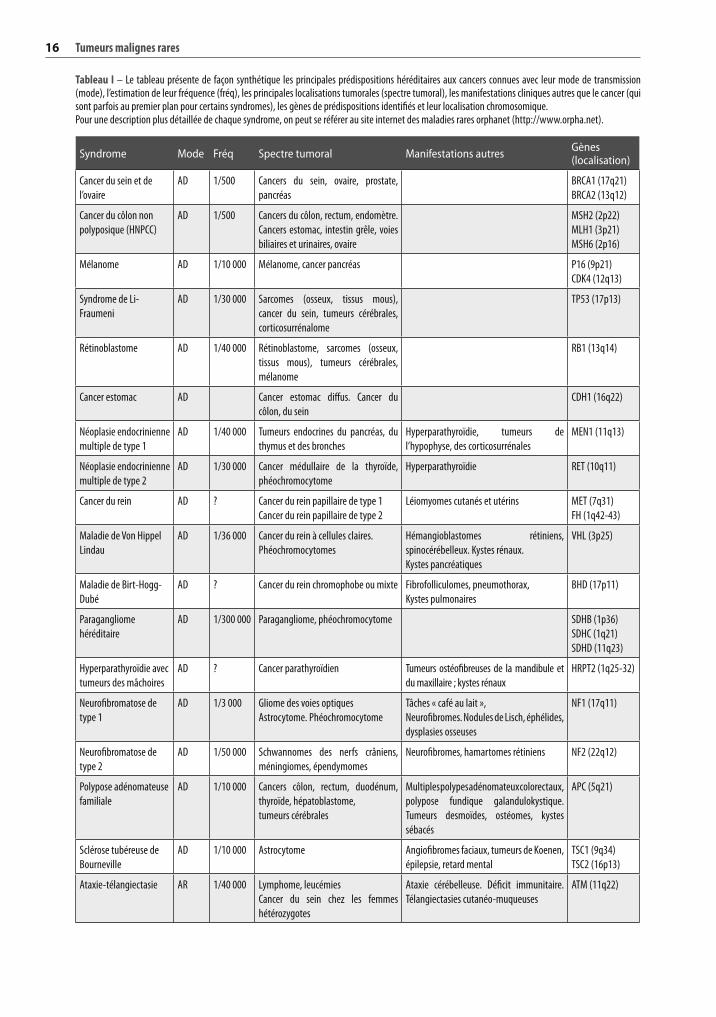
Tableau I – Le tableau présente de façon synthétique les principales prédispositions héréditaires aux cancers connues avec leur mode de transmission (mode), l’estimation de leur fréquence (fréq), les principales localisations tumorales (spectre tumoral), les manifestations cliniques autres que le cancer (qui sont parfois au premier plan pour certains syndromes), les gènes de prédispositions identifiés et leur localisation chromosomique.Pour une description plus détaillée de chaque syndrome, on peut se référer au site internet des maladies rares orphanet (http://www.orpha.net).
Syndrome Mode Fréq Spectre tumoral Manifestations autres Gènes (localisation)
Cancer du sein et de
l’ovaire
AD 1/500 Cancers du sein, ovaire, prostate,
pancréas
BRCA1 (17q21)
BRCA2 (13q12)
Cancer du côlon non
polyposique (HNPCC)
AD 1/500 Cancers du côlon, rectum, endomètre.
Cancers estomac, intestin grêle, voies
biliaires et urinaires, ovaire
MSH2 (2p22)
MLH1 (3p21)
MSH6 (2p16)
Mélanome AD 1/10 000 Mélanome, cancer pancréas P16 (9p21)
CDK4 (12q13)
Syndrome de Li-
Fraumeni
AD 1/30 000 Sarcomes (osseux, tissus mous),
cancer du sein, tumeurs cérébrales,
corticosurrénalome
TP53 (17p13)
Rétinoblastome AD 1/40 000 Rétinoblastome, sarcomes (osseux,
tissus mous), tumeurs cérébrales,
mélanome
RB1 (13q14)
Cancer estomac AD Cancer estomac diffus. Cancer du
côlon, du sein
CDH1 (16q22)
Néoplasie endocrinienne
multiple de type 1
AD 1/40 000 Tumeurs endocrines du pancréas, du
thymus et des bronches
Hyperparathyroïdie, tumeurs de
l’hypophyse, des corticosurrénales
MEN1 (11q13)
Néoplasie endocrinienne
multiple de type 2
AD 1/30 000 Cancer médullaire de la thyroïde,
phéochromocytome
Hyperparathyroïdie RET (10q11)
Cancer du rein AD ? Cancer du rein papillaire de type 1
Cancer du rein papillaire de type 2
Léiomyomes cutanés et utérins MET (7q31)
FH (1q42-43)
Maladie de Von Hippel
Lindau
AD 1/36 000 Cancer du rein à cellules claires.
Phéochromocytomes
Hémangioblastomes rétiniens,
spinocérébelleux. Kystes rénaux.
Kystes pancréatiques
VHL (3p25)
Maladie de Birt-Hogg-
Dubé
AD ? Cancer du rein chromophobe ou mixte Fibrofolliculomes, pneumothorax,
Kystes pulmonaires
BHD (17p11)
Paragangliome
héréditaire
AD 1/300 000 Paragangliome, phéochromocytome SDHB (1p36)
SDHC (1q21)
SDHD (11q23)
Hyperparathyroïdie avec
tumeurs des mâchoires
AD ? Cancer parathyroïdien Tumeurs ostéofibreuses de la mandibule et
du maxillaire ; kystes rénaux
HRPT2 (1q25-32)
Neurofibromatose de
type 1
AD 1/3 000 Gliome des voies optiques
Astrocytome. Phéochromocytome
Tâches « café au lait »,
Neurofibromes. Nodules de Lisch, éphélides,
dysplasies osseuses
NF1 (17q11)
Neurofibromatose de
type 2
AD 1/50 000 Schwannomes des nerfs crâniens,
méningiomes, épendymomes
Neurofibromes, hamartomes rétiniens NF2 (22q12)
Polypose adénomateuse
familiale
AD 1/10 000 Cancers côlon, rectum, duodénum,
thyroïde, hépatoblastome,
tumeurs cérébrales
Multiples polypes adénomateux colorectaux,
polypose fundique galandulokystique.
Tumeurs desmoïdes, ostéomes, kystes
sébacés
APC (5q21)
Sclérose tubéreuse de
Bourneville
AD 1/10 000 Astrocytome Angiofibromes faciaux, tumeurs de Koenen,
épilepsie, retard mental
TSC1 (9q34)
TSC2 (16p13)
Ataxie-télangiectasie AR 1/40 000 Lymphome, leucémies
Cancer du sein chez les femmes
hétérozygotes
Ataxie cérébelleuse. Déficit immunitaire.
Télangiectasies cutanéo-muqueuses
ATM (11q22)
Tumeurs rares et prédisposition génétique 17
réserve du maintien de la qualité de vie et d’une es-pérance de vie d’au moins dix ans. Elle relève d’un choix personnel dans un processus de décision par-tagée. La chimioprévention par anti-aromatases est en cours d’évaluation. Au niveau ovarien, le manque d’efficacité du dépistage par échographie endova-ginale annuel (et/ou du dosage du CA125) conduit à conseiller l’annexectomie bilatérale après 40 ans pour les femmes n’ayant plus de projet parental. Elle peut être différée à 45 ans quand le gène BRCA2 est impliqué (1, 4).
Prédispositions héréditaires au cancer du côlon sans polypose (HNPCC)
Environ 5 % des cancers du côlon ou du rectum sont d’origine héréditaire et surviennent en dehors d’un contexte de polypose colique, définissant ain-si le syndrome HNPCC (pour Hereditary Non-Poly-posis Colorectal Cancer) (OMIM # 120435). Trois gènes principaux sont identifiés : MSH2, MLH1 et MSH6 qui font partie du système de réparation des mésappariements de l’ADN (gènes MMR pour MisMatch Repair).Classiquement, la définition du syndrome HNPCC repose sur la réunion de trois critères dits d’Amster-dam : (i) au moins 3 individus atteints de cancers appartenant au spectre étroit du syndrome (can-cers colorectaux, cancers de l’endomètre, de l’intes-tin grêle, des voies urinaires) et histologiquement prouvés, (ii) unis deux à deux par un lien de paren-té au premier degré sur deux générations, (iii) un des cancers au moins diagnostiqué avant 50 ans. Un gène MMR muté est identifié dans 70 % des familles possédant l’ensemble des critères (35 % MSH2, 25 % MLH1 et 2 % MSH6) mais également dans 30 % à 50 % de celles pour lesquelles les cri-tères sont incomplets (2 individus atteints seule-
ment, lien de parenté de second degré, etc.). En l’absence de ces critères (complets ou incomplets), un syndrome HNPCC doit aussi être évoqué chez un patient atteint d’un cancer du spectre étroit si l’âge au diagnostic est de moins de 40 ans ou s’il a déjà un antécédent personnel d’un cancer de ce spectre. Si l’âge au diagnostic du cancer se situe entre 40 et 60 ans ou s’il existe un antécédent fa-milial au 1er degré de cancer du spectre, il convient de rechercher sur la tumeur une instabilité des mi-crosatellites (test MSI, pour MicroSatellite Instabi-lity). Si ce stigmate au niveau tumoral du défaut de fonctionnement des gènes MMR est retrouvé chez le patient, la probabilité d’identifier une mutation germinale d’un gène MMR est élevée. La mise en évidence par immunohistochimie (test IHC) d’un défaut d’expression d’une des protéines MMR peut orienter sur le gène MMR responsable (MSH2 et MSH6 notamment) (5).Par manque de spécificité, ces tests ne peuvent être utilisés pour le diagnostic d’un syndrome HNPCC chez les patients atteints de cancer colorectal. En effet, l’instabilité des microsatellites est fréquente dans les cancers sporadiques (ceux du côlon droit et/ou diagnostiqués après 60 ans surtout) car elle résulte de l’inactivation somatique des deux allèles d’un gène MMR (MLH1 en particulier). Le test MSI n’est donc pas spécifique du syndrome HNPCC et pour évaluer de façon pertinente la probabilité d’un tel syndrome, il convient toujours de confronter le résultat des tests tumoraux MSI et/ou IHC à l’histoi-re personnelle et familiale de cancers en s’appuyant sur les critères d’Amsterdam. Une seule situation in-dividuelle reste évocatrice : l’observation d’un cancer du côlon gauche ou du rectum diagnostiqué avant 60 ans avec un test MSI positif et une extinction de la protéine MSH2 ou MSH6 en IHC (5, 6).En présence d’un gène MMR muté, le risque de dé-velopper un cancer avant 70 ans est estimé à 60 %
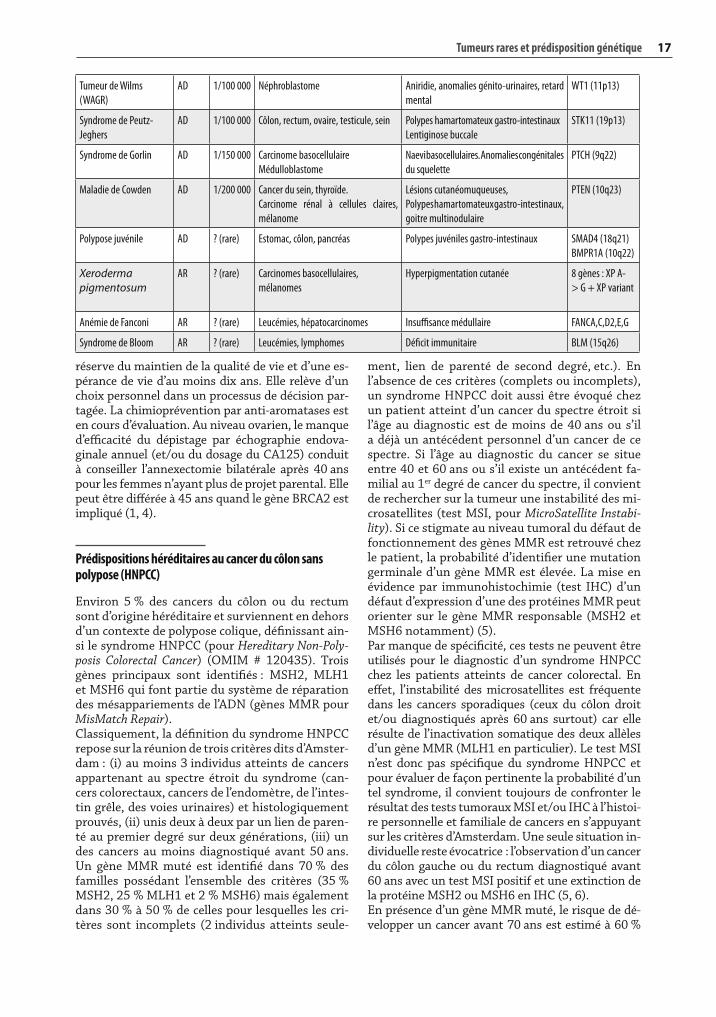
Tumeur de Wilms
(WAGR)
AD 1/100 000 Néphroblastome Aniridie, anomalies génito-urinaires, retard
mental
WT1 (11p13)
Syndrome de Peutz-
Jeghers
AD 1/100 000 Côlon, rectum, ovaire, testicule, sein Polypes hamartomateux gastro-intestinaux
Lentiginose buccale
STK11 (19p13)
Syndrome de Gorlin AD 1/150 000 Carcinome basocellulaire
Médulloblastome
Naevi basocellulaires. Anomalies congénitales
du squelette
PTCH (9q22)
Maladie de Cowden AD 1/200 000 Cancer du sein, thyroïde.
Carcinome rénal à cellules claires,
mélanome
Lésions cutanéomuqueuses,
Polypes hamartomateux gastro-intestinaux,
goitre multinodulaire
PTEN (10q23)
Polypose juvénile AD ? (rare) Estomac, côlon, pancréas Polypes juvéniles gastro-intestinaux SMAD4 (18q21)
BMPR1A (10q22)
Xeroderma pigmentosum
AR ? (rare) Carcinomes basocellulaires,
mélanomes
Hyperpigmentation cutanée 8 gènes : XP A-
> G + XP variant
Anémie de Fanconi AR ? (rare) Leucémies, hépatocarcinomes Insuffisance médullaire FANCA,C,D2,E,G
Syndrome de Bloom AR ? (rare) Leucémies, lymphomes Déficit immunitaire BLM (15q26)

18 Tumeurs malignes rares
pour le cancer colorectal (70 % chez l’homme et 40 % chez la femme) et à 40 % pour le cancer de l’en-domètre. Un sur-risque est identifié pour les cancers de l’ovaire, de l’intestin grêle, de l’urothélium (voies urinaires), des voies biliaires et de l’estomac avec des risques avant 70 ans de moins de 10 % (7).Chez les individus porteurs d’un gène MMR muté, il est conseillé de débuter entre 20 et 25 ans une surveillance par coloscopies utilisant une colo-ration type indigo carmin (chromo coloscopies), à répéter tous les deux ans en cas de normalité. Chez les femmes, une surveillance de l’endomètre annuelle, uniquement clinique ou associée à une échographie endovaginale est conseillée après 30 ans. La chirurgie prophylactique colorectale n’est pas recommandée (5).Le niveau de risque peu élevé pour les autres can-cers du syndrome HNPCC ne justifie d’aucune sur-veillance particulière en dehors de protocoles de recherche ; d’autant plus qu’il n’existe pas de mé-thodes de dépistage dont l’efficacité soit démon-trée. C’est en particulier le cas pour le cancer de l’ovaire ou des voies urinaires (5).À signaler, la possibilité d’observer dans certaines familles la présence de deux gènes MMR mutés à l’état homozygote ou hétérozygote composite confé-rant des risques majorés de cancer chez l’enfant, en particulier pour les hémopathies malignes (8).
Prédispositions héréditaires à des cancers rares
Mélanomes familiaux
Le mélanome cutané (survenant de novo ou sur nævus bénin préexistant) est héréditaire dans 8 à 12 % des cas. Deux gènes de prédisposition à trans-mission autosomique dominante ont été identi-fiés : CDKN2A (situé en 9p21) et CDK4 (situé en 12q13). Ils interviennent dans le cycle cellulaire. CDKN2A code pour deux protéines (par épissage alternatif) : INK4A (ou p16, voie Rb) et ARF (ou p14, voie P53). CDK4 inhibée par p16 intervient dans l’activation du cycle cellulaire (9, 10).La proportion de patients avec un mélanome ayant au moins un parent de premier degré atteint varie de 4 à 18 % selon les zones géographiques, témoin de taux d’incidence différents du mélanome. Un risque héréditaire pour le mélanome doit être évo-qué quand deux personnes apparentées au premier degré ont eu un mélanome ou quand trois person-nes ont eu un mélanome quel que soit leur degré de parenté.La fréquence des mutations du gène CDKN2A dans les familles ayant un contexte familial de mélano-mes évocateur est d’environ 20 %, variant de 25
à 50 % selon le nombre de cas (� 6), de patients avec mélanomes primitifs multiples (� 2) et l’âge précoce au diagnostic (< 40 ans). Elle n’est que de 0,2 % dans les mélanomes dits sporadiques (sans contexte familial) et de 5 à 10 % pour les cas de survenue précoce et/ou multiple. Les mutations germinales du gène CDK4 sont très rares, pro-duisant un « oncogène dominant » qui code pour une protéine résistant à l’effet inhibiteur de p16. Le déterminisme génétique reste inconnu dans une famille à mélanomes sur deux (9). Le risque cumulé de mélanome chez les porteurs du gène CDKN2A muté (appelé pénétrance) est estimé à 30 % à 50 ans et 70 % à 80 ans dans les familles avec au moins trois cas. Il existe des variations géo-graphiques (respectivement 13 % et 58 % en Euro-pe contre 50 % et 76 % aux États-Unis et 32 % et 91 % en Australie) qui sont corrélées à la fréquence du mélanome donc au niveau d’exposition au so-leil. D’autres facteurs de risque de mélanome tels les caractéristiques cutanées (réactions au soleil) et pigmentaires (nombre de nævi, nævus dyspla-siques) augmentent également la pénétrance. Par ailleurs, selon un effet propre, des variants du gène MC1R (impliqué dans le processus de pigmenta-tion) et en particulier ceux associés au phénotype RHC « red hair colour » (cheveux roux, peau claire, tâches de rousseur) modifient la pénétrance du gène CDKN2A (de 50 % à 84 % en Australie ; de 18 % à 35 % ou 55 % en Hollande) et induisent un âge au diagnostic plus jeune : 37 ans en moyenne contre 58 ans (9, 10). Le risque de cancer du pan-créas est augmenté chez les personnes porteuses d’un gène CDKN2A muté mais la pénétrance est encore mal estimée et pourrait être liée aux muta-tions impliquant ARF (11).Quand le mélanome familial est diagnostiqué, les patients atteints ainsi que leurs apparentés au pre-mier degré doivent bénéficier d’un examen de la peau annuel à partir de dix ans. Cette surveillance permet efficacement l’exérèse de lésions à risque ou de mélanomes de stade précoce dont semblent témoigner la moindre fréquence de second méla-nome chez les patients déjà atteints et l’impact sur la survie (10).En cas d’identification d’un gène de prédisposition muté (CDKN2A ou CDK4), la pratique de tests pré-symptomatiques dans la famille est possible mais reste controversée en raison d’un bénéfice encore incertain (12). En effet, les risques de cancer doi-vent être précisés. Par ailleurs, l’existence (démon-trée ou hautement probable) de facteurs modifica-teurs du risque de mélanome – génétiques (tels les variants du gène MC1R ou d’autres encore incon-nus) ou cliniques (phénotype RHC) – peut exposer les personnes non porteuses du gène CDKN2A ou CDK4 muté à un sur-risque persistant de mé-lanome. En conséquence, la surveillance doit être

Tumeurs rares et prédisposition génétique 19
maintenue chez ces personnes. Aussi, la pratique des tests génétiques de prédisposition héréditaire au mélanome doit-elle être effectuée au sein des consultations d’oncologie génétique dédiées et multidisciplinaires avec des protocoles de prise en charge bien établis et évalués.
Cancers du rein familiaux
Environ 2 à 3 % des tumeurs rénales entrent dans le cadre d’une prédisposition génétique (13). La nature histologique de la tumeur va orienter sur le ou les gènes de prédisposition analysables. Ainsi le cancer du rein à cellules claires affecte 40 à 60 % des patients atteints de maladie de von Hippel Lindau (VHL, OMIM 193300). C’est une maladie à transmission autosomique dominante, liée aux mutations germinales du gène suppresseur de tu-meur VHL (14). L’incidence est estimée à un nou-veau cas pour 36 000 naissances. Les manifesta-tions cliniques apparaissent habituellement entre 18 et 30 ans mais peuvent aussi se révéler dès l’en-fance. L’âge moyen de diagnostic du cancer du rein est de 39 ans. Il s’agit toujours de cancers du rein à cellules claires, le plus souvent bilatéraux et mul-tifocaux. La maladie de VHL prédispose aussi les sujets qui en sont porteurs aux kystes du rein, aux hémangioblastomes du système nerveux central et de la rétine, aux kystes et tumeurs endocrines du pancréas, aux phéochromocytomes et aux tumeurs du sac endolymphatique. Un dépistage génétique peut être proposé pour permettre la mise en place d’un bilan de surveillance annuel chez les porteurs du gène VHL muté. En dehors d’un examen clini-que approfondi, ce bilan doit comporter un fond d’œil, une échographie abdominale et un dosage des dérivés méthoxylés plasmatiques et/ou urinai-res à partir de l’âge de 5 ans. À partir de 15 ans, une IRM du système nerveux central tous les 3 à 5 ans est associée à ce bilan.Le cancer rénal papillaire peut être observé dans deux pathologies :– le cancer héréditaire papillaire de type 1 (OMIM
605074) de transmission autosomique domi-nante est dû à des mutations constitutionnelles activatrices du proto oncogène MET, qui code un récepteur tyrosine kinase (15). Il est caractérisé par le développement de carcinomes papillaires de type 1 bilatéraux et multifocaux qui se déve-loppent classiquement dans la 5e décade, mais peuvent être de survenue plus précoce. Une échographie rénale annuelle est recommandée chez les sujets à risque à partir de 30 ans ;
– la léiomyomatose héréditaire cutanée et utérine (OMIM 605839) est une affection à transmission autosomique dominante, due aux mutations inactivatrices du gène FH qui code la fumarate
hydratase (16). Cette maladie prédispose aux cancers du rein papillaire de type 2 qui sont des tumeurs de haut grade, de très mauvais pronos-tic, souvent métastatique au diagnostic. Dans la majorité des cas, la tumeur du rein est solitaire et unilatérale. L’âge moyen au diagnostic est de 44 ans, avec plusieurs cas décrits avant 30 ans. Le risque cumulé de cancers du rein en présence d’une mutation délétère du gène FH varie selon les études, entre 15 % et 24 %. Les léiomyomes cutanés et utérins qui caractérisent la maladie permettent un diagnostic vers 30 ans. Une sur-veillance annuelle doit être proposée chez les patients porteurs d’une mutation du gène FH avec un examen clinique dermatologique et gy-nécologique et une échographie abdominale à partir de 20 ans.
Le cancer du rein chromophobe isolé ou associé à un autre type tumoral dans une tumeur mixte doit faire évoquer un syndrome de Birt-Hogg-Dubé (OMIM 135150). Ce syndrome rare, à transmis-sion autosomique dominante, est dû aux muta-tions du gène suppresseur BHD ou FLCN (17) Le syndrome de Birt-Hogg-Dubé est caractérisé par des tumeurs cutanées bénignes du follicule pileux à type de fibrofolliculomes. Les autres manifesta-tions cliniques sont des kystes pulmonaires, des pneumothorax spontanés et des tumeurs rénales de différents types histologiques (cancers chromo-phobes le plus souvent, mais aussi oncocytomes et formes hybrides chromophobes-oncocytomes). Les cancers du rein à cellules claires et papillaires sont plus rares. Le risque de cancer du rein est multiplié par 7, l’âge de survenue est variable. Les tumeurs peuvent être uniques ou le plus souvent bilatérales et multifocales. Après un premier scan-ner thoracique lors du diagnostic, une surveillance pulmonaire clinique et rénale par une échographie abdominale annuelle doit être mise en place chez les patients porteurs d’une mutation du gène BHD à partir de l’âge de 18-20 ans.
Cancers de l’estomac familiaux
L’incidence du cancer de l’estomac est en constante diminution, liée principalement à une modification du mode de conservation des aliments et probable-ment à une baisse de la prévalence des infections par Helicobacter pylori. Elle ne concerne que le type dit intestinal, dans la classification de Lauren alors que le type diffus reste stable (18). Environ 5 à 10 % des cancers de l’estomac peuvent être expliqués par une prédisposition héréditaire. Il existe un risque majoré de cancer de l’estomac, essentiellement de type intestinal, dans plusieurs syndromes hérédi-taires bien caractérisés : le syndrome de Lynch ou syndrome HNPCC prédisposant aux cancers co-

20 Tumeurs malignes rares
lorectaux et aux cancers de l’endomètre, la polypose adénomateuse familiale, le syndrome de Peutz-Jeghers. Une augmentation de risque a été égale-ment décrite dans le syndrome de Li-Fraumeni. Il existe également des agrégations familiales de can-cer gastrique diffus, de survenue souvent précoce (âge moyen : 38 ans), histologiquement peu diffé-rentié et de haut grade, définissant un syndrome de prédisposition héréditaire de transmission autosomique dominante, le cancer gastrique dif-fus héréditaire (OMIM # 137215). Ses critères de reconnaissance ont été définis par un consortium international (International Gastric Cancer Linkage Consortium) et sont : (i) au moins deux cas de can-cer gastrique diffus chez des apparentés au premier ou au second degré, avec l’un des cas diagnostiqué avant 50 ans, ou (ii) au moins trois cas de cancer gastrique diffus chez des apparentés au premier ou au second degré, quel que soit l’âge au diagnostic (19). Une mutation germinale du gène CDH1, loca-lisé en 16q22.1 et codant pour la E-Cadhérine, une glycoprotéine transmembranaire intervenant dans l’adhésion des cellules épithéliales, est retrouvée dans 25 à 40 % des familles vérifiant les critères. Il est probable que d’autres gènes majeurs de prédis-position restent à découvrir. Une analyse du gène CDH1 doit être proposée à ces familles et conduite initialement chez les sujets atteints de cancer gas-trique diffus. Si une mutation est identifiée, les apparentés qui le souhaitent auront accès à un test pré-symptomatique visant à connaître leur statut génétique. Une gastrectomie totale prophylactique peut se discuter chez les porteurs de la mutation familiale, devant une pénétrance élevée des muta-tions de CDH1, le risque cumulé de cancer de l’esto-mac diffus est estimé à 70 % (20), et devant la diffi-culté à dépister en endoscopie ce cancer de mauvais pronostic qui se développe en sous-muqueux. Il s’agit néanmoins d’une intervention chirurgicale qui se grève d’une lourde morbidité. L’alternative est un dépistage par gastroscopie avec biopsies à réaliser tous les six mois à partir de l’âge de 20 ans (21). Chez la femme ayant une mutation de CDH1, un risque majoré de cancer du sein et notamment de carcinome lobulaire, dont le risque cumulé à 70 ans est estimé à 30 %, justifie de proposer une surveillance mammaire étroite avec un début du dépistage par mammographies et échographies voire IRM mammaires annuelles à 35 ans.
Sarcomes et syndrome de Li-Fraumeni
Le syndrome de Li-Fraumeni (LFS) est une prédis-position héréditaire rare exposant à une grande variété de tumeurs de survenue précoce ; elle est de transmission autosomique dominante (OMIM # 151623).
Les sarcomes osseux ou des tissus mous, les can-cers du sein chez la femme jeune, les tumeurs cé-rébrales et les corticosurrénalomes prédominent (spectre étroit) mais d’autres tumeurs peuvent se rencontrer, comme les leucémies et lymphomes, les tumeurs des gonades, les cancers pulmonaires, les cancers de l’estomac et les cancers colorectaux.Les critères cliniques de reconnaissance de ce syn-drome ont été initialement définis par l’associa-tion d’un cas de sarcome chez un sujet de moins de 45 ans, d’un cas de cancer avant 45 ans chez un apparenté au premier degré et d’un cas de cancer avant 45 ans ou un cas de sarcome quel que soit l’âge au diagnostic chez un apparenté au premier ou second degré. Ces critères très restrictifs ont été par la suite élargis et le groupe de travail français sur le LFS (sous l’égide du Groupe Génétique et Cancer de la FNCLCC) a proposé les critères sui-vants dits « critères de Chompret » : (i) cas index atteint d’une tumeur du spectre étroit avant l’âge de 36 ans et au moins un apparenté au premier ou au second degré atteint d’une tumeur du spectre étroit (sauf cancer du sein si le 1er cas en est un) avant l’âge de 46 ans ou (ii) cas index atteint de tumeurs primitives multiples dont deux appar-tiennent au spectre étroit et la première dévelop-pée avant l’âge de 36 ans ou (iii) cas index atteint d’un corticosurrénalome dans l’enfance (22). Ces critères définissent également les indications de recherche en biologie moléculaire.Une mutation germinale du gène TP53 localisé sur le chromosome 17p13.1 est retrouvée dans environ 70 % des familles évocatrices vérifiant les critères initiaux et 30 % des familles vérifiant les critères de Chompret (23). TP53 est un gène suppresseur de tumeur codant pour un facteur de transcription qui, en présence de lésions de l’ADN, va activer la synthèse de protéines impliquées dans la régula-tion du cycle cellulaire, l’apoptose et la réparation de l’ADN. Une origine maternelle ou paternelle de la mutation est le plus fréquemment retrouvée mais il existe des néomutations. Les pénétrances, ou risque cumulé sur la vie de développer un can-cer, sont élevées, estimées à 70 % chez l’homme, et 100 % chez la femme en raison du risque de cancer du sein (24). Le risque de développer un deuxième cancer, notamment radio-induit est élevé. Une grande variabilité d’expression du syndrome est observée, inter- et intra-familles, plaidant en fa-veur de facteurs génétiques modificateurs. L’iden-tification d’une mutation de TP53 permet de pro-poser des tests génétiques présymptomatiques aux apparentés majeurs qui le souhaitent ; ces tests ne devant pas être réalisés chez les enfants indemnes en raison d’un intérêt médical limité et du reten-tissement psychologique potentiel (25). La prise en charge médicale des sujets porteurs d’une mu-tation de TP53 est malheureusement limitée car

Tumeurs rares et prédisposition génétique 21
la plupart des tumeurs du spectre ne sont pas dé-tectables précocement par les examens d’image-rie classique, en dehors du cancer du sein chez la femme.Chez l’enfant, une surveillance clinique annuelle par un pédiatre connaissant le syndrome peut être proposée, afin d’éviter tout retard au diagnostic. À l’âge adulte, ce suivi clinique peut être poursuivi, en y associant chez la femme à partir de 20 ans un dépistage par IRM et échographies mammaires. La mammographie ne doit être qu’un examen de 2e intention en raison d’une densité mammaire im-portante chez les femmes jeunes limitant sa valeur prédictive et d’une radiosensibilité possible confé-rée par une mutation de P53.La précocité et la gravité de certaines tumeurs du LFS, pour lesquelles il n’existe pas d’examens de dépistage, justifient de proposer un diagnostic pré-natal aux sujets porteurs d’une mutation de TP53 ayant un projet parental.
Rétinoblastomes héréditaires
Le rétinoblastome est une tumeur embryonnaire maligne rare de la rétine, qui touche environ un en-fant sur 20 000 le plus souvent avant l’âge de 5 ans et unilatérale dans 60 % des cas (25). Le dévelop-pement de cette tumeur est dû à l’inactivation dans un rétinoblaste du gène RB1, premier gène suppresseur de tumeur cloné en 1986 et localisé sur le chromosome 13q14 (OMIM # 180200). C’est un élément régulateur négatif important du cycle cellulaire qui agit en réprimant la transcription des gènes nécessaires à la phase S.Environ 45 % des cas de rétinoblastome sont non héréditaires, liés à l’acquisition de mutations so-matiques au niveau des deux allèles du gène RB1 qui est alors inactivé. Ces événements sont très rares et, dans ce cas, la tumeur est unilatérale et unifocale, sporadique (sans contexte familial) et d’âge médian au diagnostic égal à 2 ans (26).Dans près de 55 % des cas, le rétinoblastome s’ex-plique par une prédisposition héréditaire de trans-mission autosomique dominante liée à une muta-tion germinale (constitutionnelle) d’un des deux allèles du gène RB1. L’âge au diagnostic est plus précoce, médiane de un an, et l’on observe volon-tiers des tumeurs bilatérales et/ou multifocales. Cette mutation germinale s’est produite d’une part lors de la formation du zygote : il s’agit alors d’une néomutation (près de 50 % des mutations germi-nales de RB1 surviennent de novo), qui ne sera pas retrouvée chez les parents de l’enfant, mais ce der-nier aura une probabilité de 50 % de la transmettre à sa descendance. D’autre part, cette mutation peut avoir été transmise par l’un des deux parents et l’on observe habituellement un contexte fami-
lial. La pénétrance ou le risque de développer un rétinoblastome si l’on est porteur d’une mutation germinale de RB1 est très élevée, de l’ordre de 90 % et elle est due à l’acquisition d’une altération so-matique sur le second allèle dans une ou plusieurs cellules de la rétine.Un conseil génétique doit être proposé devant tout cas de rétinoblastome, quel que soit l’âge de surve-nue et même en l’absence d’histoire familiale car 10 à 15 % des formes unilatérales, unifocales et non familiales sont d’origine héréditaire. Une ex-ploration complète du gène RB1 vise à rechercher une anomalie délétère afin d’apporter la preuve moléculaire d’une prédisposition et permettre ain-si la réalisation de tests présymptomatiques chez les apparentés (notamment dans la fratrie) et un diagnostic postnatal ou prénatal, voire préimplan-tatoire, en cas de nouveau projet parental des pa-rents ; et ce, même si la mutation n’est pas détectée chez les deux parents, car de rares cas de mosaïque germinale ont été rapportés entraînant un risque de récurrence dans la fratrie.La surveillance ophtalmologique des sujets por-teurs d’une mutation est très contraignante pen-dant les premières années de vie, elle se fait par fond d’œil (le plus souvent sous AG) en centre spé-cialisé et vise à augmenter les chances d’un traite-ment conservateur de l’œil et d’une préservation de la vision. Un examen est fait dès la 1re semaine de vie puis de façon mensuelle pendant 18 mois, puis tous les 3 mois jusqu’à 4 ans, tous les 6 mois jusqu’à 18 ans puis annuellement. Une surveillance clinique au long terme doit être également propo-sée compte tenu des risques élevés de développer une seconde tumeur comme un ostéosarcome ou un sarcome des tissus mous (risque de 10 % 15 ans après la première tumeur et de 20 % 30 ans après) voire un mélanome, une tumeur cérébrale ou un pinéaloblastome ; les risques sont accrus après une radiothérapie.Une analyse négative du gène RB1, sans mutation identifiée chez le cas index ne permet pas d’éli-miner une prédisposition héréditaire (sensibilité des techniques d’analyses < 100 % et possibilité de mosaïques somatiques rendant l’analyse san-guine négative). Le risque résiduel est estimé à environ 1 % en cas de rétinoblastome unilatéral et unifocal et justifie de proposer une surveillance ophtalmologique pour les frères et sœurs jusqu’à l’âge de 4 ans (26). Une analyse moléculaire indi-recte (étude des marqueurs du gène RB1) permet parfois de lever la surveillance lorsque le frère ou la sœur n’a aucun allèle en commun avec le cas in-dex (probabilité de 25 %). En revanche, le risque est de 100 % devant une tumeur bilatérale, mul-tifocale ou familiale, qui est à considérer comme une forme héréditaire et la surveillance de la fra-trie se poursuit à vie.

22 Tumeurs malignes rares
Prédispositions héréditaires aux tumeurs endocrines
La tumeur endocrine qui doit faire évoquer systé-matiquement une prédisposition héréditaire est le cancer médullaire de la thyroïde qui se présente dans 30 % des cas sous une forme familiale (27). Il s’intègre alors dans la néoplasie endocrinienne multiple de type 2 (NEM-2, OMIM 193300) qui prédispose aussi au développement de phéochro-mocytome et d’adénomes ou d’une hyperplasie des parathyroïdes. La NEM-2 est due à des muta-tions du proto-oncogène RET. Il existe une forte corrélation génotype-phénotype avec une forme plus précoce et grave de la maladie (NEM-2B) liée à une mutation dans l’exon 16 jusqu’à des formes isolées et plus tardives de cancer médullaire de la thyroïde isolées liées à des mutations des exons 13, 14 et 15. Un dépistage génétique est possible dès le plus jeune âge, en particulier pour les formes sé-vères : une thyroïdectomie est recommandée avant l’âge de 1 an aux porteurs de mutation de l’exon 16 et entre 2 et 6 ans pour les mutations des exons 10 et 11. Pour les autres mutations peuvent être dis-cutés une thyroïdectomie de principe vers 5 ans, 10 ans ou à l’adolescence ou une surveillance an-nuelle avec un dosage de calcitonine. Dans tous les cas, un bilan annuel sera fait à la recherche d’un phéochromocytome ou d’une hyperparathyroïdie à partir de l’âge de 15 ans.Le corticosurrénalome malin est rare et peut être associé à différents syndromes de predisposition (28) : au syndrome de Li-Fraumeni (mutations du gène TP53), au syndrome de Wiedemann-Bec-kwith (mutations du gène CDKNC1 ou unidisomie de 11p15), à une néoplasie endocrinienne multi-ple de type 1 (mutations du gène MEN1) ou à une polypose adénomateuse familiale (mutations du gène APC). En cas d’antécédents personnels ou fa-miliaux évocateurs de l’un de ces syndromes, une analyse génétique doit être effectuée à la recherche de mutations des gènes correspondants.Le phéochromocytome a une origine génétique dans 25 % des cas. Il s’intègre dans certains syn-dromes de prédisposition : la NEM-2, la maladie de VHL (voir le paragraphe sur les tumeurs rénales) et la neurofibromatose de type 1 (mutations du gè-ne NF1) où il est généralement associé à l’atteinte d’autres organes. Il peut aussi être présent de fa-çon isolée dans le cadre des paragangliomes fami-liaux dus aux mutations des gènes SDHB, SDHC ou SDHD. Même en l’absence d’histoire familiale ou d’autre lésion, une mutation est retrouvée chez 10 à 15 % des patients et une analyse génétique peut être proposée à tous les patients porteurs de phéochromocytome, quel que soit leur âge (29). Cette analyse sera orientée suivant le phénotype, les tumeurs malignes orientant en particulier vers
le gène SDHB. La surveillance recommandée sera fonction du syndrome familial identifié.Les tumeurs endocrines duodéno-pancréatiques sont l’apanage de la néoplasie endocrinienne multiple de type 1 (NEM-1) où elles sont obser-vées chez 50 % des patients et sont responsables de 50 % des décès (30). Les tumeurs les plus fré-quentes sont les tumeurs endocrines non fonc-tionnelles, les gastrinomes souvent multiples puis les insulinomes. La prévalence de la NEM-1 dans la population générale est estimée à 1/30 000. Quatre-vingt-dix pour-cent des patients ont une hyperparathyroïdie, 40 % un adénome hypophy-saire, 20 % une atteinte de la corticosurrénale (plus souvent une hyperplasie ou un adénome) et 5 à 10 % des tumeurs endocrines thymiques et bronchiques souvent malignes. Un dépistage gé-nétique peut être proposé dans les familles où une mutation du gène MEN1 a été identifiée à partir de l’âge de 10 ans. Un bilan métabolique et hormonal sera proposé aux sujets à risque adapté à l’âge du patient et aux signes cliniques présentés. Des tu-meurs pancréatiques affectent aussi 12 % des pa-tients avec une maladie de VHL.L’hyperparathyroïdie primaire est le plus souvent due à des adénomes ou une hyperplasie des para-thyroïdes qui peuvent être liés à une NEM-1 ou 2 ; dans 2 % des cas, elle est due à un carcinome parathyroïdien qui peut être dû aux mutations du gène suppresseur HRPT2 (31) aussi responsable d’ostéofibromes mandibulaires et de kystes du rein (OMIM 145001). Un diagnostic génétique peut être proposé.
Références
1. Eisinger F, Bressac B, Castaigne D et al. (2004) [Identification and management of hereditary predisposition to cancer of the breast and the ovary (update 2004)]. Bull Cancer 91: 219-37
2. Antoniou A, Pharoah PD, Narod S et al. (2003) Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 72: 1117-30
3. Palacios J, Robles-Frias MJ, Castilla MA, et al. (2008) The molecular pathology of hereditary breast cancer. Pathobiology 75: 85-94
4. Narod SA, Offit K (2005) Prevention and management of hereditary breast cancer. J Clin Oncol 23: 1656-63
5. Olschwang S, Bonaiti C, Feingold J et al. (2004) [Identification and management of HNPCC syndrome (hereditary non polyposis colon cancer), hereditary predisposition to colorectal and endometrial adenocarcinomas]. Bull Cancer 91: 303-15
6. Evans DG, Walsh S, Hill J, McMahon RT (2007) Strategies for identifying hereditary nonpolyposis colon cancer. Semin Oncol 34: 411-17
7. Alarcon F, Lasset C, Carayol J et al. (2007) Estimating cancer risk in HNPCC by the GRL method. Eur J Hum Genet 15: 831-6

Tumeurs rares et prédisposition génétique 23
8. Kruger S, Kinzel M, Walldorf C et al. (2008) Homozygous PMS2 germline mutations in two families with early-onset haematological malignancy, brain tumours, HNPCC-associated tumours, and signs of neurofibromatosis type 1. Eur J Hum Genet 16: 62-72
9. Bishop JN, Harland M, Randerson-Moor J, Bishop DT (2007) Management of familial melanoma. Lancet Oncol 8: 46-54
10. Hansson J (2008) Familial melanoma. Surg Clin North Am 88: 897-916
11. Goldstein AM, Chan M, Harland M et al. (2006) High-risk melanoma susceptibility genes and pancreatic cancer, neural system tumors, and uveal melanoma across GenoMEL. Cancer Res 66: 9818-28
12. Hansen CB, Wadge LM, Lowstuter K, et al. (2004) Clinical germline genetic testing for melanoma. Lancet Oncol 5: 314-9
13. Richard S, Lidereau R, Giraud S (2004) The growing family of hereditary renal cell carcinoma. Nephrol Dial Transplant 19: 2954-8
14. Lonser RR, Glenn GM, Walther M, et al. (2003) von Hippel-Lindau disease. Lancet 361: 2059-67
15. Schmidt LS, Nickerson ML, Angeloni D, et al. (2004) Early onset hereditary papillary renal carcinoma: germline missense mutations in the tyrosine kinase domain of the met proto-oncogene. J Urol 172: 1256-61
16. Grubb RL, III, Franks ME, Toro J et al. (2007) Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J Urol 177: 2074-9
17. Toro JR, Wei MH, Glenn GM et al. (2008) BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dube syndrome: a new series of 50 families and a review of published reports. J Med Genet 45: 321-31
18. Lynch HT, Grady W, Suriano G, Huntsman D (2005) Gastric cancer: new genetic developments. J Surg Oncol 90: 114-33
19. Park JG, Yang HK, Kim WH, et al. (2000) Report on the first meeting of the International Collaborative Group on Hereditary Gastric Cancer. J Natl Cancer Inst 92: 1781-2
20. Pharoah PD, Guilford P, Caldas C (2001) Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 121: 1348-53
21. Kaurah P, MacMillan A, Boyd N et al. (2007) Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA 297: 2360-72
22. Frebourg T, Abel A, Bonaiti-Pellie C et al. (2001) [Li-Fraumeni syndrome: update, new data and guidelines for clinical management]. Bull Cancer 88: 581-7
23. Bougeard G, Sesboue R, Baert-Desurmont S et al. (2008) Molecular basis of the Li-Fraumeni syndrome: an update from the French LFS families. J Med Genet 45: 535-8
24. Chompret A, Brugieres L, Ronsin M et al. (2000) P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer 82: 1932-7
25. Aerts I, Lumbroso-Le RL, Gauthier-Villars M, et al. (2006) Retinoblastoma. Orphanet J Rare Dis 1: 31
26. Doz F. (2006) [Retinoblatoma: a review]. Arch Pediatr 13: 1329-37
27. Niccoli-Sire P, Conte-Devolx B (2007) [Medullary thyroid carcinoma]. Ann Endocrinol 68: 325-31
28. Groussin L, Bertherat J, Gicquel C, et al. (2007) Insights into the molecular biology of adrenocortical tumors. Exp Clin Endocrinol Diabetes 115: 175-8
29. Amar L, Bertherat J, Baudin E et al. (2005) Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol 23: 8812-8
30. Chanson P, Murat A, Cadiot C, Calender A (2007) La néoplasie endocrinienne multiple de type 1. Traité d’Endocrinologie. Flammarion Médecine Sciences, Paris
31. Wang PF, Tan MH, Zhang C, et al. (2005) HRPT2, a tumor suppressor gene for hyperparathyroidism-jaw tumor syndrome. Horm Metab Res 37: 380-3





























